Bioorganic & Medicinal Chemistry ( IF 3.3 ) Pub Date : 2017-09-05 , DOI: 10.1016/j.bmc.2017.08.051 Melody D. Fulton , Laura E. Hanold , Zheng Ruan , Sneha Patel , Aaron M. Beedle , Natarajan Kannan , Eileen J. Kennedy
|
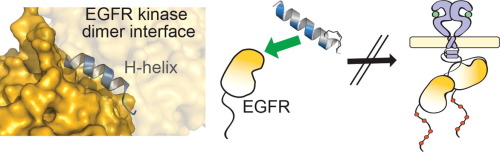
Although EGFR is a highly sought-after drug target, inhibitor resistance remains a challenge. As an alternative strategy for kinase inhibition, we sought to explore whether allosteric activation mechanisms could effectively be disrupted. The kinase domain of EGFR forms an atypical asymmetric dimer via head-to-tail interactions and serves as a requisite for kinase activation. The kinase dimer interface is primarily formed by the H-helix derived from one kinase monomer and the small lobe of the second monomer. We hypothesized that a peptide designed to resemble the binding surface of the H-helix may serve as an effective disruptor of EGFR dimerization and activation. A library of constrained peptides was designed to mimic the H-helix of the kinase domain and interface side chains were optimized using molecular modeling. Peptides were constrained using peptide “stapling” to structurally reinforce an alpha-helical conformation. Peptide stapling was demonstrated to notably enhance cell permeation of an H-helix derived peptide termed EHBI2. Using cell-based assays, EHBI2 was further shown to significantly reduce EGFR activity as measured by EGFR phosphorylation and phosphorylation of the downstream signaling substrate Akt. To our knowledge, this is the first H-helix-based compound targeting the asymmetric interface of the kinase domain that can successfully inhibit EGFR activation and signaling. This study presents a novel, alternative targeting site for allosteric inhibition of EGFR.
中文翻译:
构象受限的肽靶向变构激酶二聚体界面并抑制EGFR激活
尽管EGFR是一个非常受欢迎的药物靶标,但抑制剂的耐药性仍然是一个挑战。作为激酶抑制的另一种策略,我们试图探索是否可以有效地破坏变构激活机制。EGFR的激酶结构域通过头尾相互作用形成非典型的不对称二聚体,并且是激酶激活的必要条件。激酶二聚体界面主要由衍生自一种激酶单体的H-螺旋和第二单体的小叶形成。我们假设设计成类似于H-螺旋结合表面的肽可以作为EGFR二聚化和激活的有效破坏者。设计约束肽库以模拟激酶结构域的H-螺旋,并使用分子模型优化界面侧链。使用肽“钉合”限制肽以在结构上增强α-螺旋构象。肽钉被证明显着增强了称为EHBI2的H螺旋衍生肽的细胞渗透。使用基于细胞的测定法,进一步显示EHBI2可显着降低EGFR活性,如通过EGFR磷酸化和下游信号传递底物Akt的磷酸化所测量的。据我们所知,这是第一个靶向激酶结构域不对称界面的基于H螺旋的化合物,可成功抑制EGFR激活和信号传导。这项研究为EGFR的变构抑制提供了一个新颖的替代性靶向位点。肽钉被证明显着增强了称为EHBI2的H螺旋衍生肽的细胞渗透。使用基于细胞的测定法,进一步显示EHBI2可显着降低EGFR活性,如通过EGFR磷酸化和下游信号传递底物Akt的磷酸化所测量的。据我们所知,这是第一个靶向激酶结构域不对称界面的基于H螺旋的化合物,可成功抑制EGFR激活和信号传导。这项研究为EGFR的变构抑制提供了一个新颖的替代性靶向位点。肽钉被证明显着增强了称为EHBI2的H螺旋衍生肽的细胞渗透。使用基于细胞的测定法,进一步显示EHBI2可显着降低EGFR活性,如通过EGFR磷酸化和下游信号传递底物Akt的磷酸化所测量的。据我们所知,这是第一个靶向H结构域的化合物,该化合物靶向激酶结构域的不对称界面,可以成功抑制EGFR激活和信号传导。这项研究为EGFR的变构抑制提供了一个新颖的替代靶向位点。这是第一个针对激酶结构域不对称界面的基于H螺旋的化合物,该化合物可成功抑制EGFR激活和信号传导。这项研究为EGFR的变构抑制提供了一个新颖的替代性靶向位点。这是第一个针对激酶结构域不对称界面的基于H螺旋的化合物,该化合物可成功抑制EGFR激活和信号传导。这项研究为EGFR的变构抑制提供了一个新颖的替代性靶向位点。










































 京公网安备 11010802027423号
京公网安备 11010802027423号