当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
First-Principles Simulation of the (Li–Ni–Vacancy)O Phase Diagram and Its Relevance for the Surface Phases in Ni-Rich Li-Ion Cathode Materials
Chemistry of Materials ( IF 8.6 ) Pub Date : 2017-09-13 00:00:00 , DOI: 10.1021/acs.chemmater.7b02546 Hena Das 1, 2 , Alexander Urban 1, 2 , Wenxuan Huang 2, 3 , Gerbrand Ceder 1, 2, 3
Chemistry of Materials ( IF 8.6 ) Pub Date : 2017-09-13 00:00:00 , DOI: 10.1021/acs.chemmater.7b02546 Hena Das 1, 2 , Alexander Urban 1, 2 , Wenxuan Huang 2, 3 , Gerbrand Ceder 1, 2, 3
Affiliation
|
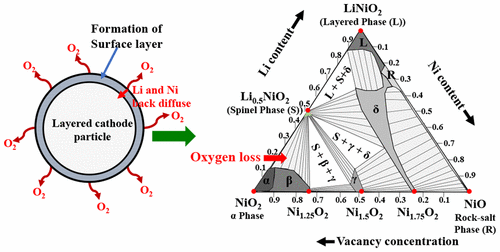
Despite several reports on the surface phase transformations from a layered to a disordered spinel and a rock-salt structure at the surface of the Ni-rich cathodes, the precise structures and compositions of these surface phases are unknown. The phenomenon, in itself, is complex and involves the participation of several contributing factors. Of these factors, transition metal (TM) ion migration toward the interior of the particle and hence formation of TM-densified surface layers, triggered by oxygen loss, is thermodynamically probable. Here, we simulate the thermodynamic phase equilibria as a function of TM ion content in the cathode material in the context of lithium nickel oxides, using a combined approach of first-principles density functional calculations, the cluster expansion method, and grand canonical Monte Carlo simulations. We developed a unified lattice Hamiltonian that accommodates not only rock-salt like structures but also topologically different spinel-like structures. Also, our model provides a foundation to investigate metastable cation compositions and kinetics of the phase transformations. Our investigations predict the existence of several Ni-rich phases that were, to date, unknown in the scientific literature. Our simulated phase diagrams at finite temperature show a very low solubility range of the prototype spinel phase. We find a partially disordered spinel-like phase with far greater solubility that is expected to show very different Li diffusivity compared to that of the prototype spinel structure.
中文翻译:

富镍锂离子正极材料中(Li-Ni-空位)O相图的第一性原理模拟及其与表面相的相关性
尽管关于富镍阴极表面从层状尖晶石到无序尖晶石和岩石盐结构的表面相转变的报道,这些表面相的精确结构和组成是未知的。这种现象本身是复杂的,并且涉及多个促成因素的参与。在这些因素中,过渡金属(TM)离子向颗粒内部迁移并因此在热力学上可能是由氧流失引起的TM致密化表面层的形成。在这里,我们使用第一原理密度函数计算,簇扩展方法和大经典蒙特卡洛模拟的组合方法,模拟了锂镍氧化物背景下热力学相平衡与阴极材料中TM离子含量的关系。 。我们开发了一个统一的晶格哈密顿量,它不仅可以容纳类似盐岩的结构,而且还可以容纳拓扑上不同的尖晶石状的结构。同样,我们的模型为研究亚稳阳离子组成和相变动力学提供了基础。我们的研究预测了迄今在科学文献中还未知的几个富镍相的存在。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们的模型为研究亚稳阳离子组成和相变动力学提供了基础。我们的研究预测了迄今在科学文献中还未知的几个富镍相的存在。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们的模型为研究亚稳阳离子组成和相变动力学提供了基础。我们的研究预测了迄今在科学文献中还未知的几个富镍相的存在。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。
更新日期:2017-09-14
中文翻译:
富镍锂离子正极材料中(Li-Ni-空位)O相图的第一性原理模拟及其与表面相的相关性
尽管关于富镍阴极表面从层状尖晶石到无序尖晶石和岩石盐结构的表面相转变的报道,这些表面相的精确结构和组成是未知的。这种现象本身是复杂的,并且涉及多个促成因素的参与。在这些因素中,过渡金属(TM)离子向颗粒内部迁移并因此在热力学上可能是由氧流失引起的TM致密化表面层的形成。在这里,我们使用第一原理密度函数计算,簇扩展方法和大经典蒙特卡洛模拟的组合方法,模拟了锂镍氧化物背景下热力学相平衡与阴极材料中TM离子含量的关系。 。我们开发了一个统一的晶格哈密顿量,它不仅可以容纳类似盐岩的结构,而且还可以容纳拓扑上不同的尖晶石状的结构。同样,我们的模型为研究亚稳阳离子组成和相变动力学提供了基础。我们的研究预测了迄今在科学文献中还未知的几个富镍相的存在。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们的模型为研究亚稳阳离子组成和相变动力学提供了基础。我们的研究预测了迄今在科学文献中还未知的几个富镍相的存在。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们的模型为研究亚稳阳离子组成和相变动力学提供了基础。我们的研究预测了迄今在科学文献中还未知的几个富镍相的存在。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。我们在有限温度下的模拟相图显示出原型尖晶石相的溶解度范围非常低。我们发现部分无序的尖晶石样相具有更高的溶解度,与原型尖晶石结构相比,有望表现出截然不同的Li扩散率。


























 京公网安备 11010802027423号
京公网安备 11010802027423号