
Abstract
Acute lymphoblastic leukemia is the most common pediatric cancer characterized by a heterogeneous genomic landscape with copy number aberrations occurring at various stages of pathogenesis, disease progression, and treatment resistance. In this study, disease-relevant copy number aberrations were profiled in bone marrow samples of 91 children with B- or T-cell precursor acute lymphoblastic leukemia using digital multiplex ligation-dependent probe amplification (digitalMLPATM). Whole chromosome gains and losses, subchromosomal copy number aberrations, as well as unbalanced alterations conferring intrachromosomal gene fusions were simultaneously identified with results available within 36 hours. Aberrations were observed in 96% of diagnostic patient samples, and increased numbers of copy number aberrations were detected at the time of relapse as compared with diagnosis. Comparative scrutiny of 24 matching diagnostic and relapse samples from 11 patients revealed three different patterns of clonal relationships with (i) one patient displaying identical copy number aberration profiles at diagnosis and relapse, (ii) six patients showing clonal evolution with all lesions detected at diagnosis being present at relapse, and (iii) four patients displaying conserved as well as lost or gained copy number aberrations at the time of relapse, suggestive of the presence of a common ancestral cell compartment giving rise to clinically manifest leukemia at different time points during the disease course. A newly introduced risk classifier combining cytogenetic data with digitalMLPATM-based copy number aberration profiles allowed for the determination of four genetic subgroups of B-cell precursor acute lymphoblastic leukemia with distinct event-free survival rates. DigitalMLPATM provides fast, robust, and highly optimized copy number aberration profiling for the genomic characterization of acute lymphoblastic leukemia samples, facilitates the decipherment of the clonal origin of relapse and provides highly relevant information for clinical prognosis assessment.
Similar content being viewed by others

Introduction
Acute lymphoblastic leukemia is the most common malignancy in childhood and includes several subtypes characterized by recurrent genetic alterations [1]. Whole chromosome gains and losses as well as subchromosomal copy number aberrations are frequently acquired during leukemogenesis and disease progression [2,3,4,5]. Numerical chromosome aberrations most commonly occur in patients with high-hyperdiploid karyotype (≥51 chromosomes) and predominantly confer gains of chromosomes X, 4, 6, 10, 14, 17, 18, and 21 [6, 7]. Subchromosomal lesions frequently involve key regulators of cell cycle control, tumor suppression, as well as lymphoid cell development/differentiation [2, 3], and were shown to be associated with clinical outcome leading to the recent introduction of copy number aberration-based risk stratification classifiers (IKZF1plus and UKALL-CNA) in B-cell precursor acute lymphoblastic leukemia [8,9,10]. Although individual patients with acute lymphoblastic leukemia harbor a limited number of somatic copy number aberrations as determined by previous molecular array-based and next-generation sequencing studies [2, 11], comprehensive and cost-efficient screening of disease-relevant aberrations requires highly multiplexed, targeted approaches due to the relatively wide repertoire of recurrently altered genomic loci that can potentially be affected. In addition, the frequently variable and small size of subchromosomal aberrations hamper the sensitive detection of copy number aberrations by using conventional methods, such as karyotyping, fluorescence in situ hybridization, or off-the-shelf array-based assays [12].
Digital multiplex ligation-dependent probe amplification (digitalMLPATM) is a recently developed technique combining conventional multiplex ligation-dependent probe amplification with high-throughput next-generation sequencing readout. In the first digitalMLPATM study, cell lines and samples of 65 patients with acute lymphoblastic leukemia were successfully analyzed and the analytical performance of the method demonstrated a diagnostic sensitivity of ~99% and a specificity of ~98% as compared with more conventional copy number aberration screening methods [13].
In this study, we performed a comprehensive profiling of disease-relevant copy number aberrations in an independent, larger cohort of patients diagnosed with pediatric B- or T-cell precursor acute lymphoblastic leukemia using digitalMLPATM. By comparing the copy number aberration profiles of matching diagnostic and relapse samples, we revealed distinct clonal relationships of leukemic cell populations prevailing at different time points during the disease course. Complementary time-resolved analysis of the abundant immunoglobulin heavy-chain (IGH) gene rearrangements (variable–diverse–joining) in patients with progressive B-cell precursor acute lymphoblastic leukemia allowed us to further scrutinize the clonal origin of relapse and to assess the added value provided by digitalMLPATM in this regard. Finally, prognostic value of digitalMLPATM results was investigated based on criteria of previous risk stratification classifiers, and a refined prognostic classifier has been introduced, which includes cytogenetic data and copy number status of an extended set of genomic loci interrogated by digitalMLPATM.
Materials and methods
Patient samples
Bone marrow samples from 91 children diagnosed with B-cell (n = 76) or T-cell (n = 15) precursor acute lymphoblastic leukemia at age 1–16 years (average: 6.8 years) were analyzed (Supplementary Table 1). Diagnoses were made based on morphological, immunophenotypical, and genotypical criteria [14], in the Department of Pathology, University of Pécs or in the 1st Department of Pathology and Experimental Cancer Research, Semmelweis University between 2004 and 2014. Patients were risk stratified and treated according to BFM protocols, such as the ALL IC-BFM 2002, ALL-REZ BFM 2002, and ALL IC-BFM 2009. In addition to the diagnostic samples of 91 patients, 14 samples from 12 patients drawn at the time of first or second relapse were also investigated. On average, 81% (range: 35–100%) blasts were measured across the specimens by flow cytometry. Ethical Committee approval and written informed consent from the patients’ parents or guardians were obtained for the study, which was conducted in accordance with the Declaration of Helsinki.
