
Abstract
Computational codes are necessary tools for geochemical modeling of the alteration of minerals due to their ability to handle key mechanisms, such as dissolution, precipitation, diffusion, and convection at many temporal and spatial resolutions. Modeling glass corrosion specifically requires a description of the amorphous layer that forms on the surface of the glass and its effect on glass alteration kinetics. The objective of the GRAAL model (glass reactivity in allowance of the alteration layer) is both to provide a simple implementation of the passivation process in a reactive transport code and to provide data relative to the composition and the solubility of the amorphous layer. The size and properties of the protective amorphous layer drives the glass alteration rate, with regard to passivation; the greater the quantity of the protective amorphous layer, the lower the dissolution rate of the primary mineral. Here, concepts, equations, and implementation of GRAAL are reported. Simple glass alteration experiments are used to apply the model and measure parameters. The International Simple Glass used for nuclear glass long-term behavior studies is at the center of the glass compositions studied.
Similar content being viewed by others


Introduction
Whether addressing global cycling of elements or the transport of groundwater nutrients and contaminants, research in the field of water-rock interactions relies on the description of mineral surfaces to understand the mechanisms involved. Many multi-oxide mineral surfaces exposed to aqueous solutions exhibit metal-depleted proton-enriched compositions.1 A large number of multi-oxide minerals are affected by this mechanism, including labradorite feldspar2,3,4,5 and forsterite.6 Oelkers et al.7 reported that metal-depleted proton-enriched layers can play a major role in controlling the alteration kinetics of many multi-oxide silicate minerals, including glasses. Theoretical considerations based on Monte Carlo simulations led Zhang and Luttge8 to assert that an interfacial dissolution–reprecipitation mechanism dominates feldspar dissolution. The role of precipitation mechanisms on mineral dissolution kinetics is increasingly reported.5,7,9,10,11
In the field of glass alteration, condensation mechanisms forming amorphous layers have also long been a subject of interest.12,13,14,15 Today it is considered that a saturation state can only be expressed with respect to a hydrated layer.16 That layer may passivate the underlying fresh glass.16,17,18,19 This passivation is rarely fully effective and, consequently, transport by diffusion must be added to classical affinity laws20 to accurately model glass alteration. Without taking this transport into account, it is impossible to explain the dependence of sodium and boron concentrations on the surface area to solution-volume ratio, and therefore impossible to account for pH.21
Coupling transport within a passivating layer with dissolution of this layer is the objective of a model named GRAAL (glass reactivity in allowance for the alteration layer).22 The GRAAL model has been implemented within the CHESS/HYTEC reactive transport code.23 The interesting point is that the GRAAL equations do not explicitly model the nanometer-scale transport phenomena within the amorphous layer but only the resulting continuum flux. This simplification enables the code to focus on mass balance and fluid chemistry. Early implementation of GRAAL was proposed in ref. 21, but both the implementation of GRAAL and the amorphous layer’s chemistry were very simple at that time. Describing the reasons and methods behind subsequent improvements to both the implementation of the code and the amorphous layer’s chemical and physical description is the aim of the present paper.
Geochemical models describing glass alteration have been proposed in the past.14,24,25,26,27,28,29,30,31 In contrast to simple analytical models,32 geochemical models are able to describe a detailed chemistry. Their main difficulty is to take account of the compositional variations of the alteration products of glass, i.e., secondary crystalized minerals and the amorphous layer.
Secondary crystallized phases can precipitate but their precipitation is generally limited by the availability of key elements, pH, redox potential, or simply their precipitation kinetics.22 For example, the precipitation of smectite requires elements such as Mg, Zn, Ni, Mn, or Fe in addition to Si and potentially Al;22 zeolites and hydrated calcium silicates may require pH above 10 at 90 °C or higher temperatures;33,34,35,36,37 and quartz and kaolinite are examples of minerals whose precipitation can be extremely slow at room temperature.
In case of ISG containing only Si, B, Na, Al, Ca, and Zr, altered in initially pure water for which pH is eventually close to 9 at 90 °C, the alteration layer is entirely amorphous after >15 years of alteration.38 Amorphous and low crystalline alteration layers have been shown to exist for millions of years: they are termed “palagonites” when observed on natural basaltic glasses.39,40 In alignment with the purpose of modeling simple glass alteration experiments, the current state of the art is relative to the amorphous alteration layer and the application of the GRAAL model to account for variation in rate with time and conditions.
There are few chemical and thermodynamical data for amorphous hydrated silicates. These layers are difficult to characterize: they are thin and can have various compositions, and have aspects of both glass and fluid compositions. In batch experiments, their average composition can also change with time: the longer the duration, the more elements reach an apparent steady state in the fluid and are incorporated in the amorphous layer. Some authors21,31 reported that, in simulations, steady-state fluid compositions meant to be in equilibrium with the amorphous layer are oversaturated with respect to crystallized phases available in their databases. Therefore, modeling experimental fluid compositions requires the way in which the amorphous layer can control the fluid chemistry to be defined.
When the fluid composition eventually reaches a steady state and a dense amorphous layer can form, the glass dissolution rate is shown to dramatically decrease, typically by four orders of magnitude for French nuclear reference glass, relative to the forward rate measured in deionized water.22 From a mathematical point of view, the affinity law formalism can account for that rate decrease. Therefore, one simple way to model fluid chemistry is to attempt to define a solubility product for the amorphous layer.16 The task of defining the temperature-dependent solubility of a multi-elemental solid solution is significant. Daux et al.29 used a “hydrated basaltic glass” (HBG) containing Si, Al, and Fe in the same proportions as their original glass. The HBG glass solubility product was calculated using the fluid composition at an apparent steady state. Munier et al.30 built various solid solutions with Al, Na, Ca, Zr, and Si. The end-members were chosen among minerals available in the database. The best agreement was often achieved when hydroxides were chosen as end-members along with chalcedony for Si. Although it is appealing to use available data and to compare fluid saturation states with respect to minerals in a database, the potential for secondary phases to be chosen as end-members should be considered since potential secondary phases may precipitate over geological timescales. Gysi and Stefansson31 also approximated the amorphous layer by a sum of amorphous SiO2 and Al(OH)3.
Though the definition of a solubility for the amorphous layer is mathematically quite efficient, many questions have been raised regarding that modeling assumption. Indeed, defining a solubility assumes the existence of a backward precipitation reaction. However, the formation of the amorphous layer is more complex than a simple precipitation from the bulk aqueous species. The amorphous layer arises both from the underlying glass structure to which it is intimately related and from dissolution and condensation reactions that may occur locally. Researchers41,42,43,44,45 have tried to understand which bonds are inherited from the glass structure and which undergo the dissolution and condensation process. Defining steady-state conditions can be challenging if activities slowly but continually increase over extensive timescales.38 Si transport processes in the amorphous layer has been proposed as a mechanism for explaining rising Si concentrations.46 Frugier et al.21 proved that speciation could explain a significant component of the Si concentration increase. The increase in remaining Si in the long term is not yet well understood.38 It could be due to reorganization of the amorphous layer,45,47 either from a physical point of view, that is homogenization by transport from the inner part of the layer toward the outer part of the layer, or from a chemical point of view due to preferential chemical interactions breaking apart the homogeneity of the amorphous layer and changing the fluid concentrations accordingly.
