
Abstract
The wheat leaf rust fungus, Puccinia triticina, is found in the major wheat growing regions of the world and is a leading cause of yield loss in wheat. Populations of P. triticina are highly variable for virulence to resistance genes in wheat and adapt quickly to resistance genes in wheat cultivars. The objectives of this study were to determine the genetic relatedness of worldwide collections of P. triticina using restriction site associated genotyping by sequencing. A total of 558 isolates of P. triticina from wheat producing regions in North America, South America, Europe, the Middle East, Ethiopia, Russia, Pakistan, Central Asia, China, New Zealand, and South Africa were characterized at 6745 single nucleotide loci. Isolates were also tested for virulence to 20 near-isogenic lines that differ for leaf rust resistance genes. Populations that were geographically proximal were also more closely related for genotypes. In addition, groups of isolates within regions that varied for genotype were similar to groups from other regions, which indicated past and recent migration across regions. Isolates from tetraploid durum wheat in five different regions were highly related with distinct genotypes compared to isolates from hexaploid common wheat. Based on a molecular clock, isolates from durum wheat found only in Ethiopia were the first to diverge from a common ancestor form of P. triticina that is found on the wild wheat relative Aegilops speltoides, followed by the divergence of isolates found worldwide that are virulent to durum wheat, and then by isolates found on common wheat.
Similar content being viewed by others

Introduction
Diseases that affect the major staple crops of the world are a serious constraint on agricultural productivity. Modern agricultural production systems emphasize large production fields of crops at the same growth and development stage that are often genetically uniform and planted across millions of hectares. These conditions provide highly favorable environments for plant pathogens to adapt and overcome disease resistance genes in host genotypes and to spread within and across fields, often resulting in epidemics that affect entire continental regions. On a global level, yield losses in wheat caused by disease average 21.5% (Savary et al. 2019).
Many plant pathogens have a global distribution, resulting in yield losses in different climatic and host environments. Plant pathogenic fungi in particular are highly adapted for invasion and dissemination across large land masses and between continents (Gladieux et al. 2014) and are rarely endemic to a single region (Bebber et al. 2014). Leaf rust caused by the fungus Puccinia triticina Erikss. is found almost wherever wheat is grown in non-arid conditions (Saari and Prescott 1985; Samborski 1985) and is the leading cause of yield loss in wheat due to disease (Savary et al. 2019) on a global level. Puccinia triticina is a macrocyclic heteroecious rust with five spore stages that requires two taxonomically unrelated hosts to complete the sexual cycle. The economically important disease phase is caused by infections of dikaryotic urediniospores on hexaploid common wheat, Triticum aestivum L.; tetraploid durum wheat, T. turgidum L.; and wild emmer wheat T. dicoccoides L. (Bolton et al. 2008). Urediniospores are clonally produced and uredinial infections can cycle indefinitely on susceptible wheat plants. Urediniospores are highly adapted for wind dissemination and are deposited on wheat fields by rainfall. The spores germinate, penetrate the leaves through the stomata and form specialized infection structures that ramify intercellular tissue below the epidermal and palisade layer of cells. Uredinial infections can produce new urediniospores within 7–10 days after initial infection. Each uredinial infection can produce thousands of urediniospores, and there may be hundreds of uredinial infections on a single leaf, resulting in a very large population of infective spores that have the potential to be carried hundreds to thousands of kilometers on the prevailing winds. Dikaryotic teliospores are produced in maturing uredinial infections on wheat, undergo a brief diploid phase, and then germinate to produce haploid basidiospores that infect the alternate host. The most compatible alternate host of P. triticina is Thalictrum speciosissimum L, common meadow rue. Haploid spermagonia are formed on the upper leaf surface and these produce spermatia that differ for a mating type allele, which then fertilize sexually compatible spermagonia. The dikaryotic condition is restored after fertilization which leads to the formation of aecial cups on the lower leaf surface that mature to produce dikaryotic aeciospores that can infect only the uredinial-telial hosts. Susceptible alternate hosts of P. triticina are found mostly in southern Europe and parts of west Asia but are not important in the spread of the disease. In North America and many other wheat producing regions the native Thalictrum spp. are not highly susceptible to infection by P. triticina basidiospores (Saari et al. 1968).