Digital multiplex ligation-dependent probe amplification
Forty nanograms of genomic DNA was subjected to each digitalMLPATM reaction using the D007 ALL probemix (version D007-X2–0516), which has recently been developed by the MRC Holland [13]. The probemix consisted a total of 598 probes including (i) 274 target-specific probes for regions recurrently altered by copy number aberrations in B- and/or T-cell precursor acute lymphoblastic leukemia (Supplementary Table 2); (ii) 196 digital karyotyping probes covering all chromosome arms for detection of gross chromosomal aberrations and to be used as reference probes for data normalization; as well as (iii) 128 internal control probes for sample identification and quantity/quality assessment [13].
DigitalMLPATM reactions were carried out as previously described [13, 15]. Briefly, the protocol started with mixing each individual sample DNA with a unique barcode solution followed by sample denaturation and the addition of digitalMLPATM probes with digitalMLPATM buffer to the mixture. Each probe comprised two or three oligonucleotides with a specific 25–50 bp hybridizing sequence. Probe oligonucleotides binding to a specific locus was designed to hybridize adjacently; therefore, if perfectly hybridized, could be ligated into a single molecule using ligase-65 enzyme. Ligated probes were then amplified by a universal primer pair compatible with Illumina sequencing platforms. After amplification, sample-specific polymerase chain reaction products (libraries) from different reactions were pooled, diluted, denatured, and loaded into a MiSeq v3 standard flow cell (Illumina) for 115 bp single-read sequencing.
After quality assessment of the exported FASTQ files, assignment of the reads to digitalMLPATM probes and subsequent data analysis were performed in two main consecutive steps using an in-house software [13]. First, read count for each probe was normalized by the median read count generated from reference probes hybridizing to usually copy number stable regions in the same genome (intrasample normalization). The relative read count generated for each probe was then compared with the matching values obtained in all reference samples (intersample normalization with at least one reference sample per seven patient samples). The final probe ratio value, called “dosage quotient,” was around 1.0 if the region of interest was unaffected by copy number aberration, while an increased or decreased value indicated the presence and level of gain or loss, respectively. Leukemic cell purity as measured by flow cytometry was also considered at the interpretation of the results. Copy number aberrations were interpreted as being subclonal if multiple consecutive probes had dosage quotients clearly outside the normal range but without reaching the expected level of monoallelic loss as calculated based on sample purity and also, as compared with other affected loci within the same specimen. Detailed general laboratory and bioinformatic protocols have previously been published [13, 15].
Conventional multiplex ligation-dependent probe amplification
Multiplex ligation-dependent probe amplification results for validation were either available from previous studies or acquired as previously described [16, 17]. Briefly, 100–150 ng input genomic DNA extracted from mononuclear cells was denatured and hybridized using one or more of the following SALSA probemixes: P202-A1, P202-B2, P335-A4, P335-B2, and P383-A2 (MRC Holland, Amsterdam, NL). Genes and regions specifically analyzed in B-cell precursor acute lymphoblastic leukemia cases included PAR1 region genes, IKZF2, EBF1, IKZF1, MIR31, CDKN2A/B, PAX5, ETV6, BTG1, RB1, 14q32.33 region genes, and IKZF3, while STIL-TAL1 fusion, LEF1, CASP8AP2, MYB, EZH2, CDKN2A/B, MTAP, MLLT3, NUP214-ABL1 fusion, PTEN, LMO1, LMO2, NF1, SUZ12, PTPN2, and PHF6 were investigated in patients with T-cell precursor acute lymphoblastic leukemia (Supplementary Table 2). The reactions, also including negative control samples, were performed according to the manufacturer’s recommendations. The amplified probes were analyzed using an ABI 3730 DNA analyzer (Life Technologies, Bleiswijk, The Netherlands) and the Coffalyser.Net software (MRC Holland). After intrasample and intersample normalizations, copy number status at each locus was assessed as described previously [16,17,18,19], also considering the proportion of leukemic cells in the sample as measured by flow cytometry.
IGH gene rearrangement profiling
Abundant clonal junctions of variable, diverse, and joining IGH gene segments were screened by targeted amplicon sequencing in matching diagnostic and relapse samples of patients with B-cell precursor acute lymphoblastic leukemia. Next-generation sequencing libraries were prepared using the LymphoTrack IGH FR1/2/3 assay (Invivoscribe, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, 50–100 ng genomic DNA was subjected to multiplex polymerase chain reaction with primers binding to conserved regions of variable and joining IGH gene segments. Depending on sample availability and considering options to combine various sequencing barcodes, framework region 1 (FR1) and/or FR2 specific primers were used in independent reactions for the targeted amplification of IGH variable–diverse–joining junctions. After purification with Agencourt Ampure XP magnetic beads (Beckman Coulter, Brea, CA, USA), libraries were normalized and pooled for sequencing on MiSeq Nano v2 flow cells with 250 bp paired-end chemistry (Illumina). FASTQ files were processed and results were evaluated using the LymphoTrack software (Invivoscribe) dedicated to identify clonotypes in the analyzed sample and rank those based on abundance. In accordance with the manufacturer’s recommendation, only rearrangements with an abundance of ≥2.5% or ≥5% as compared with the total read count were considered as clonotypes in case the total read count exceeded 20,000 or 10,000, respectively. In addition, all clonotype sequences were manually analyzed and interpreted using the ImMunoGeneTics V-Quest tool (www.imgt.org). Finally, patient-specific clonotypes identified in matching bone marrow samples drawn at the time of diagnosis and relapse were compared.
