
Abstract
The dearomative functionalization of aromatic compounds represents a fascinating but challenging transformation, as it typically needs to overcome a great kinetic barrier. Here, a catalyst-free dearomative rearrangement of o-nitrophenyl alkyne is successfully established by leveraging the remote oxygen transposition and a weak N-O bond acceleration. This reaction features high atom-, step- and redox-economy, which provides a divergent entry to a series of biologically important benzazepines and bridged polycycloalkanones. The reaction is proposed to proceed through a tandem oxygen transfer cyclization/(3 + 2) cycloaddition/(homo-)hetero-Claisen rearrangement reaction. The resulting polycyclic system is richly decorated with transformable functionalities, such as carbonyl, imine and diene, which enables diversity-oriented synthesis of alkaloid-like polycyclic framework.
Similar content being viewed by others
Introduction
The dearomative functionalization of aromatic compounds represents a fascinating transformation, which could rapidly build up stereocomplex 3-D alicyclic architectures with transformable C=C bonds from planar arenes in one step. Consequently, many successful approaches have been established during the last few decades1,2,3,4,5. From the perspective of arene substrates, most methods typically rely on low aromaticity arenes, such as phenol6,7, π-extended (hetero)arenes8,9, and hapto-coordinated arenes10. In terms of the thermodynamic profiles, highly active species such as cation11, carbene12,13,14,15, radical16, photoexcited intermediates17,18, and allyl organometallics;19,20 were usually applied in order to compensate the enthalpic penalty in breaking an aromatic system. In this vein, catalytic asymmetric dearomatization has become a powerful method in achieving optically pure molecules from arenes2,21,22,23,24. Additionally, redox manipulation of the arene could also lead the loss of aromaticity, but stoichiometric strong oxidants7,25,26 or reductants27,28,29 were required in many cases. Strangely, in spite of these achievements, there are only a few reports of dearomative arene functionalization by leveraging an aromatic [3,3]-sigmatropic rearrangement strategy5,30,31,32,33,34,35, which proceed through a dearomative intermediate. Several inherent challenges might result in the scarcity of such a dearomative [3,3]-rearrangement (Fig. 1a). Firstly, it is well-known that dearomatization processes have to overcome a prominent activation barrier (Fig. 1a, ΔG1 and ΔG3), which could not be sufficiently compensated from the enthalpy of sigma bond interchange during [3,3]-sigmatropic rearrangement. Secondly, the potential regioselectivity issues should be considered in developing such a reaction (Fig. 1a, paths a and b), so symmetrical or reactivity-differentiated substrates were usually applied36,37,38. Finally, the transiently formed dearomative intermediates are unstable and would easily collapse via rearomatization or consecutive [3,3]-sigmatropic rearrangement (Fig. 1a, product IV and VI)39. Recently, several excellent studies aiming to address the activity issues were achieved by developing new catalytic model8,32 or using energetic intermediates and substrates37,38,40,41. For example, Peng group disclosed a dearomative dual functionalization of aryl iodanes by integration a iodonio-Claisen rearrangement with a subsequent nucleophilic interception of the dearomative intermediate (Fig. 1b)38. Anderson and coworkers reported a tandem of (3 + 2) cycloaddition and hetero-Claisen rearrangement for the dearomative synthesis of spirocyclic 1-pyrrolines by exploiting the activity of nitrones and arynes (Fig. 1c)40. However, to the best of our knowledge, access to a bridged polycyclic system via dearomative [3,3]-sigmatropic rearrangement is still a yet unmet challenge. Inspired by these intriguing achievements and our successful experiences in dearomative Claisen rearrangement42,43,44, we intend to use the weak N-O bond (N-O: 53 kcal/mol vs C-O: 86 kcal/mol) as leverage to design the dearomative [3,3]-sigmatropic rearrangement, aiming for the synthesis of bridged alicyclic systems45.

a Analysis of the challenges in dearomative [3,3]-rearrangement. b Dual functionalization of aryl iodane. c Spirocyclic pyrroline synthesis. d Our design: dearomative [3,3]-rearrangement enabled bridged polycycle synthesis. PG = protective group.
In the past few decades, the (3 + 2) cycloadditions of nitrone have become a valid approach to assemble the N-O bond containing rings45, especially integrated with the alkyne chemistry46,47,48,49,50,51. For example, the nitroalkyne-based oxygen transfer could efficiently generate the nitrone, which can then be intercepted by an intermolecular alkene to give a polycyclic system48,49,50. Similarly, we surmised that an in-situ generated nitrone bearing a well-situated allene would undergo selective cycloaddition and the conformation of the newly formed ring would ensure the consequent dearomative sigmatropic rearrangement occurring selectively to give a stereochemically and functionally rich bridged polycyclic system (Fig. 1d).
In this work, we disclose a catalyst-free dearomative [3,3]-rearrangement of o-nitrophenyl alkyne for the divergent synthesis of benzazepines and bridged polycycloalkanones via remote oxygen transposition (over 11 atoms). This reaction features high atom-, step- and redox-economy. In addition, the resulting polycyclic system is constituted by four rings, two bridge-head quaternary centers, and transformable functionalities of cyclohexadiene and cyclodiketone, which enables the diversity-oriented synthesis of alkaloid-like fused polycyclic frameworks.
