当前位置:
X-MOL 学术
›
Mol. Biosyst.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An efficient method to transcription factor binding sites imputation via simultaneous completion of multiple matrices with positional consistency
Molecular BioSystems Pub Date : 2017-07-06 00:00:00 , DOI: 10.1039/c7mb00155j Wei-Li Guo 1, 2, 3, 4, 5 , De-Shuang Huang 1, 2, 3, 4, 5
Molecular BioSystems Pub Date : 2017-07-06 00:00:00 , DOI: 10.1039/c7mb00155j Wei-Li Guo 1, 2, 3, 4, 5 , De-Shuang Huang 1, 2, 3, 4, 5
Affiliation
|
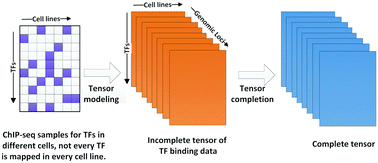
Transcription factors (TFs) are DNA-binding proteins that have a central role in regulating gene expression. Identification of DNA-binding sites of TFs is a key task in understanding transcriptional regulation, cellular processes and disease. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) enables genome-wide identification of in vivo TF binding sites. However, it is still difficult to map every TF in every cell line owing to cost and biological material availability, which poses an enormous obstacle for integrated analysis of gene regulation. To address this problem, we propose a novel computational approach, TFBSImpute, for predicting additional TF binding profiles by leveraging information from available ChIP-seq TF binding data. TFBSImpute fuses the dataset to a 3-mode tensor and imputes missing TF binding signals via simultaneous completion of multiple TF binding matrices with positional consistency. We show that signals predicted by our method achieve overall similarity with experimental data and that TFBSImpute significantly outperforms baseline approaches, by assessing the performance of imputation methods against observed ChIP-seq TF binding profiles. Besides, motif analysis shows that TFBSImpute preforms better in capturing binding motifs enriched in observed data compared with baselines, indicating that the higher performance of TFBSImpute is not simply due to averaging related samples. We anticipate that our approach will constitute a useful complement to experimental mapping of TF binding, which is beneficial for further study of regulation mechanisms and disease.
中文翻译:

通过同时完成多个具有位置一致性的矩阵来转录因子结合位点归因的有效方法
转录因子(TFs)是DNA结合蛋白,在调节基因表达中起着核心作用。TFs DNA结合位点的鉴定是理解转录调控,细胞过程和疾病的关键任务。染色质免疫沉淀后再进行高通量测序(ChIP-seq),可在体内进行全基因组鉴定TF结合位点。但是,由于成本和生物材料的可获得性,仍然难以在每个细胞系中绘制每个TF的图谱,这对基因调控的综合分析构成了巨大的障碍。为了解决此问题,我们提出了一种新颖的计算方法TFBSImpute,用于通过利用来自可用ChIP-seq TF绑定数据的信息来预测其他TF绑定配置文件。TFBSImpute将数据集融合到三模式张量,并通过以下方式插补丢失的TF绑定信号同时完成多个具有位置一致性的TF绑定矩阵。我们显示,通过评估针对观察到的ChIP-seq TF结合谱的插补方法的性能,我们的方法预测的信号与实验数据总体相似,并且TFBSImpute明显优于基线方法。此外,基序分析表明,与基线相比,TFBSImpute预制棒在捕获丰富的观察数据中的结合基序方面更好,与基线相比,这表明TFBSImpute的更高性能不仅仅是简单地对相关样本取平均值。我们预期我们的方法将为TF结合的实验作图提供有用的补充,这对进一步研究调节机制和疾病是有益的。
更新日期:2017-08-22
中文翻译:
通过同时完成多个具有位置一致性的矩阵来转录因子结合位点归因的有效方法
转录因子(TFs)是DNA结合蛋白,在调节基因表达中起着核心作用。TFs DNA结合位点的鉴定是理解转录调控,细胞过程和疾病的关键任务。染色质免疫沉淀后再进行高通量测序(ChIP-seq),可在体内进行全基因组鉴定TF结合位点。但是,由于成本和生物材料的可获得性,仍然难以在每个细胞系中绘制每个TF的图谱,这对基因调控的综合分析构成了巨大的障碍。为了解决此问题,我们提出了一种新颖的计算方法TFBSImpute,用于通过利用来自可用ChIP-seq TF绑定数据的信息来预测其他TF绑定配置文件。TFBSImpute将数据集融合到三模式张量,并通过以下方式插补丢失的TF绑定信号同时完成多个具有位置一致性的TF绑定矩阵。我们显示,通过评估针对观察到的ChIP-seq TF结合谱的插补方法的性能,我们的方法预测的信号与实验数据总体相似,并且TFBSImpute明显优于基线方法。此外,基序分析表明,与基线相比,TFBSImpute预制棒在捕获丰富的观察数据中的结合基序方面更好,与基线相比,这表明TFBSImpute的更高性能不仅仅是简单地对相关样本取平均值。我们预期我们的方法将为TF结合的实验作图提供有用的补充,这对进一步研究调节机制和疾病是有益的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号