As a pathogen, P. triticina is well suited for population genetic studies as it has broad environmental adaptation and there are many variants that differ for virulence to leaf rust resistance genes in wheat. In the United States P. triticina populations are highly variable for virulence with over 50 races identified annually. In the Great Plains of the U.S. wheat cultivars with race specific leaf rust resistance genes have continuously selected virulent races of P. triticina for the last 40 years (Kolmer and Hughes 2018). This has resulted in a constant turnover of the predominant races, with many wheat cultivars losing their effective resistance within a few years. Populations of P. triticina in North America (Ordoñez and Kolmer 2009) and other regions (Goyeau et al. 2007) have been characterized using virulence and various molecular markers as technologies have advanced. Information concerning the frequency of races with virulence to resistance genes can be used to select resistant wheat germplasm. DNA based markers have provided further insight into the genetic relationship between race groups and the origins and spread of new races and genotypes within (Ordoñez and Kolmer 2009) and between continental regions (Ordoñez et al. 2010). The objectives of this study were to determine the genetic relationships between populations of P. triticina from major wheat producing regions worldwide, using the current technology of restriction site associated genotyping by sequencing (GBS) (Poland and Rife 2012) in order to generate a large number of single nucleotide polymorphic (SNP) markers. The genetic and evolutionary relationships between geographic and host-associated groups of P. triticina can be more directly assessed using SNP markers compared to previous studies (Kolmer et al. 2019) which employed simple sequence repeat (SSR) or microsatellite markers. We also wished to determine if isolates that had nearly identical genotypes were restricted to certain geographical regions or if these isolates were widely distributed across regions, which would support the hypothesis of migration of clonally produced urediniospores. Furthermore, we examined the divergence of host-associated forms of P. triticina from a common ancestor using a molecular clock approach, to determine if the leaf rust pathogen tracked wheat during its evolution from diploid to tetraploid and hexaploid forms.
Materials and methods
Isolates of P. triticina
The isolates used in this study came from field surveys of infected wheat fields and collections in national laboratories in North America, South America, the Middle East, Central Asia, Europe, East Africa, Russia, China, Pakistan, New Zealand, and South Africa (Supplemental Table 1). Most of the isolates were collected from the early 2000s up to 2014. The collections from New Zealand and South Africa were from the mid-1990s. Collections from the late 1980s to mid-1990s were also obtained for most regions. Collections of leaf rust were made from both durum wheat and common wheat. The single-uredinial isolates were tested for virulence to a set of 20 near-isogenic lines of Thatcher wheat that have leaf rust resistance genes Lr1, Lr2a, Lr2c, Lr3a, Lr9, Lr16, Lr24, Lr26, Lr3ka, Lr11, Lr17, Lr30, LrB, Lr10, Lr14a, Lr18, Lr3bg, Lr14b, Lr20, Lr28, and Thatcher as a susceptible control as previously described (Kolmer et al. 2019). The virulence phenotypes were described using a five-letter code, where each letter described virulence/avirulence to four Thatcher differential lines adapted from the hexadecimal nomenclature used to describe virulence in P. triticina (Long and Kolmer 1989). Each isolate was also given a 20-digit binary number based on the avirulent/virulent response to the Thatcher differential lines. Differences in virulence among isolates within groups was calculated using GenAlex 6.502 (Peakall and Smouse 2012) with the Distance-Genetic-binary haploid function. Urediniospores from two isolates of P. triticina collected in Israel from the wheat progenitor Aegilops speltoides L. (Yehuda et al. 2004) were also increased.
Genotyping by sequencing
DNA was extracted from urediniospores using the Omniprep kit (G-Biosciences, St. Louis MO), and diluted to 25 ng/μl in 30 μl. The DNA was processed for GBS at the University of Minnesota Genomics Center. DNA was digested with BtgI and BamHI and ligated with barcode adapters. The DNA libraries were sequenced in 1.5 lanes using the Illumina HiSeq 2500 Rapid mode 100-single end read method (Illumina, San Diego, CA). The barcodes were removed from the raw reads using SCYTHE with default settings. The cleaned reads were mapped to the ASM15152v1 reference genome of P. triticina (available at https://fungi.ensembl.org/Puccinia_triticina/Info/Index) with minimap2 (Li 2018), using default parameters. A genome catalog was created and genotype data extracted using the gstacks and populations modules, respectively, from the STACKS v2.3b software package (Catchen et al. 2013). The –ordered_export flag in the populations module was used to ensure that GBS loci with overlapping physical positions were collapsed upon export. A total of 558 isolates from 11 different regions were genotyped.
Nucleotide diversity
In order to calculate both intra- and inter-population average pairwise nucleotide diversity values (π and dXY, respectively), the full sequence (i.e. variant and invariant positions) for each locus was exported with genotyping rate as above in.fasta format. The average pairwise diversity for all combinations of haplotypes was calculated using a custom Python script. Given either individual isolate names or groupings of isolates, average values within and between defined groups were then calculated using GNU datamash (version 1.3).