Results and discussion
Serendipitous discovery and rationalization
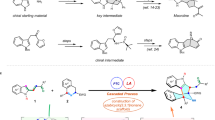
To verify our hypothesis, we set out the investigation with the easily available ynamide 1a as model substrate. Surprisingly, under thermolysis conditions, the desired bridged cycloalkanone was not detected, but an unknown compound with one carbon atom less (identified by NMR) was isolated in 81% yield (Fig. 2a, entry 3). The temperature has great impact on the reaction yields (Fig. 2a). The structure was tentatively assigned to benzazepine 2a by analysing of the NMR spectra, which was further confirmed later by the X-ray crystal diffraction analysis of the analogue 2k (Fig. 3). The formation of benzazepine 2a can be rationalized as shown in Fig. 2b. The thermal-triggered intramolecular oxygen transfer48,49,50 of 1a initially gave the nitrone III (Fig. 2b), which could be effectively trapped by the tethered allene with complete diastereoselectivity. The resulting (3 + 2) cycloaddition adduct IV then underwent the dearomative [3,3]-sigmatropic rearrangement to afford the bridged cycloalkanone 2a’, one molecule of carbon monoxide (CO) was further extruded through a cheletropic decarbonylation reaction (retro (4 + 1) reaction for simplicity)52,53,54,55,56 to regenerate the benzene ring and followed by isomerization to give the thermodynamically favourable product 2a. It is noteworthy that the CO gas could be detected by GC analysis of the gas in the reaction flask (see SI for details). The extrusion of CO was presumably due to the high tendency of rearomatization and high ring strain of the intermediate 2a’.

a Serendipitous discovery and reaction optimization. b Rationalization of the mechanism of the serendipitously observed 2a. c Selected representative drug molecules and important bioactive benzazepine scaffolds. Ts = p-toluenesulfonyl. Bn = benzyl.

aReaction conditions: The solution of 1 in THF (0.03 M) was stirred under N2 atmosphere at 90 oC for 6 h. bHeating the resulting filtrate of 1 after the ynamide formation. PG = protective group. Ts = p-toluenesulfonyl. Ms = methanesulfonyl.
Scope of the benzazepines
Encouraged by our initial discovery and the important bioactivity of benzazepines (Fig. 2c)57,58,59,60, we then started to test the generality of this reaction under the optimized conditions (Fig. 2a, entry 3). As shown in Fig. 3, the electronic nature of the arene has little influence on the formation of the benzazepines (2a-n) except the methylenedioxy-substituted product 2i. Both electron-rich and electron-deficient benzazepines were isolated in similar yields (about 70%). It is worth noting that the sterically hindered product 2n could obtained in 59% yield as well. Interestingly, the otherwise difficult to access pyridine-fused azepine 2o was also achieved in moderate yield (52%). In addition, the internal allene tethered substrates were subjected to the standard reaction conditions as well, giving the desired benzazepines 2p and 2q in 47% and 75% yield, respectively. Finally, the piperidine-fused benzazepine 2r and methylsulfonyl-protected benzazepine 2s could also be obtained in satisfactory yields.
Having realized the formation of benzazepines, we wondered if the originally desired bridged polycyclic system could be accessed through the stabilization of the transient dearomatized intermediate via minimization of ring strain (Fig. 4). As the ring strain will drastically reduce with the increasing of ring size, therefore, we tried to extend the [3.2.1]-bridged system to a [n.2.1]-bridged one (n > 3). Inspired by the homo-Nazarov cyclization reaction61,62,63 and also the mechanistic fact that the [3,3]-sigmatropic rearrangement might involve a diradical species39, replacing one C=C bond of the allene with a cyclopropane moiety64 may enable a homo-Claisen rearrangement and lead to the desired [4.2.1]-bridged polycyclic product 4 without CO extrusion. It is worthy to note that a radical mediated Brandi rearrangement65,66,67, in addition to the competitive retro (4 + 1) addition reaction, might be involved as well, which further complicated the reaction system.

The homo-hetero-Claisen rearrangement postulation inspired by mechanistic insight of Claisen rearrangement and the homo-Nazarov cyclization. PG = protective group.