Describing the thermodynamics of the amorphous layers is a first step, whichever kinetic model of glass dissolution is to be used. However, glass alteration models often consider the kinetics first. Regarding modeling, the debate lies in the role played by the alteration layer on the glass alteration kinetics. What makes the amorphous layer protective? What is the link between its structure and its transport properties? Does its structure depend more on the initial glass structure or on the condensation processes? What are the criteria to shift from a non-protective residual skeleton to a dense protective layer? Which of these properties need to be modeled, at least from a kinetic point of view?
The complexity of the amorphous layer problem has led to diverse terminology. A low density, highly silica-depleted, and zirconium-rich amorphous layer can form in a solution that is far from saturation.22 To avoid ambiguity, it should be named differently to the high density, silica-rich amorphous layer that eventually forms under steady-state conditions. The first can be considered as a sink of matter with no passivation properties, much like any crystallized secondary phase; in this paper, it is referred to as a “depleted gel.” The second may passivate the glass; within the GRAAL paradigm, the place where transport limitation occurs is known as the “passivating reactive interphase” (PRI).21 The whole amorphous layer can therefore be described by the summation of a non-protective depleted gel and a protective layer (PRI). Dividing the amorphous layer in two could appear a little over-simplistic but experiments inform this choice: in case of an alteration in a saturated solution, the amorphous layer is a nanometer-thick protective layer; in the case of alteration in almost pure water, the amorphous layer is typically several microns thick and consists of a depleted gel, in addition to an extremely thin PRI. The PRI does not address the mechanisms underlying the passivation process.
In common with many others, the GRAAL model focuses on bulk solids and fluids and assumes that once steady-state conditions are achieved, the activity of elements in solution reflects a thermodynamic equilibrium between the amorphous layer and the solution. In ref. 21, the model is rudimentary: a juxtaposition of simple pure phases (SiO2, AlO(OH), Ca(OH)2, and ZrO2). Such a model is only suitable for SON68 glass within ±0.5 units of the pH value imposed by SON68 glass dissolution in initially pure water (about pH 9.0 at 90 °C). It is unsuited to modeling other pH values or other glass or fluid compositions as the chemical description of the amorphous layer needs to be improved.
Being able to distinguish the potentially protective amorphous layer from the non-protective crystallized secondary phases or depleted gel has always been a cornerstone of the GRAAL model.22 Within that paradigm, predicting glass corrosion can be achieved if chemical descriptions (compositions and solubilities) and transport properties of both the crystallized and amorphous phases are known. Precipitation of crystallized secondary phases can drive glass dissolution, as has been demonstrated for phyllosilicates48,49,50,51,52 and zeolites.33,34,36,37,53 Thus, the ability of a model to describe whether the elements are meant to be trapped in the amorphous layer or the crystallized secondary phases is a key point for the prediction of composition effects. A higher quantity of an element that tends to precipitate in crystallized secondary phases will generally lead to a higher degree of glass alteration. This is true, regardless of whether the element is supplied by the glass or the fluid. Conversely, the greater the number of elements that are retained in the amorphous layer, the denser is the amorphous layer and the slower is the transport of reactive species.12 Although water transport through the dense amorphous layer has never been measured, evidences have been provided to explain how a porosity-closure mechanism in the amorphous layer can account for the diminishing alteration rate.17,45,54,55 Secondary crystallized phases grow on a glass surface, like trees on the ground. In contrast, precipitation within amorphous layers leads to densification and may eventually lead to porosity closure.
At the time of writing, a decade has passed since the GRAAL concept was introduced22 and first implemented.21 The following sections of the paper provide the detailed presentation of the model: the kinetic equation, its implementation in a reactive transport code, a description of the chemistry of the amorphous layer, and eventually parameter selection. The last section is a discussion of the efficiency and limitations of the model.
Results
Equations for passivation
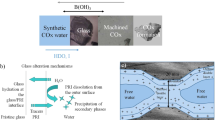
Two approaches can be considered to model a passivation process (Fig. 1). The first involves meshing the space for the explicit modeling of transport mechanisms. While useful for a deeper understanding of the transport process, such modeling becomes numerically complex when transport mechanisms in the fluid are taken into account. Indeed, the concentration profiles in the solid for each fluid cell position in space have to be held in memory by the code. The second approach only describes the transport flux arising in the fluid. Since it is numerically simpler, it enables computational effort to be focused on the effect of the transport flux on the fluid chemistry and the way in which that can drive the dissolution of the protective layer, which is the key long-term mechanism. Including the transport through the layer is essential for modeling glass dissolution experiments effectively as it has a major influence on both the evolution of pH as a function of time and glass surface area to solution-volume ratio.

Two possible approaches to modeling passivation: defining a meshing space for an explicit transport model (1) or simply modeling the flux of elements arising in the fluid (2)
In the GRAAL model, the glass alteration rate is a function of the protective layer’s transport properties, that is, its thickness and apparent diffusion coefficient. The thickness of the protective layer is function of a creation term, r1, at the interface between the primary solid and the protective layer and a dissolution term, r2, at the external face of the protective layer (Fig. 2). The coupling between these terms was solved analytically by ref. 32. A first simplification is discussed in ref. 21, but it does not allow for any feedback effect of the advancing glass hydration front on the ion diffusion rate within the protective layer. Equations and numerical resolutions are presented in the next sections.

Kinetics relative to the protective layer: Creation kinetic (r1) and dissolution kinetic (r2) of the protective layer (PL)
Experiments on borosilicate glasses show that the thicker the protective layer (PL) is, the slower is the alteration rate of the primary solid (PS)56 and that, consequently, the protective layer grows with the square root of time when not undergoing dissolution at its surface. This simple experimental observation is expressed as (Eq. 1). r1 is the primary solid alteration rate controlled by diffusion (m s−1), xPL is the protective layer’s thickness (m), and A is a constant of proportionality (m2 s−1).
Equation 2 defines r1, thus:
Combining Eqs. 1 and 2, considering x = 0 at t = 0, and assuming no dissolution at the outer surface, Eq. 3 can be stated as:
This equation is to be compared with the resolution of Fick’s second law (Eq. 4) with the following hypotheses:
-
(H1) diffusion of a glass mobile element of concentration C (g/m3),
-
(H2) in a semi-infinite media defined on one side by the glass/fluid initial interface and on the other side by the infinite glass,
-
(H3) considering that the driving force is the concentration gradient between the mobile element concentration in the pristine glass C0 (g/m3 of glass) and zero at the interface with the fluid,
-
(H4) considering a constant diffusion coefficient in the semi-infinite media D (m2 s−1).
With those initial conditions, Eq. 5 is the solution of Eq. 4.
Following Fick’s first law, derivation of Eq. 5 with respect to x and multiplication by −D enables the flux of the mobile element to be calculated. Integration with time of the flux taken in x = 0, gives the concentration in the fluid Cf(t) (g/m3 of solution) of the mobile element going through the external surface S (m2) and diluting in a perfectly steered solution of volume V (m3) (Eq. 6).
The “equivalent thickness of alteration,” with respect to a glass constituent, is given by a simple mass balance (Eq. 7). It is the thickness of glass that has been altered to explain the concentration Cf of a glass constituent in solution.