Coancestry and estimation of k groups
The coancestry among individuals was estimated using the FINERADSTRUCTURE software package (Malinsky et al. 2018). The haplotype matrix output from STACKS was converted into a format readable by the RADpainter program using the fineradinput.py script available in the FINERADSTRUCTURE-tools repository (https://github.com/edgardomortiz/fineRADstructure-tools). Firstly, the coancestry matrix was estimated using RADpainter, which determines coancestry at individual GBS loci by the number of SNP differences between haplotypes and averages those values across all GBS loci. Using the information from the coancestry matrix, clustering with FINERADSTRUCTURE was done with a burn-in of 100,000 iterations and 100,000 sampling iterations, keeping 1000 runs in total. A tree from the cluster structure was inferred again using 100,000 burn-in iterations, and 100,000 sampling iterations thinned to a total of 1000 samples. Using the populations module from STACKS, the data were also exported in VCF format, retaining a single SNP for each locus. The individuals were also grouped based on genotype using discriminate analysis of principal coordinates (DAPC) implemented in POPPR 2.02 (Kamvar et al. 2014) using SNP genotypes in VCF format. The genotypes of the isolates were transformed to 40 principal components (PCs) and then k means analysis was conducted on the PCs. The dataset was tested for values of k from 2 to 80. Change in values of the Bayesian Information Criterion (BIC) was used in an attempt to determine the number of k groups in the overall population. All isolates used in this study had been previously genotyped for allelic variation at 23 SSR loci (Kolmer et al. 2019). Based on their multi-locus genotypes (MLG) the isolates had previously been differentiated into 40 groups. These MLG groups were subsequently used to group the isolates due to problems with the FINERADSTRUCTURE clustering and the DAPC.
Estimates of genetic differentiation, heterozygosity, and linkage disequilibrium
The R software package diveRsity v1.9.90 (Keenan et al. 2013) was used to calculate Weir and Cockerham’s FST and Jost’s D between isolates grouped in their geographic region of collection, also using the SNP VCF file. Prevosti’s genetic distance between the regional populations was calculated with POPPR. The coefficient of inbreeding (FIS), expected heterozygosity (He) and observed heterozygosity (Ho) of the regional populations were calculated with diveRsity. Measures of linkage disequilibria, the index of association (Ia) and rd were calculated within the regional populations with POPPR. Unrooted neighbor joining (NJ) trees of Prevosti’s genetic distance between isolate genotypes, genotype groups, and isolates in geographical regions were done in APE (Paradis et al. 2004) with 1000 bootstrap samples.
Estimate of relative divergence time between host-associated forms
Divergence time in relative number of cell divisions between different groups of P. triticina based on host association was estimated using a genome-wide mutation rate and the π and dXY values within and between groups. The divergence time estimates were calculated for two populations as τ = (dXY−(πX + πY / 2)) / 2μ, where x and y are the respective populations being compared and μ is the genome-wide average mutation rate per base pair per cell division. Divergence times were also calculated from the branch lengths of a neighbor-joining tree by dividing those lengths by an estimate of 2μ. In both cases the estimate of μ for the basidiomycete yeast Rhodotorula toruloides of 1.9 × 10−10 per site per cell division was used (Long et al. 2016). Estimates of μ are quite consistent across the fungal kingdom; estimates for ascomycetes range from 1.63–4.1 × 10 −10 per site per generation (Farlow et al. 2015; Lynch et al. 2018; Zhu et al. 2014). The estimates should be robust with respect to variation in μ. While this simplistic estimate of τ is biased in the presence of gene flow and selection, it is at least well understood as a consequence of coalescent times and does not require optimization and parameterization of complex demographic models (Rosen et al. 2018).
Results
Genotyping of P. triticina
Among the 556 isolates from durum and common wheat, a total of 6745 SNPs across 1104 genomic contigs were retained for analysis. The SNPs were randomly distributed across the contigs, with no large gaps (Supplemental Fig. 1). Variant loci with minor allele frequency <0.01 were filtered out. The read depth varied from 30–80X for the individual isolates. Of the total isolates, 551 had <5% missing data, and 5513 SNPs had <5% missing data. The final stringency for missing data was relaxed in order to allow inclusion of the two isolates from A. speltoides in the final dataset. These divergent isolates had lower alignment to the reference genome, resulting in 4414 SNP loci. Both isolates had <36% missing data and all SNPs had <25% missing data. The average sequence diversity across all pairwise comparisons of isolates from common and durum wheat ranged from 5.0 × 10−4 to 2.0 × 10−3 (Supplemental Fig. 2). The average sequence diversity of the two isolates from A. speltoides paired with all other isolates was 6.8 × 10−3.
