当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine-Learning Methods Enable Exhaustive Searches for Active Bimetallic Facets and Reveal Active Site Motifs for CO2 Reduction
ACS Catalysis ( IF 11.3 ) Pub Date : 2017-08-31 00:00:00 , DOI: 10.1021/acscatal.7b01648 Zachary W. Ulissi 1, 2 , Michael T. Tang 1, 2 , Jianping Xiao 1, 2 , Xinyan Liu 1, 2 , Daniel A. Torelli 3, 4 , Mohammadreza Karamad 1 , Kyle Cummins 3 , Christopher Hahn 1, 2 , Nathan S. Lewis 3, 4 , Thomas F. Jaramillo 1, 2 , Karen Chan 1, 2 , Jens K. Nørskov 1, 2
ACS Catalysis ( IF 11.3 ) Pub Date : 2017-08-31 00:00:00 , DOI: 10.1021/acscatal.7b01648 Zachary W. Ulissi 1, 2 , Michael T. Tang 1, 2 , Jianping Xiao 1, 2 , Xinyan Liu 1, 2 , Daniel A. Torelli 3, 4 , Mohammadreza Karamad 1 , Kyle Cummins 3 , Christopher Hahn 1, 2 , Nathan S. Lewis 3, 4 , Thomas F. Jaramillo 1, 2 , Karen Chan 1, 2 , Jens K. Nørskov 1, 2
Affiliation
|
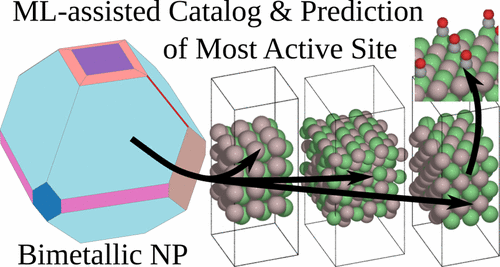
Bimetallic catalysts are promising for the most difficult thermal and electrochemical reactions, but modeling the many diverse active sites on polycrystalline samples is an open challenge. We present a general framework for addressing this complexity in a systematic and predictive fashion. Active sites for every stable low-index facet of a bimetallic crystal are enumerated and cataloged, yielding hundreds of possible active sites. The activity of these sites is explored in parallel using a neural-network-based surrogate model to share information between the many density functional theory (DFT) relaxations, resulting in activity estimates with an order of magnitude fewer explicit DFT calculations. Sites with interesting activity were found and provide targets for follow-up calculations. This process was applied to the electrochemical reduction of CO2 on nickel gallium bimetallics and indicated that most facets had similar activity to Ni surfaces, but a few exposed Ni sites with a very favorable on-top CO configuration. This motif emerged naturally from the predictive modeling and represents a class of intermetallic CO2 reduction catalysts. These sites rationalize recent experimental reports of nickel gallium activity and why previous materials screens missed this exciting material. Most importantly these methods suggest that bimetallic catalysts will be discovered by studying facet reactivity and diversity of active sites more systematically.
中文翻译:

机器学习方法可以穷举搜索活跃的双金属小面并显示活跃的中心图案以减少CO 2
双金属催化剂有望用于最困难的热和电化学反应,但是对多晶样品上许多不同的活性位点进行建模是一个开放的挑战。我们提出了一种以系统和预测的方式解决这种复杂性的通用框架。列举并分类了双金属晶体每个稳定的低折射率刻面的活性位点,产生了数百个可能的活性位点。使用基于神经网络的代理模型并行探索这些站点的活动,以在许多密度泛函理论(DFT)弛豫之间共享信息,从而使活动估计的显式DFT计算量减少了一个数量级。发现活动有趣的站点,并为后续计算提供了目标。在镍镓双金属化合物上显示2,表明大多数刻面具有与Ni表面相似的活性,但是少数暴露的Ni位置具有非常有利的顶部CO构型。该基序从预测模型中自然出现,代表一类金属间CO 2还原催化剂。这些站点合理化了最近关于镍镓活性的实验报告,以及为什么以前的材料筛选漏掉了这种令人兴奋的材料。最重要的是,这些方法表明,通过更系统地研究刻面反应性和活性位点的多样性,将发现双金属催化剂。
更新日期:2017-09-04
中文翻译:
机器学习方法可以穷举搜索活跃的双金属小面并显示活跃的中心图案以减少CO 2
双金属催化剂有望用于最困难的热和电化学反应,但是对多晶样品上许多不同的活性位点进行建模是一个开放的挑战。我们提出了一种以系统和预测的方式解决这种复杂性的通用框架。列举并分类了双金属晶体每个稳定的低折射率刻面的活性位点,产生了数百个可能的活性位点。使用基于神经网络的代理模型并行探索这些站点的活动,以在许多密度泛函理论(DFT)弛豫之间共享信息,从而使活动估计的显式DFT计算量减少了一个数量级。发现活动有趣的站点,并为后续计算提供了目标。在镍镓双金属化合物上显示2,表明大多数刻面具有与Ni表面相似的活性,但是少数暴露的Ni位置具有非常有利的顶部CO构型。该基序从预测模型中自然出现,代表一类金属间CO 2还原催化剂。这些站点合理化了最近关于镍镓活性的实验报告,以及为什么以前的材料筛选漏掉了这种令人兴奋的材料。最重要的是,这些方法表明,通过更系统地研究刻面反应性和活性位点的多样性,将发现双金属催化剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号