 柱层析(chromatography) 又称柱色谱,就是通常所说的过柱子,属于色谱法中使用最广泛的一种方法,也是有机实验中最有效的分离方法之一。但是对于很多初入此道的学生来说,过柱子经常会出现这样那样的问题。那么过柱子到底有哪些实用技巧呢?
柱层析(chromatography) 又称柱色谱,就是通常所说的过柱子,属于色谱法中使用最广泛的一种方法,也是有机实验中最有效的分离方法之一。但是对于很多初入此道的学生来说,过柱子经常会出现这样那样的问题。那么过柱子到底有哪些实用技巧呢?
 培养单晶是有机合成工作中的一个重要技能。很多朋友都觉得这一方面的知识和技术掌握起来比较困难。因此,请教一下各位有经验心得的朋友,能不能分享一下与单晶培养有关的技巧和窍门?
培养单晶是有机合成工作中的一个重要技能。很多朋友都觉得这一方面的知识和技术掌握起来比较困难。因此,请教一下各位有经验心得的朋友,能不能分享一下与单晶培养有关的技巧和窍门?
 据报道,长久以来,力致发光现象得到了科学界的广泛关注,然而在以往的尝试中,π-π堆积效应导致的聚集诱导淬灭效应严重限制了力致发光的研究进程。请问什么是力致发光?其机理是什么?
据报道,长久以来,力致发光现象得到了科学界的广泛关注,然而在以往的尝试中,π-π堆积效应导致的聚集诱导淬灭效应严重限制了力致发光的研究进程。请问什么是力致发光?其机理是什么?
 大学毕业,马上要开始研究生生活了,突然觉得很茫然,应该怎么做才能更好更快地适应呢?
大学毕业,马上要开始研究生生活了,突然觉得很茫然,应该怎么做才能更好更快地适应呢?
 编者按:本文作者是一位年轻且低调的有机化学博士,也是X-MOL的忠实用户,现在在国内大学任教。应X-MOL团队之邀,为低年级的师弟师妹们,特别是那些刚刚报道的研究生新人,分享一些他的心得和经验。X-MOL团队在此对这位不愿署名的作者表示感谢! 亲爱的师弟师妹们, 刚入学的喧闹还没有散去吧,或者说,新鲜劲还没...
编者按:本文作者是一位年轻且低调的有机化学博士,也是X-MOL的忠实用户,现在在国内大学任教。应X-MOL团队之邀,为低年级的师弟师妹们,特别是那些刚刚报道的研究生新人,分享一些他的心得和经验。X-MOL团队在此对这位不愿署名的作者表示感谢! 亲爱的师弟师妹们, 刚入学的喧闹还没有散去吧,或者说,新鲜劲还没...
 X射线衍射单晶结构分析是现代化学研究中的有力工具,也是很多化学工作者经常需要使用的手段,那么对于初入门的研究人员而言,进行单晶解析的软件主要有哪些?怎么使用?
X射线衍射单晶结构分析是现代化学研究中的有力工具,也是很多化学工作者经常需要使用的手段,那么对于初入门的研究人员而言,进行单晶解析的软件主要有哪些?怎么使用?
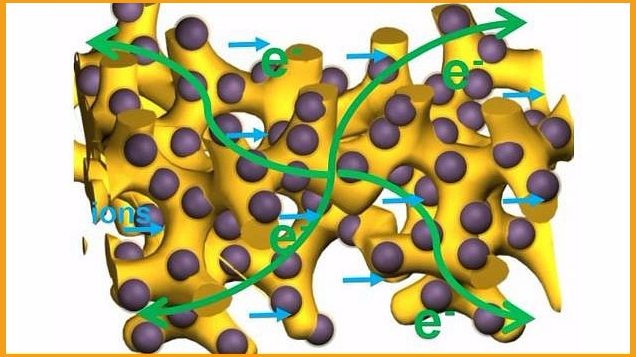 导电高分子凝胶是一种拥有广阔应用前景的新型材料,它既是一种有机导体,又继承了凝胶材料独特的三维网络结构,并由此产生独特的物理化学性质。那么请问导电高分子凝胶有哪些独特的物理化学性质?