Equation 8 follows from Eqs. 6 and 7:
Equations 8 and 3 are equivalent. However, the proportionality constant, D in Eq. 8, is more meaningful than A in Eq. 3, in agreement with hypotheses H1 to H4. Therefore, Eq. 8 is preferred for the GRAAL model. In agreement with Eq. 8, Eq. 1 becomes Eq. 9.
According to Eq. 9, the time required to create a protective layer from glass when its thickness is zero, is infinite. However, there is no physical reason for the dissolution rate of mobile ions to be infinite. Therefore, a constant kinetic, rh, known as the hydration rate, is added in order to limit the creation kinetics of the protective layer (Eq. 10). This rate is meant to be higher than the initial dissolution rate in agreement with the higher dissolution rate of mobile ions in comparison to silicon ions.
The hydration rate is negligible as soon as \(x_{\mathrm{{PL}}} > \frac{{D\frac{\pi }{2}}}{{r_{\mathrm{h}}}}\), and more so if \(x_{\mathrm{{PL}}} > \frac{{D\frac{\pi }{2}}}{{r_0}}\). Whatever the pH and temperature, numerical modeling using nuclear glass parameters shows that the hydration rate is largely negligible for a 1 nm thick layer. The conclusion is that the parameter is not measurable on nuclear glasses: whatever the value of rh > r0, modeling results after a few seconds of alteration do not change. However, the parameter is useful from a numerical point of view as it avoids the error that arises from dividing a number by zero.
Equation 11 expresses the pH and temperature dependence of the dissolution rate of the protective layer, and the exponential rate drop when the concentrations approach saturation, following the formalism proposed by Aagaard and Helgeson.20
where, k+ is the dissolution rate of the protective layer in pure water (forward rate) (m d−1), n is the pH-dependence coefficient of the initial rate, Ea is the apparent activation energy at the initial rate (J mol−1), Q is the activity product of the protective layer, K is the activity product at saturation of the protective layer, and R is the ideal gas constant (J mol−1 K−1).
Case of r1 » r2: the thickness of the diffusion zone increases with the square root of tine and the flux of mobile species released from the glass decreases with the inverse square root of time. This occurs at initial time and once concentrations of elements belonging to the protective layer approach saturation in the fluid.
Case of r1 ≥ r2: once r2 is significantly different from 0, r2 approaches r1 resulting in a roughly constant thickness xPL, then r1 has no time dependence except due to pH variations.
Implementation in a reactive transport code
The CHESS/HYTEC computational code (from versions 3.7 to 5.2) developed by MINES ParisTech was chosen for implementing the GRAAL model. One interesting feature is that the GRAAL code does not require any software improvement: the input file that describes the experimental conditions is to be written taking into account GRAAL equations. How the model equations translate into code equations is described in Table 1 and with the help of the thickness–concentration relationship and the mass balance relationship described in the following paragraphs.
The requirements for the code to handle GRAAL equations, are the availability of the Monod type equation (cf. Table 1), the ability to use the concentration of a solid as a variable of the equation, and an adaptive timescale (available in CHESS/HYTEC code since version 3.7): the time step has to be small at the beginning of the calculation when the protective layer is thin in order to prevent its dissolution within a single time step.
A spherical shrinking-core model is used to describe the reduction in surface area due to the reduction in the glass grain size, although it does not have any effect in the experimental conditions discussed in this paper.
CPL (mol m−3) is defined as the concentration of the protective layer within a calculation cell. The thickness of the protective layer xPL is proportional to CPL (Eq. 12).
where, ρPL is the protective layer’s density in mol m−3 of the protective layer, S is the protective layer’s surface area in the modeling cell (m2 m−3); the layer being thin and protective, is by definition equal to the surface area of the primary solid. V is the volume of the water in the calculation cell (m3 m−3).
The surface area to solution-volume ratio can also be written as the product of the specific surface area SSA (m2/mol) and the concentration CPS (mol m−3) of the primary solid (Eq. 13).
A mass balance can be written for each element belonging to the primary solid and the protective layer (Fig. 3 and Eq. 14).
where, Fi is the fraction of element i coming from the primary solid and recovered in the protective layer, ρPS is the primary solid’s density in mol m−3, iPS is the molar fraction of element I in the primary solid, iPL is the molar fraction of element i in the protective layer, xPS is the thickness of primary solid that has been altered, and xPL is the thickness of protective layer (Fig. 3).

Mass and volume balance in the alteration layer. PS primary solid, PL protective layer, NPL non-protective layer
Measuring the composition and the structure of the protective layer enables the calculation of iPL and ρPL. Fi can be calculated by applying the condition of mass balance in the experiment between the fluid, the non-protective layer, and the protective layer. Then, the ratio of the thickness of the protective layer to the thickness of the primary solid being altered can be deduced from Eq. 14.
Identifying the two equations of Table 1 and using equations Eqs. 13 and 14 enables the code’s parameters km (Eq. 15) and Kin (Eq. 16) to be calculated as a function of the model’s parameters.
If reaction 2 is a classical dissolution reaction, then reaction 1 occurring at the interface between the primary solid and the protective layer is a reaction that converts one solid into another plus dissolved elements. Therefore, two simple reactions occurring at the same rate are required to produce reaction 1: a congruent dissolution of the primary solid and a backward precipitation of the protective layer.
Composition of the amorphous layer
Prediction of the amorphous layer’s solubility is a priority for understanding the long-term behavior of glass as that determines the stability of the glass. Solubility is measured by fluid analysis under steady-state conditions; “apparent steady-state” refers here to the concentrations required for a reduction of at least three orders of magnitude in the alteration rate.
The former approach using separate simple end-members: SiO2, Ca(OH)2, AlO(OH), and ZrO221 had the major limitation of not including the chemical interactions between the elements. A richer and more complex model requires new experimental data. Rajmohan et al.57 enumerated a variety of phenomena directly derived from experimental data from simple glasses containing Si, B, Na, Al, Ca, and Zr. These phenomena are useful for improving the choice of end-members stoichiometry and defining the minimum number of end-members required to model those experiments (Table 2).
The composition of the amorphous layer is not only a function of the composition of the glass but also a function of the fluid composition, volume, and renewal rate. Therefore, it is meaningful to build a model applicable to any fluid or glass composition. Consequently, a single model is proposed for the six glasses studied by Rajmohan et al.,57 which is then applied to the thirty oxide glasses and the SON68 glass (Table 3) in order to verify whether it is possible to neglect the influence of minor elements.
Figure 4 represents the composition of the end-members used to describe the amorphous layer. The polyhedron defined by the end-members must, therefore, include the glass compositions being modeled. In confined media, the Si/Al/Ca/Zr stoichiometry of the amorphous layer tends toward the stoichiometry of the initial glass. This model is not a solid solution model but simply a concatenation of end-members.

Graphical representation of end-members stoichiometries
The proposed model is intended to be applied to experimental data in order to check the utility of the end-members to account for glass compositions and fluid pH variations of all available experiments and then to infer the missing parameters, mainly the logK values and diffusion coefficients of the end-members.
Parameters determination
This section presents all the parameters required in the input file of the model, starting with the experimental conditions, the glass composition, or the kinetic parameters already available in the literature. It also describes the method for determining new parameters values from the modeled experiments.