Statistical analysis
Congruency between digitalMLPATM results and multiplex ligation-dependent probe amplification data was evaluated with Fisher’s exact test. Correlation between the abundance values of IGH clonotypes as identified by FR1 and FR2 assays was assessed by calculating the Pearson’s correlation coefficient. Event-free survival was defined as the time from the start of treatment to relapse, second malignancy, or disease-related death with up to 3000 days of follow-up time. Survival rates were estimated using the Kaplan–Meier method and compared by log-rank tests. Statistical analyses were performed using SPSS 15.0 software (SPSS Inc., Chicago, IL) and R version 3.5.3 (R Foundation for Statistical Computing, Vienna, Austria).
Results
All 105 (91 diagnostic and 14 relapse) patient samples were successfully analyzed by digitalMLPATM with an average of 861 next-generation sequencing reads per probe which provided sufficient data for precise read quantification and downstream assessment of copy number status. In line with previously published data [13], analysis of a dilution series prepared from the diagnostic sample of patient #51 harboring seven mono- and two biallelic losses on autosomal chromosomes demonstrated that the method efficiently detects clonal copy number aberrations in samples with at least 30% leukemic cell purity (Supplementary Table 3); a cutoff that was exceeded by all samples analyzed in this study.
Copy number aberrations in diagnostic patient samples
In total, 502 copy number aberrations were detected in 87 (96%) diagnostic patient samples (B-cell precursor acute lymphoblastic leukemia: 73/76, T-cell precursor acute lymphoblastic leukemia: 14/15), with 458 and 44 observed in B- and T-cell precursor acute lymphoblastic leukemia, respectively. On average, 5.4 copy number aberrations were detected per patient (B-cell precursor acute lymphoblastic leukemia: 6.0, T-cell precursor acute lymphoblastic leukemia: 2.6) with a mean of 2.8 subchromosomal aberrations (B-cell precursor acute lymphoblastic leukemia: 2.9, T-cell precursor acute lymphoblastic leukemia: 2.5).
Identification of gross chromosomal aberrations was facilitated by a karyotyping subset of the probes covering all chromosomes near the telomere, centromere, and the middle of the arm. Ninety-five percent of whole chromosome alterations were observed in patients with hyperdiploid karyotype (3–14 affected chromosomes per patient) and were dominated by extra copies of chromosomes X, 4, 6, 10, 14, 17, 18, and 21; a characteristic feature of high-hyperdiploid acute lymphoblastic leukemia. Gain of multiple copies was recurrently observed at chromosomes X, 18, and 21. Modal chromosome number among the 27 patients with hyperdiploid karyotype ranged between 49 and 62 with a median of 56 (Supplementary Fig. 1).
Subchromosomal copy number aberrations were identified in 77 patients (B-cell precursor acute lymphoblastic leukemia: 63, T-cell precursor acute lymphoblastic leukemia: 14), with 218 and 33 alterations in B- and T-cell precursor acute lymphoblastic leukemia cases, respectively. Deletion of CDKN2A/B was the most common lesion occurring in 38% of B-cell precursor acute lymphoblastic leukemia and 87% of T-cell precursor acute lymphoblastic leukemia cases, besides other repeatedly altered cell cycle control, lymphoid development, signaling, or tumor suppressor genes, such as ABL1, BTG1, CASP8AP2, CD200/BTLA, ETV6, IKZF1, LEF1, MLLT3, NF1, PAX5, PHF6, PTEN, PTPN2, RB1, RUNX1, TBL1XR1, TP53, and VPREB1 as well as the PAR1 region (Table 1 and Supplementary Fig. 2). Biallelic losses were detected in diagnostic samples of 36 patients (B-cell precursor acute lymphoblastic leukemia: 23, T-cell precursor acute lymphoblastic leukemia: 13), and almost two-thirds of these aberrations affected the 9p21 region (CDKN2A/B and MLLT3). In B-cell precursor acute lymphoblastic leukemia, further biallelic losses were detected in VPREB1, RB1, IKZF2, and TBL1XR1 genes, while in T-cell precursor acute lymphoblastic leukemia, biallelic losses of PTPN2, PHF6, LEF1, and RB1 were observed (Supplementary Fig. 2).
Besides copy number aberration profiling, fusion genes occurring via copy number alterations can indirectly be detected by digitalMLPATM. Subchromosomal aberrations referring to the presence of intrachromosomal fusion genes were observed in five patients with T-cell precursor acute lymphoblastic leukemia. STIL-TAL1 gene fusion was identified in patients #77 and #78, while dosage quotient values indicating the fusion and amplification of the NUP214 and ABL1 genes were observed in patients #81, #83, and #84.
Comparative analysis of copy number aberration profiles at diagnosis and relapse
In total, 26 matching diagnostic and relapse samples were investigated from 12 patients with 8 showing B-cell precursor acute lymphoblastic leukemia phenotype and 4 diagnosed with T-cell precursor acute lymphoblastic leukemia (Table 2). Chromosomal and/or subchromosomal copy number aberrations were detected in 11 patients with 1 patient displaying normal copy number profile in all samples analyzed. Whole chromosome alterations were exclusively identified in B-cell precursor acute lymphoblastic leukemia patients harboring high-hyperdiploid chromosome set (patients #22, #23, #24, #25, and #26). Chromosomal gains were stable in three of these patients over time, while dosage quotient values referring to the gain of an extra copy of chromosome 8 were observed in patient #23 at the time of relapse. In patient #26, changes in copy numbers of six different chromosomes were observed between diagnosis and first relapse. In the 11 patients with detectable copy number aberrations, the number of subchromosomal aberrations ranged between 1 and 10 (biallelic losses considered as results of two independent events) with an increased number of copy number aberrations observed at the time of relapse as compared with diagnosis (mean: 3.5 vs 5.1). In line with this finding, an increased number of biallelic losses was observed at the time of disease progression in three (#22, #57, and #58) out of seven patients harboring this type of aberrations in any of their samples. Copy number aberrations specifically emerging at relapse affected B-cell differentiation, cell cycle control, and tumor suppressor genes, such as EBF1, ETV6, CDKN2A/B, IKZF1, PAX5, and PTEN, in patients with B-cell precursor acute lymphoblastic leukemia, while MYB gain, NUP214-ABL1 fusion and gain as well as losses of IKZF1, TP53, and PTPN2 were observed in patients with progressive T-cell precursor acute lymphoblastic leukemia.
