当前位置:
X-MOL 学术
›
Org. Chem. Front.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Density functional theoretical investigation of intramolecular proton transfer mechanisms in the derivatives of 3-hydroxychromone
Organic Chemistry Frontiers ( IF 4.6 ) Pub Date : 2017-06-13 00:00:00 , DOI: 10.1039/c7qo00367f Jin-Dou Huang 1, 2, 3, 4, 5 , Jianbin Zhang 5, 6, 7, 8 , Dengyi Chen 5, 7, 8, 9 , Huipeng Ma 5, 7, 8, 9
Organic Chemistry Frontiers ( IF 4.6 ) Pub Date : 2017-06-13 00:00:00 , DOI: 10.1039/c7qo00367f Jin-Dou Huang 1, 2, 3, 4, 5 , Jianbin Zhang 5, 6, 7, 8 , Dengyi Chen 5, 7, 8, 9 , Huipeng Ma 5, 7, 8, 9
Affiliation
|
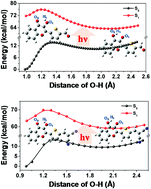
In this work, the intramolecular proton transfer (IPT) mechanisms of 5-(3-hydroxy-4-oxo-4H-chromen-2-yl)thiophene-2-carbaldehyde (3-HTCA) and 2-((5-(3-hydroxy-4-oxo-4H-chromen-2-yl)thiophen-2-yl)methylene)malononitrile (3-HTC-DiCN) have been systematically investigated. The constructed potential energy curves (PECs) of 3-HTCA and 3-HTC-DiCN in the ground state (S0) and first-excited singlet electronic state (S1) indicate that the ground state intramolecular proton transfer process is difficult to take place due to the high potential barriers (13.72 kcal mol−1 and 13.25 kcal mol−1 respectively); in comparison, the intramolecular proton transfer reaction occurs more readily with the H atom removing from the O atom of the O–H moiety to the O atom of the C![[double bond, length as m-dash]](http://www.rsc.org/images/entities/char_e001.gif) O moiety after photo-excitation. The relative size of the reaction barriers of 3-HTCA (4.24 kcal mol−1) and 3-HTC-DiCN (6.43 kcal mol−1) in the S1 state well explains the difference in their experimental fluorescence spectra. Based on the stable structures on PECs, the electronic spectra were simulated by the time-dependent density functional theory (TDDFT) method. The experimental absorption spectrum and fluorescence spectrum were well reproduced by calculating the vertical excitation energies of 3-HTCA and 3-HTC-DiCN. Moreover, the charge redistribution and charge-transfer character during photo-excitation were discussed in detail through the frontier molecular orbitals (FMOs) and natural bond orbital (NBO) analysis, which rationalizes the changes in the hydrogen bond strength and the ESIPT process of 3-HTCA and 3-HTC-DiCN.
O moiety after photo-excitation. The relative size of the reaction barriers of 3-HTCA (4.24 kcal mol−1) and 3-HTC-DiCN (6.43 kcal mol−1) in the S1 state well explains the difference in their experimental fluorescence spectra. Based on the stable structures on PECs, the electronic spectra were simulated by the time-dependent density functional theory (TDDFT) method. The experimental absorption spectrum and fluorescence spectrum were well reproduced by calculating the vertical excitation energies of 3-HTCA and 3-HTC-DiCN. Moreover, the charge redistribution and charge-transfer character during photo-excitation were discussed in detail through the frontier molecular orbitals (FMOs) and natural bond orbital (NBO) analysis, which rationalizes the changes in the hydrogen bond strength and the ESIPT process of 3-HTCA and 3-HTC-DiCN.
中文翻译:

3-羟基色酮衍生物分子内质子转移机理的密度泛函理论研究
在这项工作中,5-(3-hydroxy-4-oxo-4 H -chromen-2-yl)thiophene-2-carbaldehyde(3-HTCA)和2-((5-已经系统地研究了(3-羟基-4-氧代-4 H-铬-2-基)噻吩-2-基)亚甲基)丙二腈(3-HTC-DiCN)。在基态(S 0)和第一激发单重电子态(S 1)下建立的3-HTCA和3-HTC-DiCN的势能曲线(PEC)表明基态分子内质子转移过程难以进行由于高势垒(13.72 kcal mol -1和13.25 kcal mol -1分别); 相比之下,分子内质子转移反应更容易发生,其中光激发后H原子从OH部分的O原子移至C O部分的O原子。S 1中3-HTCA(4.24 kcal mol -1)和3-HTC-DiCN(6.43 kcal mol -1)的反应势垒的相对大小状态很好地解释了他们实验荧光光谱的差异。基于PECs上的稳定结构,通过时变密度泛函理论(TDDFT)方法模拟了电子光谱。通过计算3-HTCA和3-HTC-DiCN的垂直激发能,可以很好地再现实验吸收光谱和荧光光谱。此外,通过前沿分子轨道(FMO)和自然键轨道(NBO)分析,详细讨论了光激发过程中的电荷重新分布和电荷转移特性,合理化了氢键强度和ESIPT过程的变化[3]。 -HTCA和3-HTC-DiCN。
更新日期:2017-08-23
O moiety after photo-excitation. The relative size of the reaction barriers of 3-HTCA (4.24 kcal mol−1) and 3-HTC-DiCN (6.43 kcal mol−1) in the S1 state well explains the difference in their experimental fluorescence spectra. Based on the stable structures on PECs, the electronic spectra were simulated by the time-dependent density functional theory (TDDFT) method. The experimental absorption spectrum and fluorescence spectrum were well reproduced by calculating the vertical excitation energies of 3-HTCA and 3-HTC-DiCN. Moreover, the charge redistribution and charge-transfer character during photo-excitation were discussed in detail through the frontier molecular orbitals (FMOs) and natural bond orbital (NBO) analysis, which rationalizes the changes in the hydrogen bond strength and the ESIPT process of 3-HTCA and 3-HTC-DiCN.
中文翻译:
3-羟基色酮衍生物分子内质子转移机理的密度泛函理论研究
在这项工作中,5-(3-hydroxy-4-oxo-4 H -chromen-2-yl)thiophene-2-carbaldehyde(3-HTCA)和2-((5-已经系统地研究了(3-羟基-4-氧代-4 H-铬-2-基)噻吩-2-基)亚甲基)丙二腈(3-HTC-DiCN)。在基态(S 0)和第一激发单重电子态(S 1)下建立的3-HTCA和3-HTC-DiCN的势能曲线(PEC)表明基态分子内质子转移过程难以进行由于高势垒(13.72 kcal mol -1和13.25 kcal mol -1分别); 相比之下,分子内质子转移反应更容易发生,其中
光激发后H原子从OH部分的O原子移至C O部分的O原子。S 1中3-HTCA(4.24 kcal mol -1)和3-HTC-DiCN(6.43 kcal mol -1)的反应势垒的相对大小状态很好地解释了他们实验荧光光谱的差异。基于PECs上的稳定结构,通过时变密度泛函理论(TDDFT)方法模拟了电子光谱。通过计算3-HTCA和3-HTC-DiCN的垂直激发能,可以很好地再现实验吸收光谱和荧光光谱。此外,通过前沿分子轨道(FMO)和自然键轨道(NBO)分析,详细讨论了光激发过程中的电荷重新分布和电荷转移特性,合理化了氢键强度和ESIPT过程的变化[3]。 -HTCA和3-HTC-DiCN。










































 京公网安备 11010802027423号
京公网安备 11010802027423号