当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Study of Halide Perovskite-Derived A2BX6 Inorganic Compounds: Chemical Trends in Electronic Structure and Structural Stability
Chemistry of Materials ( IF 7.2 ) Pub Date : 2017-09-11 00:00:00 , DOI: 10.1021/acs.chemmater.7b02013 Yao Cai 1 , Wei Xie 1 , Hong Ding 1 , Yan Chen 2, 3 , Krishnamoorthy Thirumal 2, 3 , Lydia H. Wong 2, 3 , Nripan Mathews 2, 3 , Subodh G. Mhaisalkar 2, 3 , Matthew Sherburne 1 , Mark Asta 1
Chemistry of Materials ( IF 7.2 ) Pub Date : 2017-09-11 00:00:00 , DOI: 10.1021/acs.chemmater.7b02013 Yao Cai 1 , Wei Xie 1 , Hong Ding 1 , Yan Chen 2, 3 , Krishnamoorthy Thirumal 2, 3 , Lydia H. Wong 2, 3 , Nripan Mathews 2, 3 , Subodh G. Mhaisalkar 2, 3 , Matthew Sherburne 1 , Mark Asta 1
Affiliation
|
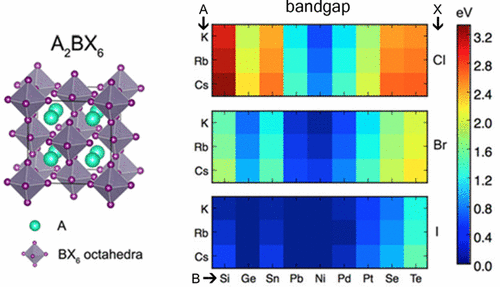
The electronic structure and energetic stability of A2BX6 halide compounds with the cubic and tetragonal variants of the perovskite-derived K2PtCl6 prototype structure are investigated computationally within the frameworks of density-functional-theory (DFT) and hybrid (HSE06) functionals. The HSE06 calculations are undertaken for seven known A2BX6 compounds with A = K, Rb, and Cs; and B = Sn, Pd, Pt, Te, and X = I. Trends in band gaps and energetic stability are identified, which are explored further employing DFT calculations over a larger range of chemistries, characterized by A = K, Rb, Cs, B = Si, Ge, Sn, Pb, Ni, Pd, Pt, Se, and Te; and X = Cl, Br, I. For the systems investigated in this work, the band gap increases from iodide to bromide to chloride. Further, variations in the A site cation influences the band gap as well as the preferred degree of tetragonal distortion. Smaller A site cations such as K and Rb favor tetragonal structural distortions, resulting in a slightly larger band gap. For variations in the B site in the (Ni, Pd, Pt) group and the (Se, Te) group, the band gap increases with increasing cation size. However, no observed chemical trend with respect to cation size for band gap was found for the (Si, Sn, Ge, Pb) group. The findings in this work provide guidelines for the design of halide A2BX6 compounds for potential photovoltaic applications.
中文翻译:

卤化物钙钛矿衍生的A 2 BX 6无机化合物的计算研究:电子结构和结构稳定性的化学趋势
在密度泛函理论(DFT)和杂化理论(HSE06)的框架内,通过计算研究了具有钙钛矿衍生的K 2 PtCl 6原型结构的立方和四方变体的A 2 BX 6卤化物的电子结构和能量稳定性。功能。针对七个已知的A 2 BX 6进行HSE06计算A = K,Rb和Cs的化合物; B = Sn,Pd,Pt,Te和X =I。确定了带隙和能量稳定性的趋势,并通过在更大范围内以D = A,K,Rb,Cs, B = Si,Ge,Sn,Pb,Ni,Pd,Pt,Se和Te;X = Cl,Br,I。对于这项工作中研究的系统,带隙从碘化物到溴化物再到氯化物增加。此外,A位阳离子的变化影响带隙以及四方畸变的优选程度。较小的A位阳离子(例如K和Rb)倾向于四方结构变形,从而导致带隙略大。对于(Ni,Pd,Pt)组和(Se,Te)组中B位的变化,带隙随阳离子尺寸的增加而增加。然而,对于(Si,Sn,Ge,Pb)组,未发现关于带隙阳离子大小的化学趋势。这项工作的发现为卤化物A的设计提供了指导。用于潜在光伏应用的2种BX 6化合物。
更新日期:2017-09-11
中文翻译:
卤化物钙钛矿衍生的A 2 BX 6无机化合物的计算研究:电子结构和结构稳定性的化学趋势
在密度泛函理论(DFT)和杂化理论(HSE06)的框架内,通过计算研究了具有钙钛矿衍生的K 2 PtCl 6原型结构的立方和四方变体的A 2 BX 6卤化物的电子结构和能量稳定性。功能。针对七个已知的A 2 BX 6进行HSE06计算A = K,Rb和Cs的化合物; B = Sn,Pd,Pt,Te和X =I。确定了带隙和能量稳定性的趋势,并通过在更大范围内以D = A,K,Rb,Cs, B = Si,Ge,Sn,Pb,Ni,Pd,Pt,Se和Te;X = Cl,Br,I。对于这项工作中研究的系统,带隙从碘化物到溴化物再到氯化物增加。此外,A位阳离子的变化影响带隙以及四方畸变的优选程度。较小的A位阳离子(例如K和Rb)倾向于四方结构变形,从而导致带隙略大。对于(Ni,Pd,Pt)组和(Se,Te)组中B位的变化,带隙随阳离子尺寸的增加而增加。然而,对于(Si,Sn,Ge,Pb)组,未发现关于带隙阳离子大小的化学趋势。这项工作的发现为卤化物A的设计提供了指导。用于潜在光伏应用的2种BX 6化合物。










































 京公网安备 11010802027423号
京公网安备 11010802027423号