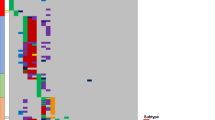
Comparative scrutiny of the matching copy number aberration profiles with the aim to reveal the relationship between clones dominating at diagnosis and relapse uncovered three different patterns: (i) completely identical copy number aberrations were observed in one patient (#88) at diagnosis and relapse (Fig. 1a); (ii) all copy number aberrations identified in the diagnostic sample were present at disease progression, plus additional aberrations indicated clonal evolution in six patients (#22, #23, #25, #57, #58, and #89; Fig. 1b); and (iii) in four patients (#24, #26, #76, and #91), only a subset of copy number aberrations detected in the diagnostic sample was retained at relapse suggesting that a common ancestral cell compartment gave rise to clinically manifest leukemia at different time points during the disease course (Fig. 1c). Samples at three different time points were available from two patients allowing for the analysis of copy number aberration profiles at the time of a second relapse. In patient #26, the leukemic cell population occurring at the second relapse most likely arose from an ancestral clone while in patient #91, the profiles observed at the first and second relapses suggested a direct clonal evolutionary relationship (Fig. 1c).

a Monoallelic and biallelic losses on chromosome 9 (9p21; CDKN2A) and monoallelic loss on chromosome 11 (11p12-13; RAG2, SLC1A2 and CD44) observed in patient #88 with T-cell precursor acute lymphoblastic leukemia at diagnosis and at relapse (month 5). b Hyperdiploid karyotype with extra chromosomes 6, 10, 14, 17, 18, 21, and X as well as a monoallelic gain on chromosome 1 (1q21-44; several genes) and a monoallelic loss on chromosome 9 (9p21; CDKN2A) detected in patient #22 with B-cell precursor acute lymphoblastic leukemia at diagnosis. At the time of relapse (month 37), additional mono- and bi-allelic losses occurred on chromosome 9 due to a larger deletion on the second allele (9p13-21; several genes including CDKN2A/B and PAX5). c In patient #91 diagnosed with T-cell precursor acute lymphoblastic leukemia, monoallelic losses were detected on chromosome 9 (9p21; CDKN2A), chromosome 13 (13q14; RB1), and chromosome X (Xq26, PHF6) as well as a biallelic loss on chromosome 9 (9p21; CDKN2A and CDKN2B) at the time of diagnosis. At the first relapse (month 29), additional monoallelic losses occurred on chromosomes 7 (7p12; IKZF1) and 18 (18p11; PTPN2). At the time of the second relapse (month 50), monoallelic gain on chromosome 9 (9q21-34; several genes including ABL1, NUP214 and NOTCH1) and monoallelic loss on chromosome 17 (17p11-13; several genes including TP53) were newly detected. The monoallelic loss identified on chromosome 13 at diagnosis was not observed in samples drawn at the first or second relapse. Red and blue arrows indicate alterations not uniformly observed in samples drawn from the same patient at different time points.
Time between diagnosis and first relapse of the three T-cell precursor acute lymphoblastic leukemia patients displaying altered copy number aberration profiles suggested a prolonged time requirement of clonal evolution (patient #89, 17 months), and of the development of manifest leukemia from an ancestral clone (patient #91, 29 months) compared with the quick return of an identical clone at the time of relapse (patient #88, 5 months) (Fig. 2). Patient #90 showing normal copy number aberration profile at both diagnosis and relapse had a rapid disease progression (month 5) based on which we speculate that a clone highly similar or identical to the one dominating at diagnosis gained selective advantage at the time of relapse. A similar trend in terms of time from diagnosis to relapse was not observed across patients with B-cell precursor acute lymphoblastic leukemia.

During the development of pediatric acute lymphoblastic leukemia, malignant transformation commonly occurs in utero and via limited clonal expansion, confers a preleukemic cell compartment with heterogeneous genomic background and branching subclonal architecture. Development of the clinically manifest leukemia is usually facilitated by secondary aberrations providing the affected cells with selective proliferative advantage. Relapse can be driven (i) by the return of the original leukemic clone, (ii) by clonal evolution, or (iii) by a cell population arising from an ancestral clone and developing in parallel with cells conferring leukemia at the time of diagnosis.
Validation of digital multiplex ligation-dependent probe amplification data
Conventional multiplex ligation-dependent probe amplification probemixes developed for the analysis of patients with B- or T-cell precursor acute lymphoblastic leukemia mostly contain probes with different ligation sites as compared with those in the D007 digitalMLPATM probemix, thus can be used for confirmation of digitalMLPATM results. Copy number aberrations identified by digitalMLPATM were validated with 121 multiplex ligation-dependent probe amplification reactions performed on 97/105 samples analyzed in this study (Supplementary Table 4). Comparison of the results obtained by the two methods demonstrated high concordance with congruency at 936/949 (98.6%) data points (Supplementary Table 5). The vast majority of discrepancies was observed in the CDKN2A/B region which was covered by an over five times higher number of digitalMLPATM probes as compared with the conventional multiplex ligation-dependent probe amplification probes used, allowing for a more robust copy number aberration detection. Routinely performed polymerase chain reaction and interphase fluorescence in situ hybridization tests confirmed the STIL-TAL1 gene fusion in patients #77 and #78 as well as the ETV6 loss, RUNX1 gain/amplification and ABL1 gain/amplification in all tested cases where digitalMLPATM detected a copy number alteration.