导电高分子凝胶是一种拥有广阔应用前景的新型材料,它既是一种有机导体,又继承了凝胶材料独特的三维网络结构,并由此产生独特的物理化学性质。那么请问导电高分子凝胶有哪些独特的物理化学性质?
polylysine guilin peptide 回答了这个问题
 1 磷酸根-PO4 2 羧基-COOH 3 羟基-OH应用聚氨基酸时常需要氢键发挥作用http://www.polylysines.com/
1 磷酸根-PO4 2 羧基-COOH 3 羟基-OH应用聚氨基酸时常需要氢键发挥作用http://www.polylysines.com/
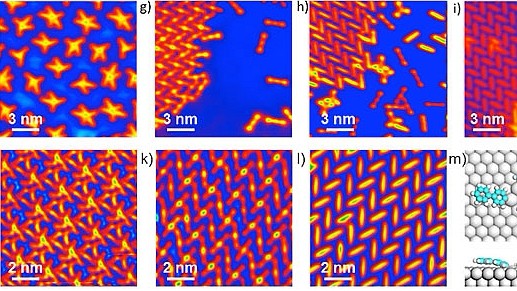 分子自组装(molecular self-assembly)广泛存在于自然界,是指分子组装基元通过非共价的弱相互作用自发形成有序结构的过程。发展分子自组装的方法学和实现分子自组装的功能化是人们在此领域开展研究的核心驱动力。那么请问目前调控分子自组装过程的方法有哪些?
分子自组装(molecular self-assembly)广泛存在于自然界,是指分子组装基元通过非共价的弱相互作用自发形成有序结构的过程。发展分子自组装的方法学和实现分子自组装的功能化是人们在此领域开展研究的核心驱动力。那么请问目前调控分子自组装过程的方法有哪些?
 一层正电荷一层负电荷一层正电荷。。。。。。http://www.polylysines.com/
一层正电荷一层负电荷一层正电荷。。。。。。http://www.polylysines.com/
 如何利用核磁共振谱法的技术来定量地计算有机反应的收率?
如何利用核磁共振谱法的技术来定量地计算有机反应的收率?
Heisen 博士研究生 广东海洋大学 回答了这个问题
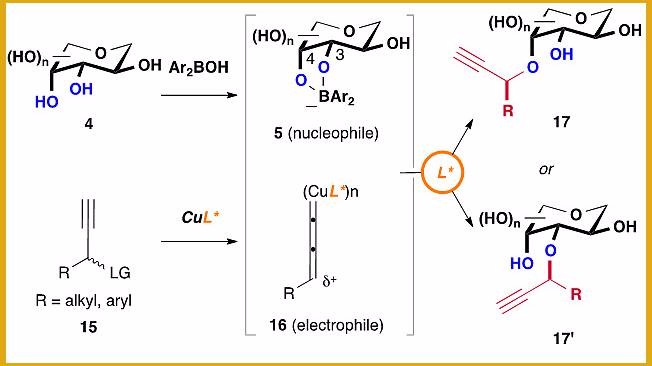 含糖化合物不仅广泛存在于自然界中,同时也是很多药物活性成分的重要组分。含糖的生物分子在分子识别、免疫、细胞凋亡以及肿瘤转移等生理学和病理学过程方面也起着重要的作用。选择性地修饰糖上特定位置的羟基,对探究糖类化合物的具体功能有着重要意义。请问如何对糖类化合物不同位点的羟基进行可调控地选择性修饰?
含糖化合物不仅广泛存在于自然界中,同时也是很多药物活性成分的重要组分。含糖的生物分子在分子识别、免疫、细胞凋亡以及肿瘤转移等生理学和病理学过程方面也起着重要的作用。选择性地修饰糖上特定位置的羟基,对探究糖类化合物的具体功能有着重要意义。请问如何对糖类化合物不同位点的羟基进行可调控地选择性修饰?
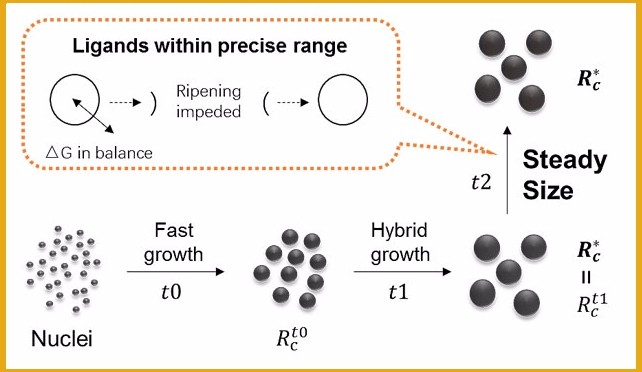 量子点作为新一代半导体纳米发光材料受到越来越多的重视,量子点材料的显示和照明产品具有更真实的色彩还原能力,合成过程中量子点发光波长的可控性是影响其生产成本的一个主要因素。请问如何在量子点生成过程中精确控制其尺寸?
量子点作为新一代半导体纳米发光材料受到越来越多的重视,量子点材料的显示和照明产品具有更真实的色彩还原能力,合成过程中量子点发光波长的可控性是影响其生产成本的一个主要因素。请问如何在量子点生成过程中精确控制其尺寸?
