当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Synthesis of Complex Glycolates by Enantioconvergent Addition Reactions
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2017-08-17 00:00:00 , DOI: 10.1021/acs.accounts.7b00263 Samuel L. Bartlett 1 , Jeffrey S. Johnson 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2017-08-17 00:00:00 , DOI: 10.1021/acs.accounts.7b00263 Samuel L. Bartlett 1 , Jeffrey S. Johnson 1
Affiliation
|
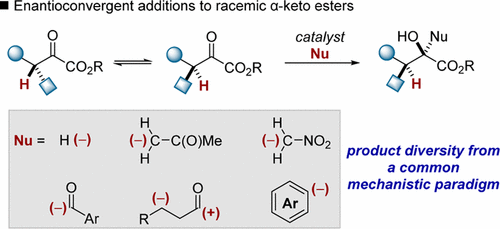
The unique role that stereochemistry plays in molecular recognition events continues to provide a driving force for synthesizing organic compounds in enantioenriched form. The tendency of enantioenriched organic compounds to revert to an entropically favored racemic state in the presence of viable racemization pathways (e.g., the enolization of stereogenic carbonyl derivatives) can sometimes interfere with this objective; however, beginning with Noyori’s foundational disclosure of a dynamic kinetic transfer hydrogenation, the ability to channel racemic, configurationally labile starting materials through stereoconvergent reaction pathways has been recognized as a powerful strategy in asymmetric synthesis. Proton transfer, retro-aldol, retro-Michael, reversible redox events, and other processes that can be deleterious to asymmetric synthesis are exploitable in enantioconvergent reactions using chiral small molecules and enzymes as asymmetric catalysts. Enantioselective reduction of configurationally labile carbonyl derivatives bearing a C–H acidic chiral center are particularly common. Because facile racemization is vital to stereocontrol in these transformations, hydrogenations of β-dicarbonyls are commonplace, while less activated substrates have been used less commonly. Our entry into enantioconvergent catalysis evolved from a long-standing interest in the synthesis of complex glycolates and began with the development of a general Noyori-type transfer hydrogenation of α-keto esters. Key innovations in this work include the identification of a new terphenylsulfonamide–Ru(II) complex, which displays unusual preference toward reduction of α-keto esters, and the observation that α-keto esters racemize under mildly basic conditions. This work was extended to the dynamic kinetic hydrogenation of racemic acyl phosphonates. Moreover, the recent recognition that the mechanistic paradigm underlying enantioconvergent hydrogenation chemistry can be extended to diverse carbon-centered nucleophiles has led to advances in the art. Our lab has developed a number of enantioconvergent tertiary alcohol syntheses. In the context of carbon-centered nucleophiles, we have focused on the use of α-keto esters; however, in the latter part of this Account, we will briefly describe our nascent efforts to develop dynamic kinetic additions of carbon-centered nucleophiles to β-oxo acid derivatives. While the enantioconvergent hydrogenation of β-keto acid derivatives is carried out on 100-ton scale annually, non-hydrogenative transformations of these compounds constitute an underexplored subclass of enantioconvergent reactions.
中文翻译:

对映体加成反应合成复杂的乙醇酸酯
立体化学在分子识别事件中发挥的独特作用继续为合成对映体富集形式的有机化合物提供驱动力。在存在可行的消旋化途径(例如,立体异构的羰基衍生物的烯醇化)的情况下,富含对映体的有机化合物趋向于恢复为熵有利的外消旋态的趋势有时会干扰这一目标。然而,从Noyori对动态动力学转移氢化的基础公开开始,通过立体收敛反应途径引导外消旋,构型不稳定的原料的能力已被认为是不对称合成的有效策略。质子转移,复古醛,复古迈克尔,可逆氧化还原事件,在使用手性小分子和酶作为不对称催化剂的对映收敛反应中,可以利用其他可能对不对称合成有害的方法。对映选择性地还原带有C–H酸性手性中心的结构不稳定的羰基衍生物。因为外消旋化对于这些转化中的立体控制至关重要,所以β-二羰基的氢化反应很普遍,而活化程度较低的底物则很少使用。我们对映体收敛催化的发展源于对复杂乙醇酸酯合成的长期关注,并始于对α-酮酯的一般Noyori型转移氢化的发展。这项工作的关键创新包括鉴定新的三苯磺酰胺-Ru(II)络合物,这显示出对还原α-酮酯的异常偏爱,并且观察到α-酮酯在温和的碱性条件下会消旋。这项工作扩展到外消旋酰基膦酸酯的动态动力学氢化。而且,最近的认识是,对映体收敛化学基础的机理范式可以扩展到各种以碳为中心的亲核试剂,这导致了本领域的进步。我们的实验室开发了许多对映体叔醇合成物。在以碳为中心的亲核试剂中,我们重点研究了α-酮酯的使用。但是,在本帐户的后半部分,我们将简要描述我们为开发将以碳为中心的亲核试剂动态动力学加成到β-氧代酸衍生物中而做出的新尝试。
更新日期:2017-08-17
中文翻译:
对映体加成反应合成复杂的乙醇酸酯
立体化学在分子识别事件中发挥的独特作用继续为合成对映体富集形式的有机化合物提供驱动力。在存在可行的消旋化途径(例如,立体异构的羰基衍生物的烯醇化)的情况下,富含对映体的有机化合物趋向于恢复为熵有利的外消旋态的趋势有时会干扰这一目标。然而,从Noyori对动态动力学转移氢化的基础公开开始,通过立体收敛反应途径引导外消旋,构型不稳定的原料的能力已被认为是不对称合成的有效策略。质子转移,复古醛,复古迈克尔,可逆氧化还原事件,在使用手性小分子和酶作为不对称催化剂的对映收敛反应中,可以利用其他可能对不对称合成有害的方法。对映选择性地还原带有C–H酸性手性中心的结构不稳定的羰基衍生物。因为外消旋化对于这些转化中的立体控制至关重要,所以β-二羰基的氢化反应很普遍,而活化程度较低的底物则很少使用。我们对映体收敛催化的发展源于对复杂乙醇酸酯合成的长期关注,并始于对α-酮酯的一般Noyori型转移氢化的发展。这项工作的关键创新包括鉴定新的三苯磺酰胺-Ru(II)络合物,这显示出对还原α-酮酯的异常偏爱,并且观察到α-酮酯在温和的碱性条件下会消旋。这项工作扩展到外消旋酰基膦酸酯的动态动力学氢化。而且,最近的认识是,对映体收敛化学基础的机理范式可以扩展到各种以碳为中心的亲核试剂,这导致了本领域的进步。我们的实验室开发了许多对映体叔醇合成物。在以碳为中心的亲核试剂中,我们重点研究了α-酮酯的使用。但是,在本帐户的后半部分,我们将简要描述我们为开发将以碳为中心的亲核试剂动态动力学加成到β-氧代酸衍生物中而做出的新尝试。










































 京公网安备 11010802027423号
京公网安备 11010802027423号