Comparative analysis of IGH clonotypes at diagnosis and relapse
IGH gene rearrangements were screened in 17 samples of 8 patients diagnosed with B-cell precursor acute lymphoblastic leukemia and showing disease progression (Supplementary Table 6). Clonotypes with abundance exceeding the detection limit of the assay were identified in seven patients with four patients harboring a single dominant rearrangement and three patients carrying 2, 3, or 4 clonal variable–diverse–joining junctions. The percentage of reads representing a clonotype was on average 40.5% with a range of 3.0–93.2% across all samples and reactions. Abundance values determined by matching FR1 and FR2 reactions performed on the same samples showed strong correlation (r = 0.9734). Comparison of the IGH rearrangements identified at diagnosis and relapse revealed identical compositions of abundant clonotypes in 6/7 patients. In patient #26, the four clonotypes detected at diagnosis most likely arose on the same allele, which assumption is supported by a shared joining segment and 13 identical N2 bases, as well as a cumulative clonotype abundance of ~44% (FR1: 41.8%, FR2: 46.2%) with a disomic chromosome 14 configuration and 87% blast content in the sample. This diversified profile suggests an ongoing recombination activity driven by RAG enzymes in this patient, which mechanism is also the most plausible explanation for the absence of detectable clonotypes in the matching samples drawn during the first and second relapses.
Genetic risk classification
Copy number aberrations detected by digitalMLPATM were used for stratifying patients with B-cell precursor acute lymphoblastic leukemia into genetic risk groups. As in previous copy number aberration-based prognostic classification studies, event-free survival was investigated as the primary end point with a median follow-up time of 72 months (range: 10–99 months). Since version of the treatment protocol (ALL IC-BFM 2002 vs 2009) had no significant influence on the event-free survival in this cohort, data from all B-cell precursor acute lymphoblastic leukemia patients were combined.
Allelic loss of the transcription factor IKZF1 was found to be associated with adverse clinical outcome in previous studies and a recent study defined a very poor prognostic subgroup called IKZF1plus comprising patients who in addition to the IKZF1 deletion, harbor PAX5, CDKN2A/B, or PAR1 deletion, without concurrent ERG loss [9]. In our cohort, patients displaying IKZF1 loss with or without fulfilling the criteria of IKZF1plus showed significantly shorter event-free survival as compared with patients with intact IKZF1 alleles (Supplementary Fig. 3).
The UKALL-CNA classifier introduced by Moorman et al. stratifies patients based on copy number aberrations in eight genes/regions (EBF1, IKZF1, CDKN2A/B, PAX5, ETV6, BTG1, RB1, and PAR1) most commonly altered in B-cell precursor acute lymphoblastic leukemia and simultaneously analyzable by commercially available conventional multiplex ligation-dependent probe amplification kits [8]. The updated version of this classifier and its combination with cytogenetic risk groups have recently been tested on a large cohort of B-cell precursor acute lymphoblastic leukemia patients across various clinical trials and study groups [10]. Compared with conventional multiplex ligation-dependent probe amplification kits, the D007 digitalMLPATM assay allows for screening copy number aberrations in additional genes/regions recurrently altered in B-cell precursor acute lymphoblastic leukemia (e.g., IKZF2, CD200/BTLA, TBL1XR1, NR3C2, ABL1, TP53, RUNX1, and ERG); thus, we investigated the potential of using an alternative classifier to stratify B-cell precursor acute lymphoblastic leukemia patients based on a more comprehensive disease-relevant copy number aberration profile. Four risk groups exclusively determined based on copy number aberrations and comprising patients with different event-free survival rates were established (Table 3 and Supplementary Fig. 4). Similar to the UKALL risk assessment strategy, we integrated our copy number aberration-based classifier relying on digitalMLPATM data with cytogenetic risk groups, with the latter ones defined as described in the UKALL system [10]. After visual inspection of the event-free survival curves derived from various combinations of copy number aberration-based and cytogenetics-defined prognostic groups, four combined genetic risk groups showing significantly different survival rates have been determined (Supplementary Table 7 and Fig. 3).

Event-free survival with patients stratified based on combined cytogenetic and digitalMLPATM-based copy number aberration profile criteria as outlined in Supplementary Table S7. CI confidence interval.
Discussion
Treatment efficacy of pediatric acute lymphoblastic leukemia has remarkably improved during the past decades with currently ~90% of patients achieving 5-year survival in several developed countries [20]. The spectacular improvement in clinical outcome can be attributed to an increasingly optimized risk stratification of patients, which among others is facilitated by the identification of recurrent genetic aberrations with predictive and prognostic impact at the time of diagnosis [21]. Owing to the recent advancement in high-throughput sequencing technologies, a great demand for comprehensive and sophisticated profiling of driver and cooperative genetic alterations has emerged in the clinical practice [22]. Hence, application of methods optimized for the efficient characterization of simultaneously present, disease-relevant genetic aberrations is highly warranted in the diagnostic and clinical workflow.