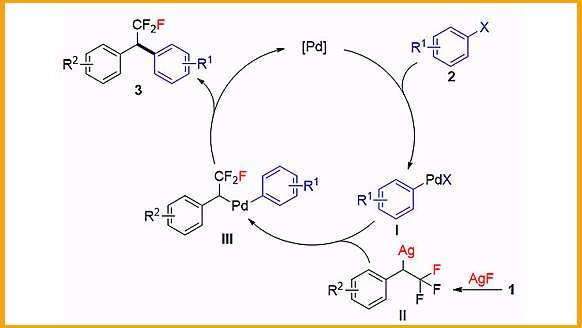 随着含氟有机化合物在医药、农药、功能材料等领域的广泛应用,建立新的策略合成含氟有机小分子已经成为化学工作者的研究热点。尤其是人们发现在化合物中引入三氟甲基可以显著改变其物理化学性质,如亲脂性、生物利用度等,因而三氟甲基化反应得到了广泛的关注。请问为什么在很多化合物中引入三氟甲基可以显著改变其物理化学性质?
随着含氟有机化合物在医药、农药、功能材料等领域的广泛应用,建立新的策略合成含氟有机小分子已经成为化学工作者的研究热点。尤其是人们发现在化合物中引入三氟甲基可以显著改变其物理化学性质,如亲脂性、生物利用度等,因而三氟甲基化反应得到了广泛的关注。请问为什么在很多化合物中引入三氟甲基可以显著改变其物理化学性质?
 细胞表面是指包围在细胞质外层的复合结构体系,是细胞与外界环境物质相互作用并产生各种复杂功能的部位。通过对细胞表面进行化学修饰,可以更好地了解细胞表面在细胞-细胞之间或者细胞-环境之间的关键作用。那么请问对细胞表面进行化学修饰的技术手段有哪些?
细胞表面是指包围在细胞质外层的复合结构体系,是细胞与外界环境物质相互作用并产生各种复杂功能的部位。通过对细胞表面进行化学修饰,可以更好地了解细胞表面在细胞-细胞之间或者细胞-环境之间的关键作用。那么请问对细胞表面进行化学修饰的技术手段有哪些?
 对各种水系电池来说,制约其寿命的短板几乎都在组成部件中的负极材料。目前用于水系电池的负极材料或多或少地存在着结构和化学稳定性的问题。那么为什么醌类有机材料作为通用负极材料能够大幅提高水系电池寿命?
对各种水系电池来说,制约其寿命的短板几乎都在组成部件中的负极材料。目前用于水系电池的负极材料或多或少地存在着结构和化学稳定性的问题。那么为什么醌类有机材料作为通用负极材料能够大幅提高水系电池寿命?
 半导体光催化材料因其能够矿化各种有机和无机的污染物而引起越来越多研究者的关注。有效提高光生电子和空穴的分离是提高半导体材料光催化性能的关键点。那么请问,如何从光催化活性物质的材料及结构的角度,提高光生电子和空穴的分离效率,从而提高光解水产氢的效率呢?
半导体光催化材料因其能够矿化各种有机和无机的污染物而引起越来越多研究者的关注。有效提高光生电子和空穴的分离是提高半导体材料光催化性能的关键点。那么请问,如何从光催化活性物质的材料及结构的角度,提高光生电子和空穴的分离效率,从而提高光解水产氢的效率呢?
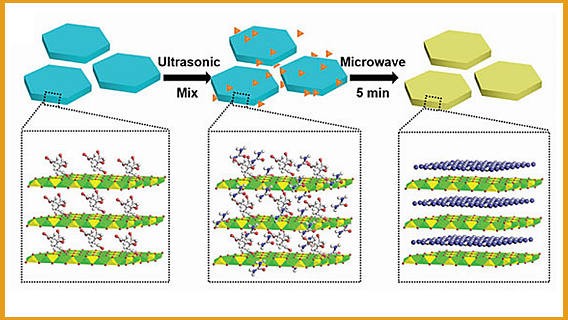 氮化碳作为一种二维碳材料具有匹配的能级结构,广泛应用于光催化领域,同时共轭的碳/氮结构使其拥有与生俱来的荧光特性。请问如何提高氮化碳材料的荧光量子产率?
氮化碳作为一种二维碳材料具有匹配的能级结构,广泛应用于光催化领域,同时共轭的碳/氮结构使其拥有与生俱来的荧光特性。请问如何提高氮化碳材料的荧光量子产率?


 京公网安备 11010802027423号
京公网安备 11010802027423号