当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Improving Visualization and Interpretation of Metabolome-Wide Association Studies: An Application in a Population-Based Cohort Using Untargeted 1H NMR Metabolic Profiling
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1021/acs.jproteome.7b00344 Raphaële Castagné , Claire Laurence Boulangé 1 , Ibrahim Karaman , Gianluca Campanella , Diana L. Santos Ferreira , Manuja R. Kaluarachchi 1 , Benjamin Lehne , Alireza Moayyeri 2 , Matthew R. Lewis 1 , Konstantina Spagou 1 , Anthony C. Dona 1 , Vangelis Evangelos 3 , Russell Tracy 4 , Philip Greenland 5 , John C. Lindon 1, 6 , David Herrington 7 , Timothy M. D. Ebbels 1, 6 , Paul Elliott , Ioanna Tzoulaki , Marc Chadeau-Hyam
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1021/acs.jproteome.7b00344 Raphaële Castagné , Claire Laurence Boulangé 1 , Ibrahim Karaman , Gianluca Campanella , Diana L. Santos Ferreira , Manuja R. Kaluarachchi 1 , Benjamin Lehne , Alireza Moayyeri 2 , Matthew R. Lewis 1 , Konstantina Spagou 1 , Anthony C. Dona 1 , Vangelis Evangelos 3 , Russell Tracy 4 , Philip Greenland 5 , John C. Lindon 1, 6 , David Herrington 7 , Timothy M. D. Ebbels 1, 6 , Paul Elliott , Ioanna Tzoulaki , Marc Chadeau-Hyam
Affiliation
|
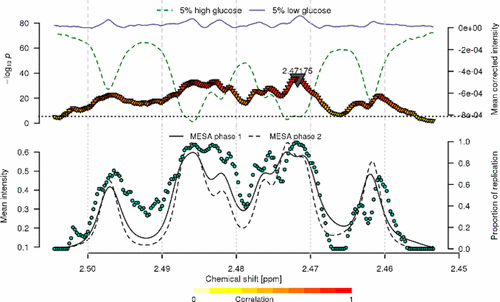
1H NMR spectroscopy of biofluids generates reproducible data allowing detection and quantification of small molecules in large population cohorts. Statistical models to analyze such data are now well-established, and the use of univariate metabolome wide association studies (MWAS) investigating the spectral features separately has emerged as a computationally efficient and interpretable alternative to multivariate models. The MWAS rely on the accurate estimation of a metabolome wide significance level (MWSL) to be applied to control the family wise error rate. Subsequent interpretation requires efficient visualization and formal feature annotation, which, in-turn, call for efficient prioritization of spectral variables of interest. Using human serum 1H NMR spectroscopic profiles from 3948 participants from the Multi-Ethnic Study of Atherosclerosis (MESA), we have performed a series of MWAS for serum levels of glucose. We first propose an extension of the conventional MWSL that yields stable estimates of the MWSL across the different model parameterizations and distributional features of the outcome. We propose both efficient visualization methods and a strategy based on subsampling and internal validation to prioritize the associations. Our work proposes and illustrates practical and scalable solutions to facilitate the implementation of the MWAS approach and improve interpretation in large cohort studies.
中文翻译:

改善代谢组学关联研究的可视化和解释:在人群中使用非靶向1 H NMR代谢谱分析的应用
生物流体的1 H NMR光谱生成可再现的数据,从而可以检测和定量大量人群中的小分子。现在已经建立了用于分析此类数据的统计模型,使用单变量代谢物组广泛关联研究(MWAS)分别研究光谱特征已成为多变量模型的一种计算有效且可解释的替代方法。MWAS依赖于代谢组宽显着性水平(MWSL)的准确估算,以用于控制家族的错误率。随后的解释需要有效的可视化和形式特征注解,进而要求对感兴趣的光谱变量进行有效的优先排序。使用人血清1来自多民族动脉粥样硬化研究(MESA)的3948名参与者的1 H NMR光谱图,我们针对血清葡萄糖水平进行了一系列的MWAS分析。我们首先提出了常规MWSL的扩展,该扩展可在不同模型参数化和结果的分布特征之间得出MWSL的稳定估计。我们提出了有效的可视化方法和基于子采样和内部验证的策略,以对关联进行优先级排序。我们的工作提出并说明了实用且可扩展的解决方案,以促进MWAS方法的实施并改善大型队列研究中的解释。
更新日期:2017-09-08
中文翻译:
改善代谢组学关联研究的可视化和解释:在人群中使用非靶向1 H NMR代谢谱分析的应用
生物流体的1 H NMR光谱生成可再现的数据,从而可以检测和定量大量人群中的小分子。现在已经建立了用于分析此类数据的统计模型,使用单变量代谢物组广泛关联研究(MWAS)分别研究光谱特征已成为多变量模型的一种计算有效且可解释的替代方法。MWAS依赖于代谢组宽显着性水平(MWSL)的准确估算,以用于控制家族的错误率。随后的解释需要有效的可视化和形式特征注解,进而要求对感兴趣的光谱变量进行有效的优先排序。使用人血清1来自多民族动脉粥样硬化研究(MESA)的3948名参与者的1 H NMR光谱图,我们针对血清葡萄糖水平进行了一系列的MWAS分析。我们首先提出了常规MWSL的扩展,该扩展可在不同模型参数化和结果的分布特征之间得出MWSL的稳定估计。我们提出了有效的可视化方法和基于子采样和内部验证的策略,以对关联进行优先级排序。我们的工作提出并说明了实用且可扩展的解决方案,以促进MWAS方法的实施并改善大型队列研究中的解释。










































 京公网安备 11010802027423号
京公网安备 11010802027423号