In this study, we used a digitalMLPATM assay specifically designed for the comprehensive screening of unbalanced genomic lesions recurrently observed in pediatric B- and T-cell precursor acute lymphoblastic leukemia. The method has (i) great scalability and high throughput with potentially up to 192 samples that can be multiplexed per sequencing run, (ii) low input requirement allowing for library preparation from as low as 20 ng DNA, and (iii) short turn-around time providing results within 36 hours [13, 15]. Since digitalMLPATM is a highly targeted approach, data processing and evaluation are computationally less demanding as compared with most next-generation sequencing methods. Due to the rationalized assay design, it provides higher throughput in an informative manner than alternative genome-wide applications aiming to infer copy number aberrations. Owing to its specific probe composition, digitalMLPATM allows for detection of both genome-wide whole chromosome alterations and large copy number aberrations as well as for targeted high-resolution interrogation of driver regions in a single reaction, which makes the technique attractive for the genomic research and diagnostics of pediatric acute lymphoblastic leukemia. Notably, the applied D007 digitalMLPATM assay screens all unbalanced aberrations listed in the National Comprehensive Cancer Network® guideline for risk stratification of pediatric B-cell precursor acute lymphoblastic leukemia (version 1.2020), a feature which further demonstrates the potential of the assay to efficiently complement other, more conventional genetic tests currently used in clinical diagnostics.
Considering the advantages and limitations of the most widely applied standard techniques, karyotyping is a genome-wide approach able to detect both balanced and unbalanced aberrations but it is time-consuming, requires dividing cells, provides a limited genomic resolution of 3–10 megabases and has low representativity with typically 20–30 metaphases analyzed per sample. In addition, karyotyping is challenging in acute lymphoblastic leukemia, especially in the hyperdiploid subgroup where abnormal metaphases typically have lower quality in terms of chromosome morphology as compared with normal metaphases observable in the same sample. DNA index measurement is fast, allows for the analysis of a high number of events and can be performed on nondividing cells but it only measures whole DNA content without providing data on specific chromosomal alterations, which in acute lymphoblastic leukemia may have relevance to risk stratification. Fluorescence in situ hybridization allows for the analysis of balanced and unbalanced aberrations in hundreds of cell nuclei, but using off-the-shelf standard probe sets, a maximum of three or four genomic loci can be analyzed at the same time, with a typical resolution of 100 kilobases to 1 megabase. DigitalMLPATM can detect unbalanced aberrations and does not provide single-cell information, but it allows for the simultaneous analysis of hundreds of genomic loci at exon-level resolution in a focused, rationalized manner. Certainly, the power and limitations of these different techniques had an influence on the genetic characterization of our patient cohort: balanced aberrations (e.g., ETV6-RUNX1, BCR-ABL1, and MLL fusions) were exclusively detected by karyotyping or fluorescence in situ hybridization tests applied in the diagnostic workflow, while the vast majority of unbalanced aberrations including whole chromosome changes, subchromosomal lesions and intrachromosomal gene fusions revealed by digitalMLPATM were not previously identified by any of the more conventional methods.
Indeed, performance as well as technical and application potential of digitalMLPATM have been demonstrated in several different ways in our study: (i) investigation of diagnostic samples revealed an average copy number aberration count per patient commensurable with values observed in previous studies [2], especially if we consider that our assay was highly focused on genomic regions expectedly being altered by driver lesions; (ii) besides the determination of modal chromosome number, the assay allowed for a robust characterization of specific whole chromosome gains in hyperdiploid patients which is impossible with DNA index assessment, commonly challenging with karyotyping, laborious with fluorescence in situ hybridization and noneconomical by using array- or alternative next-generation sequencing approaches; (iii) detailed profiling of subchromosomal driver copy number aberrations, also including alterations conferring intrachromosomal gene fusions, was efficiently achieved in B- and T-cell precursor acute lymphoblastic leukemia patients; (iv) in accordance with previous studies, comparative analysis of matching diagnostic and relapse samples revealed an increased number of copy number aberrations at the time of disease progression [3], and various distinct patterns of clonal origin of relapse which could potentially have clinical implications in the future; (v) digitalMLPATM results were found to be fully congruent with corresponding fluorescence in situ hybridization and polymerase chain reaction data collected via the routine diagnostic workflow and an additional validation performed by conventional multiplex ligation-dependent probe amplification showed a nearly 99% concordance between the two methods.
In lymphoid malignancies, immunoreceptor gene rearrangements provide cell-specific “barcodes,” which can be used as markers not only for detecting clonal proliferation in diagnostic samples and monitoring residual tumor load in follow-up specimens, but also for exploring the repertoire and heterogeneity of subclones gaining selective advantage in the course of leukemogenesis as well as for identifying shifts in clonal composition during disease progression [23,24,25,26,27]. Hence, scrutiny of these physiological, patient-specific genomic markers provided an additional independent opportunity to investigate the clonal relationship of leukemic cell populations dominating at different time points, thus being the major determinants of copy number profiles observed by digitalMLPATM. Deep-sequencing of the IGH locus in our patients with progressive B-cell precursor acute lymphoblastic leukemia revealed identical abundant IGH variable–diverse–joining junctions at diagnosis and relapse in all but one case. This finding is in line with previous IGH deep-sequencing studies performed in relapsed B-cell precursor acute lymphoblastic leukemia patients [28, 29], and suggests that even if branching evolution has been uncovered by copy number aberration profiling in a patient, relapse predominantly originated from a cell population that had already undergone complete RAG-driven IGH rearrangement and had a relatively close clonal relationship with blasts dominating the leukemia at diagnosis. The IGH gene has a high structural complexity coupled with a great recombinatorial potential, therefore IGH gene rearrangements provide highly patient-specific (typically unique) markers which in principle allow for tracking the changes of clonal composition with the highest possible resolution and precision. Still, thorough analysis of the IGH repertoire did not reveal an additional depth of clonal history in our cohort, e.g., by demonstrating the presence of an ancestral clone as the source of clonal expansion at disease progression in a patient with copy number aberration profiles suggesting direct clonal evolution between diagnosis and relapse. In contrast, digitalMLPATM provided a qualitatively novel layer of information with detecting clonal changes leading to the emergence or disappearance of various driver copy number aberrations besides unraveling features of clonal dynamics associated with relapse.