Six glass compositions were studied. The procedures for glass synthesis and preparation are described in ref. 57. Experiments were carried out in a static system at 90 °C with a glass surface area to solution-volume (S/V) ratio of 20 cm−1 (except CJ9: 48 cm−1) and with various imposed pH values (7, 8, 10) at 90 °C for ~150 days. For pH 7 and 8, 0.2 mol of tris (hydroxymethyl) aminomethane (TRIS) buffer solution was prepared. Potassium hydroxide solution was used as a buffer for pH 10 experiments. Experiments in initially pure water at free pH (near 9) and S/V = 80 cm−1 were initiated by Jegou58 and by Gin and Jegou59 and have been running since November 1996. These have been updated by Gin et al.38 Solutions were ultra-filtered to 10,000 Da before analysis. Twenty-four experiments are available for analysis: six glass compositions at four pH values (7, 8, ~9, 10).
The stoichiometry of the glasses is based on the composition of the French SON68 inactive reference glass, following the procedure initiated by Jégou, et al.58 In order to understand the effect of Ca, Al, and Zr, glasses were prepared by adding these oxides to the three major oxides of Si, B, and Na (Table 3). CJ4 is also known as the International Simple Glass ISG.60
r2 values are measured in pure water based on Si and B almost congruent release. For those glasses in those experimental conditions, protective layer’s thickness is very small compared to altered glass thickness and non-protective layer’s contribution to Si release is negligible. Therefore, Si and B release are almost congruent.
They were measured on each simple glass by Jégou et al.58 at 90 °C, and pH 9.0; n and Ea (Eq. 11) are assumed to have the same values as those measured for SON68 glass.61 This is justified here as the forward rates have a negligible effect under the experimental conditions discussed in this paper. The k+ values are calculated from rdiss pH 9, 90 °C to fit equation (Eq. 11) in order to have k+ expressed in m s−1 (Table 4).
The interdiffusion coefficient varies with temperature and pH according to a simple relation (Eq. 17) proposed by Chave et al.56:
, where n′ is the pH-dependence factor of the interdiffusion coefficient (dimensionless), Ea′ is the activation energy associated with the interdiffusion coefficient (kJ mol−1), and D0 is the interdiffusion constant (m2 s−1).
In Eq. 17, hydroxide ions are preferred to hydronium ions because the equation is applied in neutral to basic pH conditions. Choosing hydronium ions, the minor species, would introduce the water dissociation constant into the equation, which is also a function of temperature and would change the numerical value of the activation energy, accordingly.
Experimental measurement of the diffusion coefficient is based on the diffusive flux arising in solution once dissolution of the protective layer by its outer surface has stopped. This flux is proportional to D/xPL (Eq. 9). The thickness of the protective layer, xPL, depends on the alteration thickness of the glass, xPS, and the molar density of the protective layer (Eq. 14). Therefore, knowledge of the molar density of the protective layer is required to calculate D. However, only the ratio D/xPL is used in the calculations. As a consequence, the choice is made here to select the molar density of the protective layer so that xPL = xPS (Eq. 14). Collapse of the gel, as identified by small angle X-ray scattering62 and modeled by a Monte Carlo method,54 is very limited in silica-saturated media. Isovolumetric alteration enables D to be calculated by simply applying Eq. 8 to the experimental data. Consequently, the diffusion coefficients given in Table 5 are calculated with this hypothesis of an isovolumetric alteration. Should the protective layer occupy 90% of the altered glass, then the diffusion coefficient would be 0.9 of the value given in Table 5.
The measurement of the diffusion coefficient can be performed at high surface area to solution-volume ratio where the dissolution thickness due to r2 can be neglected compared to the diffusion thickness due to r1. At low surface area to solution-volume ratio, the formation of non-protective end-members will consume Si, delay saturation of PRI, and sustain glass dissolution, globally increasing the mass fraction of non-protective layer (Fig. 2).
The activation energies of the diffusion coefficients of these glasses are still unknown, though probably close to that of SON68 glass. D0 values (Table 5) are calculated from 90 °C data57 considering the same activation energy as SON68 glass.56
Uncertainties of the diffusion coefficient values come both from the precision of experimental data and the effectiveness of Eq. 17 in taking pH and temperature variations into account. The value of the diffusion coefficient from Eq. 17 was applied using the parameters of Table 5 to calculate D within a factor of two, i.e., with a precision on the quantities of altered glass equal to a factor of √2. Moreover, under these experimental conditions, a value of 2 × 10−21 m2 s−1 is near the measurement limit and should be considered as a maximum value and not as an accurate measurement of the diffusion coefficient. The measurements determined from experiments at pH 10, all of which are below 2 × 10−21 m2 s−1, were therefore excluded when calculating the regression to determine the parameters indicated in Table 5. The calculated values extrapolated to pH 10 are consistent with the experimental data obtained at pH 10.
Figure 5 to Fig. 6 give representative examples of model results for the case of ISG glass, the glass that contains all the elements. The fit-quality is discussed in the next section.

Total concentrations in solution at pH 7: comparison between model (blue line) and experiment (red circles) for ISG: pH 7, 90 °C, S/V = 20 cm−1. Real average pH of the experiment (7.2) is slightly above the set-point value (7.0). Al concentrations below-detection limits are not drawn

Total concentrations in solution at pH 9: comparison between model and experiment for ISG: unconstrained pH, 90 °C, S/V = 80 cm−1
Given the stoichiometry of the elements in the glass and in the end-members, the operation of the model can be summarized as follows:
-
The solubility products of the SiAlCa and SiAlNa end-members have a first-order effect on the Al activities in soda-lime glass and in soda glass, respectively. Al concentrations are lower in presence of Ca.
-
The solubility products of the SiZrCa and SiZrNa end-members have a first-order effect on the Zr activities in soda-lime glass and in soda glass, respectively. Given the below-detection limits concentrations of Zr in solution, the logK values indicated for these two end-members were chosen in order for the solution to remain undersaturated, with respect to zirconium oxide.
-
The Al/Ca and Zr/Ca stoichiometry determines the calcium fraction retained in the amorphous layer at pH ≤8. At pH <8, the SiCa end-member does not form. The solubility of the SiCa end-member determines the calcium activities for more alkaline pH values (e.g., pH90 °C = 9).
-
The (Al-2Ca)/Na and (Zr-Ca)/Na stoichiometry determine the sodium fraction retained in the amorphous layer in initially pure water.
-
The SiAl and Si end-members have a first-order effect on the silicon activities.
The calculations are compared with the results obtained in 18 experiments, i.e., for all six glasses at three different pH values. The pH and elemental concentrations are measured in solution for Si, B, Na, Al, and Ca. The model-experiment comparison therefore covers a hundred curves of concentrations versus time, and a thousand experimental data points. This comparison is used to select the logK values corresponding to each of the end-members indicated in Table 6.
Table 6 indicates the dissolution equations selected for the end-members, as well as the logK90 °C value for each end-member. These values are determined by applying the model to experiments at pH 7, 8 and pH free57 and using CTDP database.23
The uncertainty presented in Table 6 gives an overview of the ability of the model to reproduce experimental data (discussed in the Results section). It is calculated, so that each available data point (Si, Al, and Ca concentrations), lies within the modeling results whatever the glass, pH, or sampling time.