Scope of the bridged polyclcloalkanones
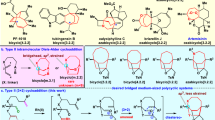
With these considerations in mind, we then set out to validate the feasibility of such hypothesis. Fortunately, when nitroalkyne 3a with a tailored methylenecyclopropane was applied as substrate, the envisioned dearomative bridged polycyclic product 4a was obtained in 74% yield, which was unambiguously confirmed by X-ray crystal diffraction analysis (Fig. 5). To the best of our knowledge, it is the first homo-hetero-Claisen rearrangement reaction. It is also noteworthy that in this cascade process the two oxygen atoms of the nitro group were transferred over 4 and 11 atoms, giving two transformable ketone functionalities in a redox-neutral manner. In line with our previous observation, the electronic properties and substituted patterns of the arene have little effect on the reaction performance (4a-p, 60-81%). For example, the nitrobenzenes with -CO2Me or -OMe group at the meta-position could deliver the desired products 4b and 4h in excellent yields. In addition, the methylenedioxy substituted product 4j could be obtained in 76% yield, which is in stark contrast to that of 2i (22%). Notably the formation of 4o needed a slightly elevated temperature and prolonged reaction time (80 oC, 36 h), which might be attributed to the steric hindrance. The practicability of the naphthalene and pyridine-based substrates were also tested to give the corresponding products 4q and 4r in satisfactory yields. The reaction was highly sensitive to the size of R2 as the yields decreased sharply when the group R2 was replaced by ethyl and phenyl, furnishing the corresponding products 4s and 4t in only 36% and 21% yield, respectively. When ynamide with a shorter tether were used as substrates, the desired products decomposed quickly upon chromatography isolation on silica gel. Fortunately, these in situ generated polycyclic diketones could be selectively reduced to the corresponding alcohols 4u’ and 4v’ in 68% and 45% yield, respectively. It is worthy to mention that the dimethyl amine substituted product 4v could be observed by NMR in 62% yield, but it was sensitive to silica gel and transformed to the ring fragmentation product 4w’ in 58% yield. In addition, methylsulfonyl- and o-nitrobenzenesufonyl protected polycyclic product 4x and 4y could also be obtained in good yield. In sharp contrast to the sulfonyl protected counterparts, the carbamate protected substrate 3z required more forcing conditions (100 oC for 120 h), delivering the desired bridged adduct 4z in 62% yield with the thermolabile tert-butoxycarbamate moiety survived. Interestingly, when the cyclobutene-containing substrate was tested, however, only the (3 + 2) adduct 4aa’ was isolated in excellent yield, which couldn’t be further transferred to the desired bridged cyclic product even elevating the reaction temperature to 200 oC, which indicated that the weak N-O bond is thermally stable and the cyclopropane is critical for the following rearrangement.

aReaction conditions: The solution of 3 in THF (0.03 M) was stirred under N2 atmosphere at 75 oC for 12 h; bHeating the resulting filtrate of 3 after the ynamide formation; c80 °C for 36 h; dNaBH4 (3 eq) and 0.5 mL MeOH were added when the rearrangement reaction mixture cooling to room temperature, then stirred for another 1 h. Note: these imine-containing bridged skeletons are quite stable, without obvious decomposition when placed under ambient atmosphere and temperature for more than 2 months. PG = protective group. Ts = p-toluenesulfonyl. Ms = methanesulfonyl. Ns = 2-nitrobenzenesulfonyl. Boc = t-butoxycarbonyl. THF = tetrahydrofuran.
The compatibility of other type substrates
To further explore the reaction scope, other type of nitroalkynes were tested as substrates as well (Fig. 6). Unexpectedly, a low conversion was achieved when the alkynyl thioethers were tested as the substrates under the standard conditions, which was in striking contrast to Verniest’s results48. When the reaction temperature was increased from 75 oC to 120 oC, the thioether 3ab exclusively afforded the endo-cycloaddition adduct 4ab’ rather than the desired bridged dione 4ab. Such selectivity inversion indicated that the steric repulsion between the Ts group and cyclopropane moiety is critical in ensuring the exo-cycloaddition and the subsequent rearrangement processes. We further envisioned that a shorter tether might alter the reaction selectivity in favoring exo-cycloaddition as the ring tension would increase dramatically for the endo-selectivity. Surprisingly, the thioether 3ac with one less carbon atom delivered a bridged-carbonyl group transposed product 4ac’ without observing 4ab’-like endo-adduct. In addition, the allene thioether 1t was also examined, which gave the benzazepines 2t and 2t’, which might arise from an asynchronous heterolysis54 of transient bridged product 4t. In addition to the substrates with heteroatom-modified and electronically biased carbon-carbon triple bond (ynamide and thioalkyne), the nitroalkynes 3ad and 3ae with carbon atom-capped carbon-carbon triple bond were explored as well. Interestingly, the substrate 3ad could transfer to 4ad’ and 4ad” upon exposure to air at room temperature over a period about two months. The structure of 4ad” was unambiguous validated by X-ray diffraction analysis. However, under thermal conditions, the substrate decomposed dramatically (>150 °C) with only trace amount of the desired products were observed by NMR analysis of the reaction mixture. The formation of the endo-adduct 4ad’ confirmed again the steric effect is very important, which is in line with the observation in 3ab. For the propiolamide 3a, the reaction is sluggish under heating conditions (150 oC for 8 h). A cooperative strategy by combined the gold catalysis49,50 and the thermolysis process was also applied. However, we could not detect the desired product 4ae, but with its descendant 4ae’ being isolated in 7% yield, which might proceed through the intermediate 4ae-Int with the assistance of gold coordination and nucleophilic chloride.

aThe solution of 3 in toluene (0.03 M) was stirred under N2 atmosphere at 110 or 120 oC for 12 h; bThe viscous oil 3ad (50 mg) in a 25 mL round bottom flask with a stopper was stood at room temperature for 60 days; cTo the solution of 3ae in PhMe (0.03 M) was added PicAuCl2 (0.2 eq) under nitrogen atmosphere, then stirred at 100 °C for 12 h. n.d. means not detected. Pic = 2-pyridinecarboxylate. Ts = p-toluenesulfonyl. Bn = benzyl.
