当前位置:
X-MOL 学术
›
Anal. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Systems-Level Annotation of a Metabolomics Data Set Reduces 25 000 Features to Fewer than 1000 Unique Metabolites
Analytical Chemistry ( IF 6.7 ) Pub Date : 2017-09-15 00:00:00 , DOI: 10.1021/acs.analchem.7b02380 Nathaniel G Mahieu 1 , Gary J Patti 1
Analytical Chemistry ( IF 6.7 ) Pub Date : 2017-09-15 00:00:00 , DOI: 10.1021/acs.analchem.7b02380 Nathaniel G Mahieu 1 , Gary J Patti 1
Affiliation
|
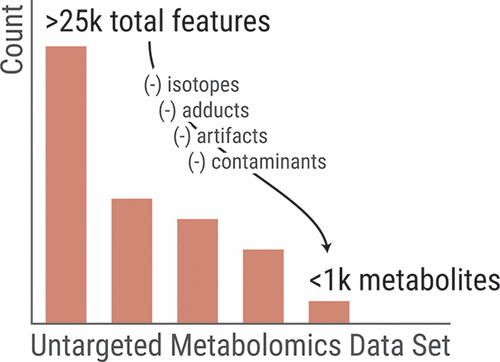
When using liquid chromatography/mass spectrometry (LC/MS) to perform untargeted metabolomics, it is now routine to detect tens of thousands of features from biological samples. Poor understanding of the data, however, has complicated interpretation and masked the number of unique metabolites actually being measured in an experiment. Here we place an upper bound on the number of unique metabolites detected in Escherichia coli samples analyzed with one untargeted metabolomics method. We first group multiple features arising from the same analyte, which we call “degenerate features”, using a context-driven annotation approach. Surprisingly, this analysis revealed thousands of previously unreported degeneracies that reduced the number of unique analytes to ∼2961. We then applied an orthogonal approach to remove nonbiological features from the data using the 13C-based credentialing technology. This further reduced the number of unique analytes to less than 1000. Our 90% reduction in data is 5-fold greater than previously published studies. On the basis of the results, we propose an alternative approach to untargeted metabolomics that relies on thoroughly annotated reference data sets. To this end, we introduce the creDBle database (http://creDBle.wustl.edu), which contains accurate mass, retention time, and MS/MS fragmentation data as well as annotations of all credentialed features.
中文翻译:

代谢组学数据集的系统级注释将 25000 个特征减少到少于 1000 个独特代谢物
当使用液相色谱/质谱 (LC/MS) 进行非靶向代谢组学时,现在可以常规检测生物样品中的数万个特征。然而,对数据的了解不足导致解释变得复杂,并掩盖了实验中实际测量的独特代谢物的数量。在这里,我们对使用一种非靶向代谢组学方法分析的大肠杆菌样品中检测到的独特代谢物的数量设定了上限。我们首先使用上下文驱动的注释方法对同一分析物产生的多个特征进行分组,我们将其称为“简并特征”。令人惊讶的是,这项分析揭示了数千种以前未报告的简并性,将独特分析物的数量减少到~2961。然后,我们采用正交方法,使用基于13 C 的认证技术从数据中删除非生物特征。这进一步将独特分析物的数量减少到不到 1000 个。我们的数据减少了 90%,比之前发表的研究多了 5 倍。根据结果,我们提出了一种依赖于彻底注释的参考数据集的非靶向代谢组学替代方法。为此,我们引入了 creDBle 数据库 (http://creDBle.wustl.edu),其中包含精确质量、保留时间和 MS/MS 碎片数据以及所有认证特征的注释。
更新日期:2017-09-15
中文翻译:
代谢组学数据集的系统级注释将 25000 个特征减少到少于 1000 个独特代谢物
当使用液相色谱/质谱 (LC/MS) 进行非靶向代谢组学时,现在可以常规检测生物样品中的数万个特征。然而,对数据的了解不足导致解释变得复杂,并掩盖了实验中实际测量的独特代谢物的数量。在这里,我们对使用一种非靶向代谢组学方法分析的大肠杆菌样品中检测到的独特代谢物的数量设定了上限。我们首先使用上下文驱动的注释方法对同一分析物产生的多个特征进行分组,我们将其称为“简并特征”。令人惊讶的是,这项分析揭示了数千种以前未报告的简并性,将独特分析物的数量减少到~2961。然后,我们采用正交方法,使用基于13 C 的认证技术从数据中删除非生物特征。这进一步将独特分析物的数量减少到不到 1000 个。我们的数据减少了 90%,比之前发表的研究多了 5 倍。根据结果,我们提出了一种依赖于彻底注释的参考数据集的非靶向代谢组学替代方法。为此,我们引入了 creDBle 数据库 (http://creDBle.wustl.edu),其中包含精确质量、保留时间和 MS/MS 碎片数据以及所有认证特征的注释。










































 京公网安备 11010802027423号
京公网安备 11010802027423号