
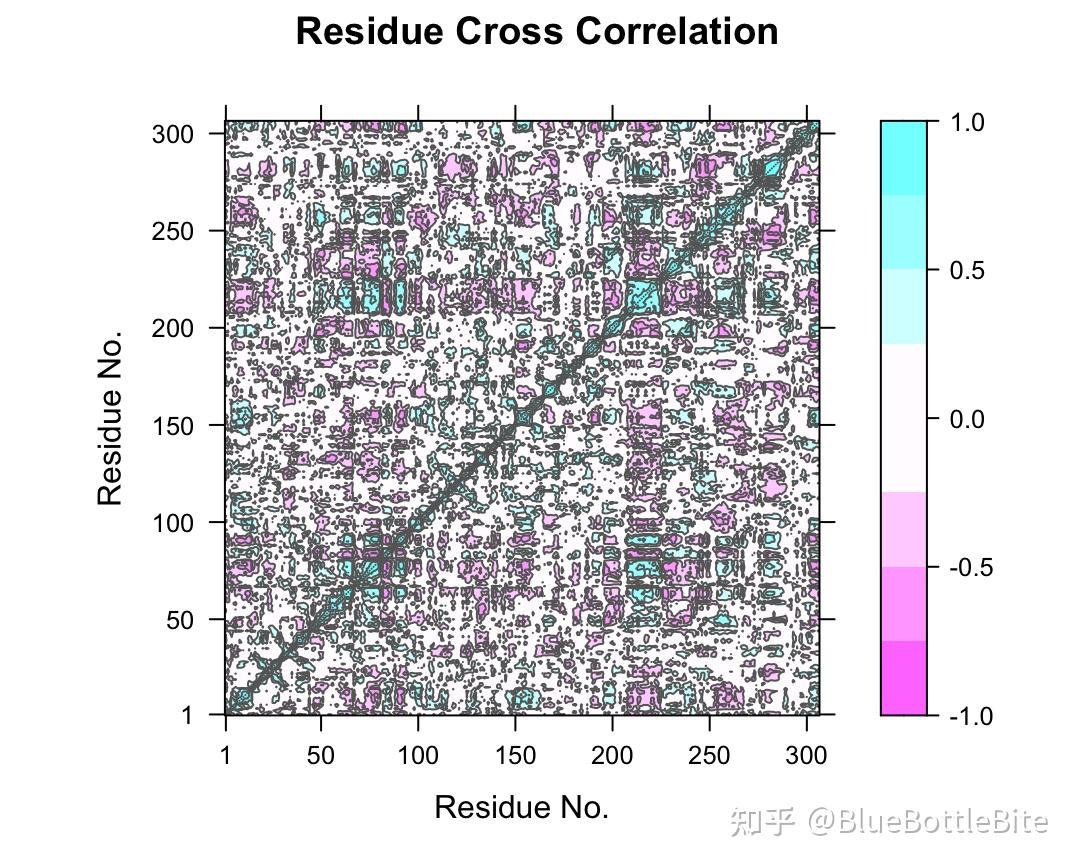
在完成一个模拟之后,如何分析和解读动力学模拟的输出文件是很重要的,事实上,解读和分析结果文件的能力与能够准备和运行模拟的能力是同等重要的。如下面的氨基酸相关图,可以用来分析不同位置的氨基酸之间是否存在关联运动,这对于理解和开发别构小分子抑制剂可提供帮助。
使用VMD等软件是可以完成分析任务的,但步骤缺琐碎,不够简约。
分析MD结果很重要的一个方面是结果的可视化,这包括了蛋白小分子互作视频的展示以及各种曲线图的制作,视频展示这里还是建议使用VMD/pymol等软件来完成。
图形的制作是很重要的一个目标,比如RMSD曲线可以告诉我们模拟是否达到平衡,RMSF曲线可以告诉我们氨基酸,特别是小分子结合位置的氨基酸的柔性信息。这些图形的制作最好既要反应模拟的科学性,也具备一定的美观性,以适于展示和发表。
这里介绍Bio3D来代替VMD,来处理MD轨迹文件, 使用R来制作所有的RMSD等曲线图。
这可以保证MD分析的流程化,简约化。兼顾图形/曲线的格式的美化需求,对于展示和发表,以及数据的可重复性等,有所帮助。
- 安装R 和 Rstudio,先安装R,再安装Rstudio,点击Rstudio打开界面
# 使用Bio3D分析分子动力学模拟的轨迹文件
# 在Rstudio界面的左侧光标闪动的位置,输入以下命令安装Bio3D
install.packages("bio3d", dependencies=TRUE)
# 测试Bio3D是否成功
library(bio3d)
help(package="bio3d")
vignette(package="bio3d")
# 在输入help 命令后,在右侧应当弹出帮助和说明,则安装成功
# 复现教程例子的曲线图,这里只复现md的图形,
# 这也是我们分析MD轨迹所需的主要的模块
# library(bio3d)
# demo("md")
# demo('pdb')
# demo只是演示,可以不用
#分析自己做的MD轨迹
library(bio3d)
# 定义自己的文件,修改到自己的文件夹下面
# Rstudio中,通过“Session -> set working directory”选择工作目录
# 然后,所有的工作操作中就直接写文件名即可
# 值得注意的是,只要在这里修改pdb和dcd文件,下面的一般不要更改,直接运行
mypdbfile <- "dynamics.z.pdb"
mydcdfile <- "dynamics.z.dcd"
# 将变量赋值给两个新的变量
dcd <- read.dcd(mydcdfile)
pdb <- read.pdb(mypdbfile)
# 如果我们想看pdb的序列信息
print(pdb)
# 打印出dcd文件
print(dcd)
# 做轨迹叠合,叠合以氨基酸的 Alpha 碳原子为基准,先选中阿尔法碳
ca.inds <- atom.select(pdb, elety="CA")
# 叠合,这里使用了一个模块,fit.xyz
xyz <- fit.xyz(fixed=pdb$xyz, mobile=dcd, fixed.inds=ca.inds$xyz, mobile.inds=ca.inds$xyz)
# 输入 dim(xyz) == dim(dcd),如果输出True 则叠合没有问题
dim(xyz) == dim(dcd)
# 输出RMSD 曲线图,
# RMSD是阿尔法碳原子坐标相对于初始坐标的根平均离散方差,
# 它反映了蛋白阿尔法碳原子在空间上随时间的扰动程度,
# 如果RMSD不再变化,说明模拟已经达到了一个相对平衡的状态
rd <- rmsd(xyz[1,ca.inds$xyz], xyz[,ca.inds$xyz])
plot(rd, typ="l", ylab="RMSD", xlab="Frame No.")
points(lowess(rd), typ="l", col="red", lty=2, lwd=2)

# RMSD 柱状分布图
hist(rd, breaks=40, freq=FALSE, main="RMSD Histogram", xlab="RMSD")
lines(density(rd), col="gray", lwd=3)

# 平均RMSD
summary(rd)
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.0000 0.3075 0.3150 0.3013 0.3340 0.3560
# RMSF 曲线图
# RMSF反应了阿尔法碳原子在一定时间内的,相对于其平均位置的离散程度
rf <- rmsf(xyz[,ca.inds$xyz])
plot(rf, ylab="RMSF", xlab="Residue Position", typ="l")