Mechanistic study
To better understand the reaction mechanism, several control reactions were then conducted. Intuitively, we think that a loose radical pair involved pathway39 might be more feasible as tremendous transannular strain will built-up in achieving a synchronic [3,3]-sigmatropic rearrangement transition state. Therefore, the cyclopropane substituted substrate 1u was applied to probe the intermediacy of such radical pair. However, no ring opening products, such as 2u’, were detected, but with the normal benzazepine 2u being isolated in good yield (Fig. 7, Eq. 1). In addition, the attempt to intercept or terminate the envisioned radical species with TEMPO and Et3SiH also failed (Eq. 2). Finally, 3a was conducted on a gram scale in order to detect the potential Brandi cyclization product 4a’, which typically proceeded through a radical pathway65,66,67. Similarly, the normal rearrangement product 4a was obtained in 65% yield, while no desired Brandi cyclization product 4a’ was isolated even in trace amount (Eq. 3). These results indicated that the postulated radical pair either reluctant to be perturbated from outside or not existed. Introducing 4 equivalents of H218O to the reaction system has no effect on the reaction outcome (72%), which further confirmed its robustness. The HRMS and 13C-NMR of resulting product revealed that no 18O was incorporated (Eq. 4), which corroborated that the carbonyl groups came from the transposition of oxygen of nitroxide rather than external water. In addition, the reaction profiles were monitored by in situ IR spectroscopy, which characterized by the stretching vibration absorption of two carbonyl groups at 1720 cm−1 and 1775 cm−1, while no obvious absorption of intermediates were accumulated throughout, indicating their metastable character. Taken together, we are preferred more to a concerted but asynchronous [3,3]-sigmatropic rearrangement process wherein the N-O cleavage more advanced to relieve the incoming transannular strain.

a Control experiments: The Eqs. 1 and 2 indicate that no radical is involved in the (homo-)hetero-Claisen rearrangement. Eq. 3 is robust and radical mediated Brandi cyclization occurred, which imply the absence of radical species in the homo-hetero-Claisen rearrangement. b Investigation of the cascade reaction profiles by the in situ IR spectroscopy. THF = tetrahydrofuran. Ts = p-toluenesulfonyl.
With the bridged polycyclic compounds in hand, efforts to demonstrate the synthetic utility of them were then conducted (Fig. 8). The stereochemically and functionally rich polycyclic systems could be successfully converted into a series of alkaloid-like frameworks (5-14) by using very simple and practical procedures (6-7 steps in total from the commercially available starting material). Taking 4a as an example, the two carbonyl groups could be selectively differentiated under the Wittig reaction conditions to give the olefin 5 in 51% yield. Interestingly, the reduction of 4a with sodium borohydride gave alcohol 6 in 2.2:1 dr, while under the otherwise identical conditions except nickel chloride, a cage product 7 was obtained in high yield, which might arise from the selective ketone reduction, C=C bond hydration and acetalation processes. In addition, under the Luche reduction conditions, a similar cage product 8 with one C=C reserved was furnished in excellent yield. Under more forcing conditions, another cage compound 9 with the bridged carbonyl group being reduced could be isolated in 80% yield. These complex architectures were confirmed by the X-ray analysis (7, 8 and 9). In addition, selective reduction of one C=C bond of the cyclohexadiene could also be achieved with the Hantzsch ester as hydride donor, giving the product 10 in 91% yield. Epoxidation of the less sterically hindered C=C bond was feasible, but accompanied by the oxidation of imine motif. The stereochemistry of the epoxide 11 was also established by X-ray diffraction analysis. Reducing the loading of m-CPBA would selectively produce the kinetic product 12 in good yield. Under basic conditions in the presence of silica gel, a ring fragmentation product 13 was obtained in moderate yield. Exposing 13 to the acid condition led to a bis-spiral system 14, which was also confirmed by X-ray diffraction analysis. Interestingly, the spiral indolone 14 could also be obtained in 77% yield by treating 4a with trifluoroacetic acid at 90 oC for 6 h. The Boc-protected product 4z was labile to acidic condition, with the bridged architecture collapse to a planar molecule 15 at room temperature in quantitative yield. Finally, further transformation of benzazepine 2a was conducted to demonstrate its versatile capacity in diversification modification the benzazepine skeleton. As many of the vaptan class of drugs contain a N-linked benzoyl moiety57,68,69, the benzoylation product 16 was synthesized preferentially. Interestingly, the peripheral pyrrole unit could shift to another position under refluxing acidic environment to give 17 in good yield.

Selective olefination and reduction of the dione to give 5 and 6. Reductive acetalation companied with entirely reduction of the diene to afford the cage product 7. Luche reduction conditions could also produce the cage product 8 with the cyclohexadiene partially hydrogenated. The bridged carbonyl group could be reduced with lithium aluminium hydride to furnish 9. One C = C double bond of the cyclohexadiene was selectively reduced by Hantzsch ester to provide 10. Epoxidation product 11 was obtained with m-CPBA. Kinetically oxidation of the imine moiety to give nitrone 12. Retro Michael addition to give the fragmentation oxindole 13, which could transform to the spiral dione 14. Under the acidic deprotection conditions the three-dimensional architecture of 4z collapsed entirely to form a unique [6-6-5-6] tetracyclic architecture 15. Furthermore, the benzoyl functionality could be easily installed on free nitrogen atom of 2a to give product 16. Interestingly, compound 2a has been structurally reorganized into a [6-7-5] tricyclic architecture 17 by refluxing in TsOH/methanol solution. m-CPBA = m-chloroperoxybenzoic acid. TEA = triethylamine. TFA = trifluoroacetic Acid. DMAP = 4-(dimethylamino)pyridine. Ms = methylsulfonyl. Ts = p-toluenesulfonyl. Boc = t-butoxycarbonyl. NaBH4 = sodium borohydride. LiAlH4 = lithium aluminum hydride.