Copy number aberrations recurrently occurring in pediatric acute lymphoblastic leukemia have not only biological but also prognostic relevance as revealed by large-scale genomic and clinical studies. [2, 30,31,32] While identification of distinct copy number aberrations with well characterized clinical significance (e.g., IKZF1 loss) has its own value, as also demonstrated in our cohort (Supplementary Fig. 3), uncovering the co-segregation of driver aberrations in individual patient samples could potentially allow for a more personalized prognosis assessment and treatment response prediction [33]. In line with this, a growing body of evidence demonstrates that the clinical impact of copy number aberrations is dependent on the broader genomic context, which also provided basis for the introduction of classifiers integrating copy number aberrations and other classes of genetic aberrations into risk stratification [8, 10, 34, 35]. Utilizing the power of the comprehensive copy number aberration profiling provided by digitalMLPATM, we tested a novel risk classifier which combines cytogenetic data with digitalMLPATM-based copy number aberration profiles and lays more emphasis on copy number aberration data than seen in previously established classifiers (Supplementary Table 7). With the newly introduced combined classifier, four distinct subgroups with significantly different clinical outcomes were distinguishable even in our relatively small group of 76 patients (Fig. 3), which was not achievable with the UKALL classifier in this case (data not shown). This suggests that the digitalMLPATM-based classification may potentially provide further improvement in the risk assessment of patients with B-cell precursor acute lymphoblastic leukemia, which therefore will be worth testing and validating on large independent patient cohorts.
In summary, digitalMLPATM is a robust tool for the reliable detection of disease-relevant copy number aberrations which determine distinct genetic subgroups of pediatric acute lymphoblastic leukemia (e.g., hyperdiploidy, iAMP21) and/or are associated with disease prognosis (e.g., IKZF1 loss and various combinations of copy number aberrations), or confer gene fusions potentially stratifying patients for targeted therapies (e.g., NUP214-ABL1) [36,37,38]. The comprehensive profiling of recurrent genomic aberrations achieved by examining a high number of different genomic loci also provides valuable information regarding the origin of relapse, not necessarily uncovered by alternative, clonality screening techniques. Our results further demonstrate the versatile applicability of digitalMLPATM and strongly support its future incorporation into the clinical research and diagnostic workflow of pediatric acute lymphoblastic leukemia.
References
Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35:975–83.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80.
Kawamata N, Ogawa S, Seeger K, Kirschner-Schwabe R, Huynh T, Chen J, et al. Molecular allelokaryotyping of relapsed pediatric acute lymphoblastic leukemia. Int J Oncol. 2009;34:1603–12.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8.
Paulsson K, Johansson B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2009;48:637–60.
Alpár D, Pajor G, Varga P, Kajtár B, Pótó L, Mátics R, et al. Sequential and hierarchical chromosomal changes and chromosome instability are distinct features of high hyperdiploid pediatric acute lymphoblastic leukemia. Pediatr Blood Cancer. 2014;61:2208–14.
Moorman AV, Enshaei A, Schwab C, Wade R, Chilton L, Elliott A, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434–44.
Stanulla M, Dagdan E, Zaliova M, Möricke A, Palmi C, Cazzaniga G, et al. IKZFplus defines a new minimal residual disease-dependent very poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36:1240–9.
Hamadeh L, Enshaei A, Schwab C, Alonso CN, Attarbaschi A, Barbany G, et al. Validation of the United Kingdom copy-number alteration classifier in 3239 children with B-cell precursor ALL. Blood Adv. 2019;3:148–57.
Ma Y, Dobbins SE, Sherborne AL, Chubb D, Galbiati M, Cazzaniga G, et al. Developmental timing of mutations revealed by whole-genome sequencing of twins with acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2013;110:7429–33.
Usvasalo A, Savola S, Räty R, Vettenranta K, Harila-Saari A, Koistinen P, et al. CDKN2A deletions in acute lymphoblastic leukemia of adolescents and young adults: an array CGH study. Leuk Res. 2008;32:1228–35.
Benard-Slagter A, Zondervan I, de Groot K, Ghazavi F, Sarhadi V, Van Vlierberghe P, et al. Digital multiplex ligation-dependent probe amplification for detection of key copy number alterations in T- and B-cell lymphoblastic leukemia. J Mol Diagn. 2017;19:659–72.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC; 2008. 167–78.
Kosztolanyi S, Kiss R, Atanesyan L, Gango A, de Groot K, Steenkamer M, et al. High-throughput copy number profiling by digital multiplex ligation-dependent probe amplification in multiple myeloma. J Mol Diagn. 2018;20:777–88.
Alpar D, de Jong D, Savola S, Yigittop H, Kajtar B, Kereskai L, et al. MLPA is a powerful tool for detecting lymphoblastic transformation in chronic myeloid leukemia and revealing the clonal origin of relapse in pediatric acute lymphoblastic leukemia. Cancer Genet. 2012;205:465–9.
Alpar D, de Jong D, Holczer-Nagy Z, Kajtar B, Savola S, Jakso P, et al. Multiplex ligation-dependent probe amplification and fluorescence in situ hybridization are complementary techniques to detect cytogenetic abnormalities in multiple myeloma. Genes Chromosomes Cancer. 2013;52:785–93.