Geochemical calculation code databases contain many mineral phases, but not all of them are likely to form at temperatures below 100 °C. Some can only be synthesized at high temperatures and cannot be observed in these experimental conditions. They are largely supersaturated in the leaching solutions and taking them into account without any kinetic limitation in the calculations would result in extremely low element concentrations in solution, far below the measured concentrations. Among the precipitating phases, it is important to discriminate between those with fast precipitation kinetics—for which the supply of a stoichiometric element from the glass or from the environment is a growth-limiting factor—and those with slow precipitation kinetics.53,63
Precipitation has been allowed for simple oxides and hydroxides whose precipitation at low temperatures has been established: aluminum hydroxide, portlandite, zirconium oxide, borax, and colemanite. However, all these minerals are undersaturated with respect to fluid experimental compositions and when modeling, precipitation of the amorphous layer’s end-members prevent their formation. Even if they do not form and, as a consequence, have no influence on the modeling results, they do deserve to be taken into account and cannot be removed from the database.
The secondary minerals selected here are those suitable for describing the laboratory experiments performed in initially pure water under oxidizing conditions. In a complex chemical environment, or under the reducing conditions expected in a geological repository, other minerals may have to be considered.
Hydrated calcium silicates and zeolites have been observed in the experiments at pH 10, at least after 150 days. Therefore, experiments at pH 10 could not be used to measure the end-members log K (cf.Table 6). Modeling of these experiments requires the kinetics of zeolite precipitation to be taken into account,34 which is beyond the scope of the paper.
Model application
In this section, the model’s usefulness and ability to account for experimental data at short- and long timescales is presented. An example of the calculation of the amorphous layer composition is given.
Below, we discuss the precision of the fit of the model to the data obtained from the experiments that were used for measuring the interdiffusion coefficient and the end-member solubilities. An examination of the results for ISG glass (Fig. 5 through Fig. 7) prompts the following remarks. These can be generalized to all the glasses and pH below 10:
-
1.
The model accounts precisely for the pH variations with the glass composition (refer to the experiments with unconstrained pH: Fig. 6). The predicted values of pH lie within the uncertainty margin of pH measurement (±0.1 unit). Under these experimental conditions (closed system and high S/V ratio), the concentrations of B, Na (at all pH values), and Ca (at pH 7 and 8) depend mainly on the value of the diffusion coefficient. Variation over time is generally satisfactory, which supports the relevance of a time-squared model. The fit between the model and experimental results is related to the precision of the law describing the variations in the diffusion coefficient versus the pH (the uncertainties are discussed in the dedicated section). For example, at pH 8, for ISG glass, the D = f (pH) law is below the experimental value (Fig. 8); therefore the modeled B concentrations are underestimated for the experiment at pH 8 (Fig. 9). At pH 7 (Fig. 5) and pH 9 (Fig. 7), however, the fit is good. B and Na concentrations are usually known within an uncertainty margin of a factor of 1.4.
-
2.
Si, Al, Zr, and pH 9-Ca concentrations depend on the amorphous layer model and on the pH. Uncertainties given for logK values in Table 6 are given based on the precision with which the concentrations are predicted by the model: the ±0.2 uncertainty in SiAl logK is equivalent to an uncertainty of a factor 1.5 in Si concentration; the ±1 uncertainty in SiAlNa logK is equivalent to an uncertainty of a factor 5 in Al concentration. This also reflects the experimental uncertainty of Al measurement, especially in alkali rich solutions obtained at pH 7.

Calculated amorphous layer composition for ISG glass: unconstrained pH, 90 °C, S/V = 80 cm−1. a End-members concentration, (b) composition of the amorphous layer, oxygen excepted, in molar percentage


Total concentrations in solution at pH 8: Comparison between model (blue line) and experiment (red circles) for ISG: at pH 8, 90 °C, S/V = 20 cm−1
Figure 7 shows the time-dependent composition of the amorphous layer. A single experiment is shown here as an example, at unconstrained pH. Due to the presence of Ca in sufficient amounts, the SiZrCa and SiAlCa end-members form at the expense of the soda end-members, SiZrNa and SiAlNa. Similarly, the presence of Al in sufficient amounts leads to the formation of the SiAl end-member and the absence of the Si end-member. Finally, as Ca is in excess with respect to the total Al/2 + Zr, and the pH is high enough, the SiCa end-member can form. The relative amounts of each end-member vary over time and thus modify the average composition of the layer (Fig. 7). Si, the most soluble of the elements that form the amorphous layer, accounts for an increasing proportion of the amorphous layer over time, tending toward the initial glass composition ratio.
Experiments at unconstrained pH were initiated several years ago to demonstrate the evolution of the reactive diffusion coefficient over time. Over the long-term, the model accurately accounts for the B concentrations for some glass compositions, but is conservative for others (e.g., ISG, Fig. 10). Experimental data55,64 and Monte Carlo models of glasses17,54,62,65 show that it is not sufficient to reach saturation with respect to Si to observe an inhibition of alteration: the gel must also reorganize. A decrease in the diffusion coefficient over time could therefore be due to a reorganization of the elements within the amorphous layer. Another hypothesis is that more Ca is incorporated in the amorphous layer,12 consistent with the slightly increasing pH. Indeed, an increase in pH causes a strong decrease in the diffusion coefficient for calcium-containing glass (Fig. 8).

Total concentrations in solution at long term: comparison between the model and long-term experiment for ISG: unconstrained pH, 90 °C, S/V = 80 cm−1
Discussion
Numerical application of the model allows us to draw the following general conclusion: each degree of freedom originally given to the model appears to be required in order to represent the observed phenomena. A simpler model would fail to address the chemical complexity of these systems. Until now, there has been no evidence that the precision of the concentration prediction is inadequate for the purpose of predicting the long-term behavior of glass. However, the limitations of the model are explored further in the next sections.
Previous authors have identified the limits of the formalism of Eq. 11.66,67,68,69 Nevertheless, the simplest first-order law was still used in this study. The amorphous layer’s chemical composition is far too complex to argue that the inability to account for the drop in the dissolution rate when conditions diverge from equilibrium is due to, only the simple formalism of Eq. 11, rather than the evolution with fluid chemistry of the composition and structure of amorphous layers with significant solubilities and transport properties.
One particular hypothesis of the GRAAL model results in a model for the amorphous layer that is independent of the composition of the glass and contributes very significantly to the simplicity of the model. This hypothesis is that the reactivity of the elements that precipitate in the amorphous layer depends primarily on their behavior within the solution and thus that the effect that the initial structure might be disregarded in a first approximation.
The model is not unique in the choice of the stoichiometry values of the end-members. More experimental data are required to improve the choice for this stoichiometry. For example, the stoichiometry of the Si/Ca end-member was defined as 2, but a value of 3 combined with a suitable logK value would give equally satisfactory fitting results. However, the possible variation ranges of the stoichiometry of the end-members are bounded. For example, it is advisable for the Si/Ca ratio to be higher than for the calcium silicate hydrates that form at a higher pH.70,71 It has therefore not been demonstrated that the model is valid, i.e., that it describes the reality of Si−Al−Ca−Zr reactivity at a molecular scale. Instead, however, it accounts for the main macroscopic effects of these interactions and thus fulfills its objective. If the predictive capabilities of the model are called into question, then the construction of a model closer to the actual chemical reality will be justified. This would require additional experimental data points in order to improve the chemical description of the amorphous layer. Finally, atomic theory modeling studies of the structure of the amorphous layer could help to provide a theoretical justification for identifying interactions and choosing the stoichiometry of end-members.72,73,74 Once the end-member stoichiometry has been defined, the logK values selected for the end-members are then imposed by the activities in solution of their constituent elements.