The antiviral activity
The approved anti-HIV drug, nevirapine, is a benzazepine analogue. Therefore, we firstly evaluated the chemotype 2 compounds (with benzazepine scaffold) using as nevirapine the positive control. As results, the chemotype 2 compounds only showed modest anti-HIV activity in SupT1 cells at 10 µM concentration (Table S3). To explore whether these unique architectures of 4 and its derivatives could serve as potential bioactive agents with novel core structure, we performed chemical structure similarity search in therapeutic target database (TTD) using the core structure of this series compound as the template, and four compounds were obtained with the Tanimoto similarity more than 0.7. The most similar compound Spiro[pyrrolidine-2,2-adamantane] (Tanimoto similarity was 0.745) was identified with the antiviral activity against influenza A virus (IAV) and the human immunodeficiency virus type 1 (HIV-1) and type 2 (HIV-2). Therefore, we explored the bioactivity of these compounds via testing the antiviral activity of IAV and HIV-1. As results, preliminary anti-HIV-1 assay indicated that compound 13 displayed the best anti-HIV effect with 97% reduction of VSV-G-pseudotyped HIV-1 virus in SupT1 cells at 10 µM concentration (Table S4). Further testing showed that 13 inhibited HIV-1 in a dose dependent manner (EC50 = 4.24 µM, Fig. 9a). However, the antiviral potency was measured in a Luc reporter assay which could not separate inhibition from the cytotoxicity. Therefore, the observed Luc reduction shown in Fig. 9a may largely reflect the cytotoxicity of 13 in SupT1 cell lines (CC50 = 8.22 µM, Fig. 9b). As for the antiviral activity against IAV, most of the compounds under preliminary test exhibited anti-IAV activity in 293T-GLUC cell at 10 µM concentration (Table S5), and 4a displayed the best anti-IAV effect with 92% reduction of IAV virus. Further activity inspection indicated 4a could inhibit the growth of IAV virus in dose dependent manner with the EC50 of 7.73 µM (Fig. 9c). Importantly, 4a exhibited low cytotoxicity (not showing cytotoxicity up to 50 µM) for 293T-GLUC cell (Fig. 9d), suggesting that the observed antiviral effect was not due to cytotoxicity. Taken together, this series compound would become a type of promising anti-virus agents with brand new scaffold.

a 13 inhibited the growth of HIV-1 virus in dose dependent manner in SupT1 cell; b 13 exhibited moderate cytotoxicity with CC50 of 8.22 µM for SupT1 cell; c 4a inhibited the growth of IAV virus in dose dependent manner in 293T-GLUC cell; d 4a exhibited low cytotoxicity for 293T-GLUC cell. (n = 3). Bar = mean. Error bars = ±SEM. Data are from at least three independent experiments and evaluated via Prism (version 5.01, GraphPad Software, San Diego, CA, USA).
In summary, we disclosed a dearomative rearrangement, which enable divergent accesses to a series of unique benzazepines and bridged polycycloalkanones in a redox-neutral, step- and atom-economic manner. The reaction was proposed to proceed through a tandem oxygen transfer cyclization/(3 + 2) cycloaddition/(homo-)hetero-Claisen rearrangement reaction, wherein an oxygen atom was eventually transposed over 11 atoms. The resulting bridged polycycloalkanones could be successfully converted into several alkaloid-like frameworks via late-stage diversification. The preliminary bioactivity investigation indicated that these unique bridged frameworks hold great potential to be a type of promising anti-virus agents with brand new scaffold.
Methods
General procedure for the synthesis of benzazepines and bridged polycycloalkanones
A dried 25 mL Schlenk tube was flushed with nitrogen three times. The solution of 1 (or 3) in the THF (0.03 M) was added to the tube under nitrogen atmosphere. The resulting mixture was put in a 90 °C (or 75 °C for 3) oil bath and stirred for 6 h (or 12 h for 3). After cooled to room temperature, the mixture was transferred to a 25 mL round bottom flask and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc as eluent) to give desired cyclization product 2 (or 4).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The materials and methods, experimental procedures, characterization data, 1H, 13C, 19F NMR spectra and mass spectrometry data are available in the Supplementary Information. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2092437 (2k), CCDC 2092438 (2u), CCDC 2092439 (4a), CCDC 2092440 (4t), CCDC 2131908 (4ad’), CCDC 2092441 (7), CCDC 2092442 (8), CCDC 2092443 (9), CCDC 2092444 (11) and CCDC 2092445 (14) and CCDC 2131952 (15). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. The chemical structure similarity search is conducted in therapeutic target database (TTD) via http://db.idrblab.net/ttd/.
References
Pape, A. R., Kaliappan, K. P. & Kündig, E. P. Transition-metal-mediated dearomatization reactions. Chem. Rev. 100, 2917–2940 (2000).
Wu, W.-T., Zhang, L. & You, S.-L. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev. 45, 1570–1580 (2016).