Schwab CJ, Jones LR, Morrison H, Rzan SL, Yigittop H, Schouten JP, et al. Evaluation of multiplex ligation-dependent probe amplification as a method for the detection of copy number abnormalities in B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2010;49:1104–13.
Kiss R, Kosztolányi S, Gángó A, Szuhai K, Bödör C, Alpár D. [Multiplex ligation-dependent probe amplification in oncohematological diagnostics and research]. Orv Hetil. 2018;159:583–92.
Pui CH, Yang JJ, Bhakta N, Rodriguez-Galindo C. Global efforts toward the cure of childhood acute lymphoblastic leukaemia. Lancet Child Adolesc Health. 2018;2:440–54.
Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7:e577.
Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. 2015;33:2938–48.
van Dongen JJ, Langerak AW, Brüggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–317.
Peham M, Konrad M, Harbott J, König M, Haas OA, Panzer-Grümayer ER. Clonal variation of the immunogenotype in relapsed ETV6/RUNX1-positive acute lymphoblasticleukemia indicates subclone formation during early stages of leukemia development. Genes Chromosomes Cancer. 2004;39:156–60.
László R, Alpár D, Kajtár B, Lacza Á, Ottóffy G, Kiss C, et al. Detection of early precursors of t(12;21) positive pediatric acute lymphoblastic leukemia during follow-up. Pediatr Blood Cancer. 2010;54:158–60.
Gawad C, Pepin F, Carlton VE, Klinger M, Logan AC, Miklos DB, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120:4407–17.
Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, et al. Clonal origins of ETV6-RUNX1+ acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia. 2015;29:839–46.
Bashford-Rogers RJ, Nicolaou KA, Bartram J, Goulden NJ, Loizou L, Koumas L, et al. Eye on the B-ALL: B-cell receptor repertoires reveal persistence of numerous B-lymphoblastic leukemia subclones from diagnosis to relapse. Leukemia. 2016;30:2312–21.
Theunissen PMJ, de Bie M, van Zessen D, de Haas V, Stubbs AP, van der Velden VHJ. Next-generation antigen receptor sequencing of paired diagnosis and relapse samples of B-cell acute lymphoblastic leukemia: clonal evolution and implications for minimal residual disease target selection. Leuk Res. 2019;76:98–104.
Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–66.
Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112:4178–83.
Szarzyńska-Zawadzka B, Kunz JB, Sędek Ł, Kosmalska M, Zdon K, Biecek P, et al. PTEN abnormalities predict poor outcome in children with T-cell acute lymphoblastic leukemia treated according to ALL IC-BFM protocols. Am J Hematol. 2019;94:E93–6.
Scheijen B, Boer JM, Marke R, Tijchon E, van Ingen Schenau D, Waanders E, et al. Tumor suppressors BTG1 and IKZF1 cooperate during mouse leukemia development and increase relapse risk in B-cell precursor acute lymphoblastic leukemia patients. Haematologica. 2017;102:541–51.
Irving JA, Enshaei A, Parker CA, Sutton R, Kuiper RP, Erhorn A, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016;128:911–22.
Steeghs EMP, Boer JM, Hoogkamer AQ, Boeree A, de Haas V, de Groot-Kruseman HA, et al. Copy number alterations in B-cell development genes, drug resistance, and clinical outcome in pediatric B-cell precursor acute lymphoblastic leukemia. Sci Rep. 2019;9:4634.
Harrison CJ, Moorman AV, Schwab C, Carroll AJ, Raetz EA, Devidas M, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia. 2014;28:1015–21.
De Keersmaecker K, Porcu M, Cox L, Girardi T, Vandepoel R, de Beeck JO, et al. NUP214-ABL1-mediated cell proliferation in T cell acute lymphoblastic leukemia is dependent on the LCK kinase and various interacting proteins. Haematologica. 2014;99:85–93.
Boer JM, van der Veer A, Rizopoulos D, Fiocco M, Sonneveld E, de Groot-Kruseman HA, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. 2016;30:32–38.
Acknowledgements
The authors are grateful to the patients and their families involved in this study. The digitalMLPATM probemix and reagents were kindly provided by MRC Holland. The study was supported by grants of the Hungarian National Research, Development and Innovation Office—NKFIH (IDs: K_16 #119950, NVKP_16-1-2016-0004, and KH17-126718), a Momentum Research Grant (ID: LP95021) and a János Bolyai Research Scholarship (ID: BO/00320/18/5) of the Hungarian Academy of Sciences as well as the New National Excellence Program of the Ministry of Human Capacities (IDs: ÚNKP-17-4-III-SE-9 and ÚNKP-19-4-SE-77).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
ABS, KdG, and SS are employees of the MRC Holland (Amsterdam, NL). The other authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kiss, R., Gángó, A., Benard-Slagter, A. et al. Comprehensive profiling of disease-relevant copy number aberrations for advanced clinical diagnostics of pediatric acute lymphoblastic leukemia. Mod Pathol 33, 812–824 (2020). https://doi.org/10.1038/s41379-019-0423-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0423-5
This article is cited by
-
PersonALL: a genetic scoring guide for personalized risk assessment in pediatric B-cell precursor acute lymphoblastic leukemia
British Journal of Cancer (2023)
-
Prider: multiplexed primer design using linearly scaling approximation of set coverage
BMC Bioinformatics (2022)
-
Prognostic Impact of Somatic Copy Number Alterations in Childhood B-Lineage Acute Lymphoblastic Leukemia
Current Oncology Reports (2021)

{kind=link}