In this paper (cf. Table 5), each glass is assigned a reactive diffusion coefficient. However, the theoretical basis of the GRAAL model states that the diffusion coefficient refers to the protective amorphous layer and not to the glass. For the experiments presented here, in initially pure water at high surface area to solution-volume ratio, the stoichiometry of the amorphous layer tends toward the stoichiometry of glass, thus the approximation to assign each glass a diffusion coefficient is justified.
Chave et al.12 presented experimental evidence that the diffusion coefficient is related to the amorphous layer and not the glass, since the apparent reactive diffusion coefficient of a glass containing no Ca drops by several orders of magnitude when a sufficient amount of Ca is supplied by the solution, especially if the pH conditions favors Ca condensation. This result, as well as the hypothesis discussed in the section relative to Model-experiment comparisons at long term, demonstrates the need for the diffusion coefficient to be a function of the composition of the amorphous layer.75,76
Several interactions are not taken into account in the model because their effects were considered too weak to justify an additional model parameter. The interactions not taken into account, and their consequences for the model, are listed below:
-
The effect of Zr on the apparent Si solubility was completely negligible: however, glasses with very high Zr content should exhibit slightly lower Si activity than what is predicted by the model.
-
The effect of Al on the apparent solubility of Si is taken into account only via the SiAl end-member, which contains neither alkalies nor alkaline earth metals; retention of the latter in the amorphous layer is taken into account via the SiAlCa and SiAlNa end-members. It might have been justified to create two silicon-rich end-members, Si+AlCa and Si+AlNa, instead of a single SiAl end-member, but a simpler model was preferred.
-
The behavior of K, Mg, and Fe, elements abundant in natural environments, warrant specific studies.
-
When the pH rises above 9.5, the effect of Ca on the apparent solubility of Si can no longer be ignored otherwise the Si activities are overestimated. For a pH above 10, zeolite and calcium silicate hydrates can precipitate and sustain glass dissolution by consuming Si. Nucleation and growth kinetics of these minerals are therefore required for modeling glass alteration at such high pH values.
A series of scientific questions were raised in the introduction on the role of the alteration layer on glass dissolution kinetics. How amorphous layer forms and how passivation occurs at a molecular level remain open questions whose answers require molecular models. However, macroscopic geochemical models relying on measurable bulk concentrations are unavoidable tools for glass experiments understanding and glass long-term behavior predictions. This is due to their ability to describe chemical interactions and mass balances, their ability to explain how elements split between the amorphous layer and secondary crystallized minerals, their ability to calculate the fluid composition and therefore the effect of a fluid renewal. The apparent solubility of the amorphous minerals is a key parameter: there is still a need to better understand their precipitation processes at low temperature (<100 °C) and their ripening in conjunction with their transport properties in order to better predict the fate of glass and minerals in natural environments.
Glasses, as well as many multi-element minerals, or even metals, exhibit non-congruent dissolution. This non-congruent dissolution is both due to the faster dissolution rates of some chemical bonds, typically those involved with alkali metals, and the ability of some elements to precipitate, typically Si and Al metal oxides. It ends up in the formation of a layer with potential passivation properties. Moreover, this non-congruent dissolution has major consequences on fluid pH and composition which affects, in turn, the dissolution of the surface layer.
A simple and very general model has been proposed for modeling passivation as part of a reactive transport code. Concepts, equations, and implementation feedback have been presented. Fluid chemistry can be predicted by the model, thus it enables us to focus on the modeling of the dissolution-condensation phenomena in solution in attempt to give a better description of the composition and solubility of the protective layer, thereby improving the understanding of chemical interactions at low temperature in an amorphous mineral.
Validation is a key step toward justifying the simplified assumptions on which the model is based. The usefulness of the model was demonstrated by applying it to other glasses, especially the French reference inactive surrogate SON68, and can be tested further by application to other solution compositions or, more generally, for modeling interactions with other solids in the glass environment.
Data availability
The authors declare that the data supporting the findings of this study are available within the article.
References
Muir, I. J. & Nesbitt, H. W. Reactions of aqueous anions and cations at the labradorite-water interface: coupled effects of surface processes and diffusion. Geochim. Cosmochim. Acta 61, 265–274 (1997).
Chardon, E. S., Livens, F. R. & Vaughan, D. J. Reactions of feldspar surfaces with aqueous solutions. Earth Sci. Rev. 78, 1–26 (2006).
Hellmann, R., Dran, J. C. & Della Mea, G. The albite water system: Part III. Characterization of leached and hydrogen - enriched layers formed at 300 °C using MeV ion Beam techniques. Geochim. Cosmochim. Acta 61, 1575–1594 (1997).
Schott, J. et al. Formation, growth and transformation of leached layers during silicate minerals dissolution: the example of wollastonite. Geochim. Cosmochim. Acta 98, 259–281 (2012).
Daval, D. et al. Influence of amorphous silica layer formation on the dissolution rate of olivine at 90 °C and elevated pCO2. Chem. Geol. 284, 193–209 (2011).
Pokrovsky, O. S. & Schott, J. Forsterite surface composition in aqueous solutions: a combined potentiometric, electrokinetic, and spectroscopic approach. Geochim. Cosmochim. Acta 64, 3299–3312 (2000).
Oelkers, E. H., Golubev, S. V., Chairat, C., Pokrovsky, O. S. & Schott, J. The surface chemistry of multi-oxide silicates. Geochim. Cosmochim. Acta 73, 4617–4634 (2009).
Zhang, L. & Luttge, A. Theoretical approach to evaluating plagioclase dissolution mechanisms. Geochim. Cosmochim. Acta 73, 2832–2849 (2009).
Casey, W. H., Banfield, J. F., Westrich, H. R. & McLaughlin, L. What do dissolution experiments tell us about natural weathering? Chem. Geol. 105, 1–15 (1993).
Hellmann, R. et al. Unifying natural and laboratory chemical weathering with interfacial dissolution–reprecipitation: a study based on the nanometer-scale chemistry of fluid–silicate interfaces. Chem. Geol. 294–295, 203–216 (2012).
Urosevic, M. et al. In situ nanoscale observations of the dissolution of dolomite cleavage surfaces. Geochim. Cosmochim. Acta 80, 1–13 (2012).
Chave, T., Frugier, P., Gin, S. & Ayral, A. Glass-water interphase reactivity with calcium rich solutions. Geochim. Cosmochim. Acta 75, 4125–4139 (2011).
Conradt, R. Chemical durability of oxide glasses in aqueous solutions: a review. J. Am. Ceram. Soc. 91, 728–735 (2008).
Crovisier, J., Atassi, H., Daux, V., Honnorez, J., Petit, J. & Eberhart, J. A New Insight into the Nature of the Leached Layers Formed on Basaltic Glasses in Relation to the Choice of Constraints for Long Term Modelling. MRS Proceedings 127, 41 (1988).
Van Iseghem, P. et al. A Critical Evaluation of the Dissolution Mechanisms of High-Level Waste Glasses in Conditions of Relevance for Geological Disposal (GLAMOR), SCK.CEN, CEA, Subatech, PNNL, and SRNL, Report No. EUR 23097, 1-164 (2007).