Sharma, U. K., Ranjan, P., Van der Eycken, E. V. & You, S.-L. Sequential and direct multicomponent reaction (MCR)-based dearomatization strategies. Chem. Soc. Rev. 49, 8721–8748 (2020).
Xia, Z.-L., Xu-Xu, Q.-F., Zheng, C. & You, S.-L. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem. Soc. Rev. 49, 286–300 (2020).
Wertjes, W. C., Southgate, E. H. & Sarah, D. Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev. 47, 7996–8017 (2018).
Uyanik, M., Mutsuga, T. & Ishihara, K. 4,5-dimethyl-2-Iodoxybenzenesulfonic acid catalyzed site-selective oxidation of 2-substituted phenols to 1,2-Quinols. Angew. Chem. Int. Ed. 56, 3956–3960 (2017).
Quideau, S., Pouységu, L. & Deffieux, D. Oxidative dearomatization of phenols: why, how and what for? Synlett 2008, 467–495 (2008).
Peruzzi, M. T., Lee, S. J. & Gagné, M. R. Gold(I) catalyzed dearomative claisen rearrangement of allyl, allenyl methyl, and propargyl aryl ethers. Org. Lett. 19, 6256–6259 (2017).
Yang, Z.-P., Jiang, R., Zheng, C. & You, S.-L. Iridium-catalyzed intramolecular asymmetric allylic alkylation of hydroxyquinolines: simultaneous weakening of the aromaticity of two consecutive aromatic rings. J. Am. Chem. Soc. 140, 3114–3119 (2018).
Liebov, B. K. & Harman, W. D. Group 6 dihapto-coordinate dearomatization agents for organic synthesis. Chem. Rev. 117, 13721–13755 (2017).
Ling, J., Lam, S., Low, K.-H. & Chiu, P. Dearomative intramolecular (4+3) cycloadditions of arenes with epoxy and aziridinyl enolsilanes. Angew. Chem. Int. Ed. 56, 8879–8882 (2017).
Zhu, D., Cao, T., Chen, K. & Zhu, S. Rh2(II)-catalyzed enantioselective intramolecular Büchner reaction and aromatic substitution of donor-donor carbenes. Chem. Sci. 13, 1992–2000 (2022).
Lee, E., Hwang, Y., Kim, Y. B., Kim, D. & Chang, S. Enantioselective access to spirolactams via nitrenoid transfer enabled by enhanced noncovalent interactions. J. Am. Chem. Soc. 143, 6363–6369 (2021).
Zeng, Q. et al. Divergent construction of macrocyclic alkynes via catalytic metal carbene C(sp2)–H insertion and the Buchner reaction. ACS Catal. 9, 10773–10779 (2019).
Miura, T., Funakoshi, Y. & Murakami, M. Intramolecular dearomatizing [3 + 2] annulation of α-Imino carbenoids with aryl rings furnishing 3,4-fused indole skeletons. J. Am. Chem. Soc. 136, 2272–2275 (2014).
Flynn, A. R., McDaniel, K. A., Hughes, M. E., Vogt, D. B. & Jui, N. T. Hydroarylation of arenes via reductive radical-polar crossover. J. Am. Chem. Soc. 142, 9163–9168 (2020).
Southgate, E. H., Pospech, J., Fu, J., Holycross, D. R. & Sarlah, D. Dearomative dihydroxylation with arenophiles. Nat. Chem. 8, 922–928 (2016).
Hu, N. et al. Catalytic asymmetric dearomatization by visible-light-activated [2+2] photocycloaddition. Angew. Chem. Int. Ed. 57, 6242–6246 (2018).
Zheng, C. & You, S.-L. Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products. Nat. Prod. Rep. 36, 1589–1605 (2019).
Zheng, C. & You, S.-L. Catalytic asymmetric dearomatization by transition-metal catalysis: a method for transformations of aromatic compounds. Chem 1, 830–857 (2016).
Yang, L. et al. Palladium-catalyzed dynamic kinetic asymmetric transformation of racemic biaryls: axial-to-central chirality transfer. J. Am. Chem. Soc. 137, 4876–4879 (2015).
Nan, J. et al. RuII-catalyzed vinylative dearomatization of naphthols via a C(sp2)–H bond activation approach. J. Am. Chem. Soc. 135, 17306–17309 (2013).
Wang, Y. et al. Enantioselective desymmetrization of bisphenol derivatives via Ir-catalyzed allylic dearomatization. J. Am. Chem. Soc. 142, 19354–19359 (2020).
Zhu, M., Huang, X.-L., Sun, S., Zheng, C. & You, S.-L. Visible-light-induced dearomatization of indoles/pyrroles with vinylcyclopropanes: expedient synthesis of structurally diverse polycyclic indolines/pyrrolines. J. Am. Chem. Soc. 143, 13441–13449 (2021).
Phipps, R. J. & Toste, F. D. Chiral anion phase-transfer catalysis applied to the direct enantioselective fluorinative dearomatization of phenols. J. Am. Chem. Soc. 135, 1268–1271 (2013).
Tamura, Y., Yakura, T., Tohma, H., Ki-kuchi, K. & Kita, Y. Hypervalent iodine oxidation of p-alkoxy- and related phenols: a facile and efficient synthesis of p-Quinones. Synthesis 1989, 126–127 (1989).
Lei, P. et al. A practical and chemoselective ammonia-free birch reduction. Org. Lett. 20, 3439–3442 (2018).
