当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Kinetics of Electrocatalytic Reactions from First-Principles: A Critical Comparison with the Ab Initio Thermodynamics Approach
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2017-05-02 00:00:00 , DOI: 10.1021/acs.accounts.7b00077 Kai S. Exner 1, 2 , Herbert Over 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2017-05-02 00:00:00 , DOI: 10.1021/acs.accounts.7b00077 Kai S. Exner 1, 2 , Herbert Over 1
Affiliation
|
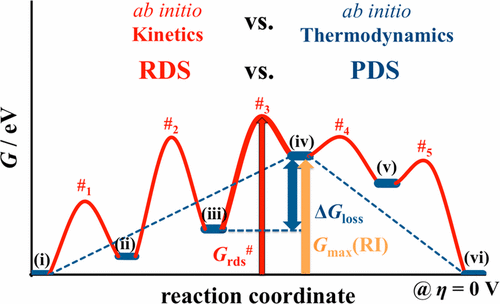
Multielectron processes in electrochemistry require the stabilization of reaction intermediates (RI) at the electrode surface after every elementary reaction step. Accordingly, the bond strengths of these intermediates are important for assessing the catalytic performance of an electrode material. Current understanding of microscopic processes in modern electrocatalysis research is largely driven by theory, mostly based on ab initio thermodynamics considerations, where stable reaction intermediates at the electrode surface are identified, while the actual free energy barriers (or activation barriers) are ignored. This simple approach is popular in electrochemistry in that the researcher has a simple tool at hand in successfully searching for promising electrode materials. The ab initio TD approach allows for a rough but fast screening of the parameter space with low computational cost. However, ab initio thermodynamics is also frequently employed (often, even based on a single binding energy only) to comprehend on the activity and on the mechanism of an electrochemical reaction. The basic idea is that the activation barrier of an endergonic reaction step consists of a thermodynamic part and an additional kinetically determined barrier. Assuming that the activation barrier scales with thermodynamics (so-called Brønsted–Polanyi–Evans (BEP) relation) and the kinetic part of the barrier is small, ab initio thermodynamics may provide molecular insights into the electrochemical reaction kinetics. However, for many electrocatalytic reactions, these tacit assumptions are violated so that ab initio thermodynamics will lead to contradictions with both experimental data and ab initio kinetics.
中文翻译:

第一性原理的电催化反应动力学:与从头算热力学方法的关键比较
电化学中的多电子过程要求在每个基本反应步骤之后,稳定电极表面的反应中间体(RI)。因此,这些中间体的结合强度对于评估电极材料的催化性能很重要。当前在现代电催化研究中对微观过程的理解主要由理论驱动,主要是基于从头算热力学的考虑,其中确定了电极表面的稳定反应中间体,而忽略了实际的自由能垒(或活化垒)。这种简单的方法在电化学中很流行,因为研究人员可以使用一个简单的工具来成功地寻找有前途的电极材料。从头开始TD方法允许以较低的计算成本对参数空间进行粗略但快速的筛选。然而,从头开始的热力学也经常被使用(通常,甚至仅基于单个结合能)来理解电化学反应的活性和机理。基本思想是,皮层反应步骤的激活壁垒由热力学部分和附加的动力学确定的壁垒组成。假设活化势垒随热力学成比例(所谓的布朗斯台德-波兰尼-埃文斯(BEP)关系),并且势垒的动力学部分很小,则从头算热力学可能会提供有关电化学反应动力学的分子见解。但是,对于许多电催化反应,
更新日期:2017-05-02
中文翻译:
第一性原理的电催化反应动力学:与从头算热力学方法的关键比较
电化学中的多电子过程要求在每个基本反应步骤之后,稳定电极表面的反应中间体(RI)。因此,这些中间体的结合强度对于评估电极材料的催化性能很重要。当前在现代电催化研究中对微观过程的理解主要由理论驱动,主要是基于从头算热力学的考虑,其中确定了电极表面的稳定反应中间体,而忽略了实际的自由能垒(或活化垒)。这种简单的方法在电化学中很流行,因为研究人员可以使用一个简单的工具来成功地寻找有前途的电极材料。从头开始TD方法允许以较低的计算成本对参数空间进行粗略但快速的筛选。然而,从头开始的热力学也经常被使用(通常,甚至仅基于单个结合能)来理解电化学反应的活性和机理。基本思想是,皮层反应步骤的激活壁垒由热力学部分和附加的动力学确定的壁垒组成。假设活化势垒随热力学成比例(所谓的布朗斯台德-波兰尼-埃文斯(BEP)关系),并且势垒的动力学部分很小,则从头算热力学可能会提供有关电化学反应动力学的分子见解。但是,对于许多电催化反应,










































 京公网安备 11010802027423号
京公网安备 11010802027423号