Bourcier, W. L., Peiffer, D. W., Knauss, K. G., McKeegan, K. D. & Smith, D. K. in Scientific Basis for Nuclear Waste Management XIII. 176 edn, Vol. 176 (eds Oversby, V. M. & Brown, P. W.) 209–216 (Mater. Res.Soc., Pittsburg, Pennsylvania, 1990).
Cailleteau, C. et al. Insight into silicate-glass corrosion mechanisms. Nat. Mater. 7, 978–983 (2008).
GinS.. & JégouC.. Theoretical review of the Aagaard-Helgeson law applied to the dissolution of minerals and glasses. Am. J. Sci 255, 1–14 (2002).
Grambow, B. & Muller, R. First-order dissolution rate law and the role of surface layers in glass performance assessment. J. Nucl. Mater. 298, 112–124 (2001).
Aagaard, P. & Helgeson, H. C. Thermodynamic and kinetic constraints on reaction rates among minerals and aqueous solutions. I. Theoretical considerations. Am. J. Sci. 282, 237–285 (1982).
Frugier, P., Chave, T., Gin, S. & Lartigue, J. E. Application of the GRAAL model to leaching experiments with SON68 nuclear glass in initially pure water. J. Nucl. Mater. 392, 552–567 (2009).
Frugier, P. et al. SON68 nuclear glass dissolution kinetics: current state of knowledge and basis of the new GRAAL model. J. Nucl. Mater. 380, 8–21 (2008).
van der Lee, J., De Windt, L., Lagneau, V. & Goblet, P. Module-oriented modeling of reactive transport with HYTEC. Comput. Geosci. 29, 265–275 (2003).
Paul, A. Chemical durability of glasses; a thermodynamic approach. J. Mater. Sci. 12, 2246–2268 (1977).
Crovisier, J. L., Fritz, B., Grambow, B. & Eberhart, J. P. in Scientific Basis for Nuclear Waste Management IX 50 edn, Vol. 50 (ed Werme, L.) 273–280 (Mater. Res. Soc., Pittsburg, Pennsylvania, 1985).
Crovisier, J. L., Honnorez, J., Fritz, B. & Petit, J. C. Dissolution of subglacial volcanic glasses from Iceland: laboratory study and modelling. Appl. Geochem. 7, 55–81 (1992).
Curti, E. Modelling the Dissolution of Borosilicate Glasses for Radioactive Waste Disposal with the PHREEQE/GLASSOL Code: Theory and Practice. Report No. PSI-Bericht Nr.86., 1–62 (Baden, Switzerland, 1991).
Advocat, T., Crovisier, J. L., Clement, A., Gerard, F. & Vernaz, E. in Scientific Basis for Nuclear Waste Management XVIII Vol. 353 (eds Murakami, T. & Ewing, R. C.) 31–38 (Mater. Res. Soc., Pittsburg, Pennsylvania, 1995).
Daux, V., Guy, C., Advocat, T., Crovisier, J. L. & Stille, P. Kinetic aspects of basaltic glass dissolution at 90 °C: role of aqueous silicon and aluminium. Chem. Geol. 142, 109–126 (1997).
Munier, I., Grambow, B., Fritz, B. & Clement, A. Modelling the alteration gel composition of simplified borosilicate glasses by precipitation of an ideal solid solution in equilibrium with the leachant. J. Nucl. Mater. 324, 97–115 (2004).
Gysi, A. P. & Stefansson, A. Experiments and geochemical modeling of CO2 sequestration during hydrothermal basalt alteration. Chem. Geol. 306–307, 10–28 (2012).
Minet, Y., Bonin, B., Gin, S. & Frugier, P. Analytic implementation of the GRAAL model: application to a R7T7-type glass package in a geological disposal environment. J. Nucl. Mater. 404, 178–202 (2010).
Fournier, M., Gin, S. & Frugier, P. Resumption of nuclear glass alteration: state of the art. J. Nucl. Mater. 448, 348–363 (2014).
Frugier, P., Fournier, M. & Gin, S. Modeling resumption of glass alteration due to zeolites precipitation. Procedia Earth Planet. Sci. 17, 340–343 (2017).
Gin, S. et al. The fate of silicon during glass corrosion under alkaline conditions: a mechanistic and kinetic study with the International Simple Glass. Geochim. Cosmochim. Acta 151, 68–85 (2015).
Fournier, M., Frugier, P. & Gin, S. Application of GRAAL model to the resumption of International Simple Glass alteration. npj Mater. Degrad. 2, 21 (2018).
Fournier, M., Gin, S., Frugier, P. & Mercado-Depierre, S. Contribution of zeolite-seeded experiments to the understanding of resumption of glass alteration. npj Mater. Degrad. 1, 17 (2017).
Gin, S., Beaudoux, X., Angeli, F., Jegou, C. & Godon, N. Effect of composition on the short-term and long-term dissolution rates of ten glasses of increasing complexity from 3 to 30 oxides. J. Non-Cryst. Solids 358, 2559–2570 (2012).
Crovisier, J. L., Advocat, T. & Dussossoy, J. L. Nature and role of natural alteration gels formed on the surface of ancient volcanic glasses (natural analogs of waste containment glasses). J. Nucl. Mater. 321, 91–109 (2003).
Ducasse, T. et al. Alteration of synthetic basaltic glass in silica saturated conditions: analogy with nuclear glass. Appl. Geochem. 97, 19–31 (2018).
Gin, S. et al. The controversial role of inter-diffusion in glass alteration. Chem. Geol. 440, 115–123 (2016).
Gin, S. et al. Atom-probe tomography, TEM and ToF-SIMS study of borosilicate glass alteration rim: a multiscale approach to investigating rate-limiting mechanisms. Geochim. Cosmochim. Acta 202, 57–76 (2017).
Hellmann, R. et al. Nanometre-scale evidence for interfacial dissolution-reprecipitation control of silicate glass corrosion. Nat. Mater. 14, 307–311 (2015).
Gin, S. et al. Origin and consequences of silicate glass passivation by surface layers. Nat. Commun. 6, 6360 (2015).
Gin, S. et al. Dynamics of self-reorganization explains passivation of silicate glasses. Nat. Commun. 9, 2169 (2018).
Delage, F., Ghaleb, D., Dussossoy, J. L., Chevallier, O. & Vernaz, E. A mechanistic model for understanding nuclear waste glass dissolution. J. Nucl. Mater. 190, 191–197 (1992).
Collin, M. et al. Structure of International Simple Glass and properties of passivating layer formed in circumneutral pH conditions. npj Mater. Degrad. 2, 4 (2018).
Debure, M., Frugier, P., De Windt, L. & Gin, S. Borosilicate glass alteration driven by magnesium carbonates. J. Nucl. Mater. 420, 347–361 (2012).
Jollivet, P. et al. Effect of clayey groundwater on the dissolution rate of the simulated nuclear waste glass SON68. J. Nucl. Mater. 420, 508–518 (2012).
Arena, H. et al. Impact of Zn, Mg, Ni and Co elements on glass alteration: additive effects. J. Nucl. Mater. 470, 55–67 (2016).
Aréna, H. et al. Impact of iron and magnesium on glass alteration: characterization of the secondary phases and determination of their solubility constants. Appl. Geochem. 82, 119–133 (2017).
Debure, M., Frugier, P., De Windt, L. & Gin, S. Dolomite effect on borosilicate glass alteration. Appl. Geochem. 33, 237–251 (2013).