Zimmerman, H. E. A mechanistic analysis of the birch reduction. Acc. Chem. Res. 45, 164–170 (2012).
Cole, J. P. et al. Organocatalyzed birch reduction driven by visible light. J. Am. Chem. Soc. 142, 13573–13581 (2020).
Cao, T., Deitch, J., Linton, E. C. & Kozlowski, M. C. Asymmetric synthesis of allenyl oxindoles and spirooxindoles by a catalytic enantioselective Saucy–Marbet Claisen rearrangement. Angew. Chem. Int. Ed. 51, 2448–2451 (2012).
Linton, E. C. & Kozlowski, M. C. Catalytic enantioselective Meerwein−Eschenmoser claisen rearrangement: asymmetric synthesis of allyl oxindoles. J. Am. Chem. Soc. 130, 16162–16163 (2008).
Zheng, H. et al. Stereodivergent synthesis of vicinal quaternary-quaternary stereocenters and bioactive hyperolactones. Nat. Commun. 9, 1968 (2018).
Huang, S., Kötzner, L., De, C. K. & List, B. Catalytic asymmetric dearomatizing redox cross coupling of ketones with aryl hydrazines giving 1,4-diketones. J. Am. Chem. Soc. 137, 3446–3449 (2015).
Huck, C. J. & Sarlah, D. Shaping molecular landscapes: recent advances, opportunities, and challenges in dearomatization. Chem 6, 1589–1603 (2020).
Pan, C., Guo, W. & Gu, Z. Unusual biaryl torsional strain promotes reactivity in Cu-catalyzed Sommelet–Hauser rearrangement. Chem. Sci. 9, 5850–5854 (2018).
Guo, Q., Wang, M., Liu, H., Wang, R. & Xu, Z. Visible-light-promoted dearomative fluoroalkylation of β-naphthols through intermolecular charge transfer. Angew. Chem. Int. Ed. 57, 4747–4751 (2018).
Huang, X. et al. Dearomatization of aryl sulfoxides: a switch between mono- and dual-difluoroalkylation. Chem. Sci. 11, 3048–3053 (2020).
Zhao, W. et al. Dearomative dual functionalization of aryl iodanes. Angew. Chem. Int. Ed. 58, 17210–17214 (2019).
Martín Castro, A. M. Claisen rearrangement over the past nine decades. Chem. Rev. 104, 2939–3002 (2004).
Alshreimi, A. S. et al. Synthesis of spirocyclic 1-pyrrolines from nitrones and arynes through a dearomative [3,3′]-sigmatropic rearrangement. Angew. Chem. Int. Ed. 59, 15244–15248 (2020).
Shi, J. et al. Benzyne 1,2,4-trisubstitution and dearomative 1,2,4-trifunctionalization. J. Am. Chem. Soc. 143, 10530–10536 (2021).
Cao, T., Kong, Y., Luo, K., Chen, L. & Zhu, S. Cascade Claisen rearrangement: rapid synthesis of polysubstituted salicylaldehydes and total syntheses of hemigossypol and gossypol. Angew. Chem. Int. Ed. 57, 8702–8707 (2018).
Cao, T., Chen, L. & Zhu, S. Mechanism-guided scaffold diversification: perturbing and trapping the intermediates of maltol-type cascade Claisen rearrangement. Org. Lett. 21, 90–94 (2019).
Cao, T., Shi, Q. & Zhu, S. Benzene-free synthesis of multisubstituted catechol via oxidative dearomatic reorganization. Org. Lett. 23, 1411–1415 (2021).
Murahashi, S.-I. & Imada, Y. Synthesis and transformations of nitrones for organic synthesis. Chem. Rev. 119, 4684–4716 (2019).
Ye, L.-W. et al. Nitrene transfer and carbene transfer in gold catalysis. Chem. Rev. 121, 9039–9112 (2021).
Xiao, J. & Li, X. Gold α-Oxo carbenoids in catalysis: catalytic oxygen-atom transfer to alkynes. Angew. Chem. Int. Ed. 50, 7226–7236 (2011).
Marien, N. et al. Metal-free cyclization of ortho-Nitroaryl Ynamides and Ynamines towards spiropseudoindoxyls. Angew. Chem. Int. Ed. 57, 5660–5664 (2018).
Marien, N., Brigou, B., Pinter, B., De Proft, F. & Verniest, G. Synthesis of 2-spiropseudoindoxyls via an intramolecular nitroalkyne redox–dipolar cycloaddition cascade. Org. Lett. 17, 270–273 (2015).
Suneel Kumar, C. V. & Ramana, C. V. Tuning the regioselectivity of gold-catalyzed internal nitroalkyne redox: a cycloisomerization and [3 + 2]-cycloaddition cascade for the construction of spiro-pseudoindoxyl skeleton. Org. Lett. 16, 4766–4769 (2014).
Asao, N., Sato, K. & Yamamoto, Y. AuBr3-catalyzed cyclization of o-(alkynyl)nitrobenzenes. Efficient synthesis of isatogens and anthranils. Tetrahedron Lett. 44, 5675–5677 (2003).
Xiao, X. & Hoye, T. R. The domino hexadehydro-Diels–Alder reaction transforms polyynes to benzynes to naphthynes to anthracynes to tetracynes (and beyond?). Nat. Chem. 10, 838–844 (2018).
