当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab Initio Studies on Spectroscopic Constants for the HAsO Molecule
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1021/acs.jpca.7b01665 Qiushuang Xu 1 , Meishan Wang 1 , Yanliang Zhao 1 , Chuanlu Yang 1 , Xiaoguang Ma 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1021/acs.jpca.7b01665 Qiushuang Xu 1 , Meishan Wang 1 , Yanliang Zhao 1 , Chuanlu Yang 1 , Xiaoguang Ma 1
Affiliation
|
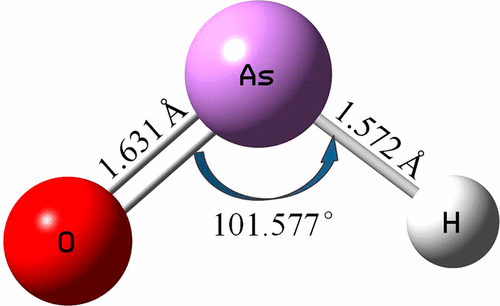
The anharmonic force fields and spectroscopic constants of the electronic ground state (X̃1A′) for the HAsO molecule are reported employing the MP2, B3LYP, B3P86, and B3PW91 methods with cc-pVQZ and cc-pV5Z basis sets. The calculated molecular geometries, rotational constants, vibrational frequencies, and anharmonic constants of the HAsO molecule are compared with the experimental data. It is found that the best agreement between the calculated results and experiment data is at the B3LYP/cc-pV5Z theoretical level. The predicted cubic and quartic force fields, vibration–rotation interaction constants, quartic and sextic centrifugal distortion constants, and Coriolis coupling constants of the HAsO molecule at the B3LYP/cc-pV5Z theoretical level are expected to be reliable.
中文翻译:

HAsO分子光谱常数的从头算研究
使用具有cc-pVQZ和cc-pV5Z基集的MP2,B3LYP,B3P86和B3PW91方法报道了HAsO分子的非基波力场和电子基态(X̃ 1 A')的光谱常数。将计算出的HAsO分子的分子几何形状,旋转常数,振动频率和非谐常数与实验数据进行比较。发现计算结果与实验数据之间的最佳一致性是在B3LYP / cc-pV5Z理论水平上。预计在B3LYP / cc-pV5Z理论水平上,HAsO分子的预测立方和四方力场,振动-旋转相互作用常数,四方和六方离心变形常数以及科里奥利耦合常数是可靠的。
更新日期:2017-09-08
中文翻译:
HAsO分子光谱常数的从头算研究
使用具有cc-pVQZ和cc-pV5Z基集的MP2,B3LYP,B3P86和B3PW91方法报道了HAsO分子的非基波力场和电子基态(X̃ 1 A')的光谱常数。将计算出的HAsO分子的分子几何形状,旋转常数,振动频率和非谐常数与实验数据进行比较。发现计算结果与实验数据之间的最佳一致性是在B3LYP / cc-pV5Z理论水平上。预计在B3LYP / cc-pV5Z理论水平上,HAsO分子的预测立方和四方力场,振动-旋转相互作用常数,四方和六方离心变形常数以及科里奥利耦合常数是可靠的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号