Ribet, S. & Gin, S. Role of neoformed phases on the mechanisms controlling the resumption of SON68 glass alteration in alkaline media. J. Nucl. Mater. 324, 152–164 (2004).
Arab, M. et al. Aqueous alteration of five-oxide silicate glasses: experimental approach and Monte Carlo modeling. J. Non-Cryst. Solids 354, 155–161 (2008).
Jollivet, P. et al. Investigation of gel porosity clogging during glass leaching. J. Non-Cryst. Solids 354, 4952–4958 (2008).
Chave, T., Frugier, P., Ayral, A. & Gin, S. Solid state diffusion during nuclear glass residual alteration in solution. J. Nucl. Mater. 362, 466–473 (2007).
Rajmohan, N., Frugier, P. & Gin, S. Composition effects on synthetic glass alteration mechanisms: Part 1. Experiments. Chem. Geol. 279, 106–119 (2010).
Jegou, C. Mise en évidence expérimentale des mécanismes limitant l’altération du verre R7T7 en milieu aqueux. Critique et proposition d'évolution du formalisme cinétique (University of Montpellier II, Montpellier, 1998).
Gin, S. & Jegou, C. in Water-Rock Interaction (ed Balkema, A. A.) 279–282 (2001).
Gin, S. et al. An international initiative on long-term behavior of high-level nuclear waste glass. Mater. Today 16, 243–248 (2013).
Advocat, T., Crovisier, J. L., Vernaz, E., Ehret, G. & Charpentier, H. in Scientific Basis for Nuclear Waste Management XIV 212 edn. Vol. 212 (eds Abrajano, T. A., Jr. & Johnson, L. H.) 57–64 (Mater. Res. Soc., Pittsburg, Pennsylvania,1991).
Spalla, O., Barboux, P., Sicard, L., Lyonnard, S. & Bley, F. Influence of insoluble elements on the nanostructure of water altered glasses. J. Non-Cryst. Solids 347, 56–68 (2004).
Sverdrup, H. & Warfvinge, P. Estimating field weathering rates using laboratory kinetics. Rev. Mineral. 3, 485–541 (1995).
Jégou, C., Gin, S. & Larché, F. Alteration kinetics of a simplified nuclear glass in an aqueous medium: effects of solution chemistry and of protective gel properties on diminishing the alteration rate. J. Nucl. Mater. 280, 216–229 (2000).
Ledieu, A., Devreux, F., Barboux, P. & Minet, Y. Contribution of Monte Carlo modeling to understanding the alteration of nuclear glasses by water. Nucl. Sci. Eng. 153, 285–300 (2006).
Gin, S., Jegou, C., Frugier, P. & Minet, Y. Theoretical consideration on the application of the Aagaard-Helgeson rate law to the dissolution of silicate minerals and glasses. Chem. Geol. 255, 14–24 (2008).
Lasaga, A. C. Transition state theory, in Kinetics of Geochemical Processes. Rev. Mineral. 8, 135–169 (1981).
Lüttge, A. Crystal dissolution kinetics and Gibbs free energy. J. Electron Spectrosc. Relat. Phenom. 150, 248–259 (2006).
Arvidson, R. S. & Luttge, A. Mineral dissolution kinetics as a function of distance from equilibrium - new experimental results. Chem. Geol. 269, 79–88 (2010).
Richardson, I. G. The calcium silicate hydrates. Cem. Concr. Res. 38, 137–158 (2008).
Sugiyama, D. & Fujita, T. A thermodynamic model of dissolution and precipitation of calcium silicate hydrates. Cem. Concr. Res. 36, 227–237 (2006).
Houston, J. R., Herberg, J. L., Maxwell, R. S. & Carroll, S. A. Association of dissolved aluminum with silica: connecting molecular structure to surface reactivity using NMR. Geochim. Cosmochim. Acta 72, 3326–3337 (2008).
Pelmenschikov, A., Strandh, H., Petterson, L. G. M. & Leszczynski, J. Lattice resistance to hydrolysis of Si-O-Si bonds of silicate minerals: ab initio calculations of a single water attack onto the (001) and (111) beta-cristobalite surfaces. J. Phys. Chem. B 104, 5779–5783 (2000).
Criscenti, L. J., Kubicki, J. D. & Brantley, S. L. Silicate glass and mineral dissolution: calculated reaction paths and activation energies for hydrolysis of a Q3 Si by H3O+ using ab initio methods. J. Phys. Chem. A 110, 198–206 (2006).
Collin, M., Fournier, M., Charpentier, T., Moskura, M. & Gin, S. Impact of alkali on the passivation of silicate glass. npj Mater. Degrad. 2, 16 (2018).
Collin, M. et al. Molecular dynamics simulations of water structure and diffusion in a 1 nm diameter silica nanopore as a function of surface charge and alkali metal counterion identity. J. Phys. Chem. C 122, 17764–17776 (2018).
Angeli, F. et al. Influence of calcium on sodium aluminosilicate glass leaching behaviour. Phys. Chem. Glasses 42, 279–286 (2001).
Angeli, F., Charpentier, T., Gin, S. & Petit, J. C. O-17 3Q-MAS NMR characterization of a sodium aluminoborosilicate glass and its alteration gel. Chem. Phys. Lett. 341, 23–28 (2001).
Angeli, F., Gaillard, M., Jollivet, P. & Charpentier, T. Influence of glass composition and alteration solution on leached silicate glass structure: a solid-state NMR investigation. Geochim. Cosmochim. Acta 70, 2577–2590 (2006).
Iler, R. K. Effect of adsorbed alumina on the solubility of amorphous silica in water. J. Colloid Interface Sci. 43, 399–408 (1973).
Oelkers, E. H. & Schott, J. The effect of aluminum, pH, and chemical affinity on the rates of aluminosilicate dissolution reactions. Geochim. Cosmochim. Acta 58, 2011–2024 (1994).
Van Bennekom, A. J., Buma, A. G. J. & Nolting, R. F. Dissolved aluminium in the Weddell-Scotia confluence and effect of Al on the dissolution kinetics of biogenic silica. Mar. Chem. 35, 423–434 (1991).
Acknowledgements
This work was funded by the French Alternative Energies and Atomic Energy Commission (CEA) and ORANO.
Author information
Authors and Affiliations
Contributions
P.F. wrote the paper. Experiments were done by N.R. Modeling was done by P.F., N.R., and Y.M. N.G. was deeply involved in the data interpretation. S.G. supervised the study. N.G. and S.G. corrected the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Frugier, P., Minet, Y., Rajmohan, N. et al. Modeling glass corrosion with GRAAL. npj Mater Degrad 2, 35 (2018). https://doi.org/10.1038/s41529-018-0056-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-018-0056-z
This article is cited by
-
The fate of Si and Fe while nuclear glass alters with steel and clay
npj Materials Degradation (2021)
-
Aqueous alteration of silicate glass: state of knowledge and perspectives
npj Materials Degradation (2021)
-
Forty years of durability assessment of nuclear waste glass by standard methods
npj Materials Degradation (2021)
-
Self-accelerated corrosion of nuclear waste forms at material interfaces
Nature Materials (2020)
-
Insights into the mechanisms controlling the residual corrosion rate of borosilicate glasses
npj Materials Degradation (2020)