Zhang, J., Ho, D. M. & Pascal, R. A. Synthesis of an extremely crowded naphthalene via a stable norbornadienone. J. Am. Chem. Soc. 123, 10919–10926 (2001).
Irie, T. & Tanida, H. Decarbonylation of 9,10-dihydro-9,10-methanoanthracen-11-one (dibenzonorbornadienone). Effects of nitro substituents and solvents. J. Org. Chem. 44, 1002–1003 (1979).
Fray, G. I., Lay, W. P., Mackenzie, K. & Miller, A. S. Thermal transformation of [π4a + π2a]cycloadducts of tetracyclone and tricyclo[4.2.2.02,5] deca-3,7-dienes: sequential decarbonylation, retro-6π electrocyclisation, and intramolecular [π4a + π2a]cycloaddition. Tetrahedron Lett. 20, 2711–2714 (1979).
Jasiński, R. & Dresler, E. A desulfonylation process as easy route for synthesis of 1,4-dinitro-1,3-dienes: mechanistic study. Phosphorus Sulfur Silicon Relat. Elem. 191, 311–315 (2016).
Liu, X., Wang, J. & Dong, G. Modular entry to functionalized tetrahydrobenzo[b]azepines via the palladium/norbornene cooperative catalysis enabled by a C7-modified norbornene. J. Am. Chem. Soc. 143, 9991–10004 (2021).
Schoen, W. R. et al. A novel 3-substituted benzazepinone growth hormone Secretagogue (L-692,429). J. Med. Chem. 37, 897–906 (1994).
Ogawa, H. et al. Orally active, nonpeptide vasopressin V2 receptor antagonists: a novel series of 1-[4-(Benzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-benzazepines and related compounds. J. Med. Chem. 39, 3547–3555 (1996).
Hoyt, S. B. et al. A novel benzazepinone sodium channel blocker with oral efficacy in a rat model of neuropathic pain. Bioorg. Med. Chem. Lett. 23, 3640–3645 (2013).
Phun, L. H., Patil, D. V., Cavitt, M. A. & France, S. A catalytic Homo-Nazarov cyclization protocol for the synthesis of heteroaromatic ring-fused cyclohexanones. Org. Lett. 13, 1952–1955 (2011).
Patil, D. V., Phun, L. H. & France, S. Indium-catalyzed Homo-Nazarov cyclizations of alkenyl cyclopropyl ketones. Org. Lett. 12, 5684–5687 (2010).
De Simone, F., Andrès, J., Torosantucci, R. & Waser, J. Catalytic formal homo-nazarov cyclization. Org. Lett. 11, 1023–1026 (2009).
Miura, T., Nakamuro, T., Liang, C.-J. & Murakami, M. Synthesis of trans-cycloalkenes via enantioselective cyclopropanation and skeletal rearrangement. J. Am. Chem. Soc. 136, 15905–15908 (2014).
Brandi, A., Cicchi, S., Cordero, F. M. & Goti, A. Heterocycles from alkylidenecyclopropanes. Chem. Rev. 103, 1213–1270 (2003).
Brandi, A. et al. Rearrangement of isoxazoline-5-spiro derivatives. 2. Synthesis and rearrangement of tetrahydroisoxazole-5-spirocyclopropanes. Preparation of precursors of quinolizine, isoquinoline, and indole alkaloids. J. Org. Chem. 53, 2430–2434 (1988).
Brandi, A., Guarna, A., Goti, A. & De Sarlo, F. Rearrangement of nitrone cycloadducts to methylene cyclopropane. Synthesis of indolizidine and quinolizidine derivatives. Tetrahedron Lett. 27, 1727–1730 (1986).
Tabata, H. et al. Atropisomerism in the vaptan class of vasopressin receptor ligands: the active conformation recognized by the receptor. Angew. Chem. Int. Ed. 50, 3075–3079 (2011).
Cordero-Vargas, A., Quiclet-Sire, B. & Zard, S. Z. A flexible approach for the preparation of substituted benzazepines: application to the synthesis of tolvaptan. Bioorg. Med. Chem. 14, 6165–6173 (2006).
Acknowledgements
We appreciate the financial support from the NSFC (21871096, 22071062, 22001077), Ministry of Science and Technology of the People’s Republic of China (2016YFA0602900), Guangdong Science and Technology Department (2018B030308007, 2021A1515012331), China Postdoctoral Science Foundation (2018M643062, 2019T120723), 111 Project (B20003), Natural Science Foundation of Zhejiang Province (Y21B020024).
Author information
Authors and Affiliations
Contributions
Q. Shi and T. Cao performed the optimization, investigated the scope of substrate, and conducted the synthetic application and mechanistic investigation experiments; Z. Liao, Z. Liu, C. Li, J. He and J. Deng participated in the reaction development and synthesized several substrates; J. Wen employed the bioactivity study; S. Cen and J. Zhou designed and supervised the bioactivity study; T. Cao wrote the paper and S. Zhu revised-reviewed & edited the paper. All the authors discussed the experimental results and commented on the manuscript. S. Zhu directed the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shi, Q., Liao, Z., Liu, Z. et al. Divergent synthesis of benzazepines and bridged polycycloalkanones via dearomative rearrangement. Nat Commun 13, 4402 (2022). https://doi.org/10.1038/s41467-022-31920-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-31920-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