An unrooted neighbor joining plot of the SNP genotypes of all isolates except the two from A. speltoides (Fig. 1), indicated that isolates from some of the regions were dispersed across different parts of the tree and were often closely related to isolates from other regions. As an example, groups of isolates from Europe, Pakistan, and the Middle East were found in many parts of the tree, and isolates from these groups were closely related to isolates from other regions. In contrast, isolates from China and Russia were not widely distributed, being found mostly in only two parts of the tree. Isolates from North America and South America were highly related and were also not widely distributed in the tree. Overall, although there was some separation of isolates based on geographic region, many isolates were closely related to isolates from other regions.

NA North America; SA South America; EA East Africa; ME Middle East; NZ New Zealand; SAF South Africa; EU Europe; PK Pakistan; CN China; CA Central Asia; RU Russia.
Differentiation of regional populations of P. triticina
The isolates from common wheat and durum wheat were initially grouped based on region of origin. Population differentiation was determined using three complementary methods; Jost’s D, FST, and Prevosti’s distance. Genetic distance as measured by Jost’s D ranged from 0.0 for the population pairs Middle East–New Zealand; Europe–Pakistan; North America–South America; and New Zealand–South Africa; to 1.1 × 10−3 for Russia–East Africa (Supplemental Table 2). Values of FST between the populations ranged from 0.0102 for Europe–Pakistan to 0.2112 for Russia–East Africa. All population pairings had values of FST significantly greater than 0.0 with p < 0.05. Based on Prevosti’s genetic distance, the Russia–East Africa populations were the most distant and the North America-South America populations were the most closely related (Supplemental Fig. 3). With respect to the neighbor-joining tree, the North America–South America and Europe–Pakistan populations were paired with bootstrap values of 100% and the Russia–Central Asia; South Africa–New Zealand; and the Europe–Pakistan populations were grouped with bootstrap values >90%. The eastern most regional populations; Russia, Central Asia, and China, were separated from the other populations with 100% confidence.
Nucleotide diversity and heterozygosity
Measures of nucleotide diversity and heterozygosity were assessed to determine if there were large differences in the distribution of variation within the regional populations. The Middle Eastern population was the most diverse for average pairwise nucleotide variation at 1.63 × 10−3, while the Russian population was the least diverse at 8.70 × 10−4 (Table 1). All populations had values of Ho greater than He (Fig. 2). Only the population from East Africa had a FIS value that was not significantly less than 0.0 (p < 0.01) (Table 1). The populations were further assessed for non-random distribution of variation. All populations had very high levels of Ia and had values of rd > 0.00 (p < 0.01) (Table 1). Taken together with the high values of Ho relative to He, the high disequilibria measures were strong indicators of non-random variation that confirmed the clonal reproduction of all the regional populations.

NA North America; SA South America; EU Europe; ME Middle East; EA East Africa; RU Russia; PK Pakistan; CA Central Asia; CN China; New Zealand; SAF South Africa.
Differentiation of P. triticina groups
The isolates were then grouped based on molecular genotypes. A total of 163 inferred clusters were derived from a heatmap of coancestry, as determined by the inferred population membership in the finestructure clustering algorithm in FINERADSTRUCTURE (Malinsky et al. 2018). This high number of groups made further analyses cumbersome, so grouping the isolates based on DAPC was then done. However, there was no indication of cluster number since the BIC values declined continuously, with no clear lower limit. Given the difficulties in finding a workable number of distinct genotype groups with both finestructure and DAPC, the isolates from common wheat and durum wheat (Table 1) were grouped within the regional populations as determined with SSR markers for the same isolates in a previous study (Kolmer et al. 2019). In this grouping, the isolates within groups had very little SSR variation, and much greater variation between groups. This also had the advantage of making it easier to infer genetic similarity or differences between groups of isolates from different regional populations, as compared to making inferences regarding isolates from different regions within arbitrary group designations. There were overall 40 MLG groups of isolates from common and durum wheat within the regional populations. Isolates from durum wheat that were found in Europe, North America, South America, the Middle East, and East Africa had distinct MLG and SNP genotypes compared to the isolates from common wheat and were placed in separate groups that were indicated with the suffix durum. Isolates from Ethiopia that were virulent to durum wheat, but highly avirulent to nearly all common wheat cultivars were placed in a separate group designated as ETH-EEEEE. Isolates from New Zealand and South Africa were not divided into additional groups since there were 10 or fewer isolates in each group.
The nucleotide diversity of the isolates within the MLG genotype groups ranged from 1.20 × 10−3 for EU5, to 5.80 × 10−4 for EA Durum (Table 1). With two exceptions, the regional populations overall had higher nucleotide diversity compared to the groups within their respective populations. The NA2 group had nucleotide diversity of 1.07 × 10−3 that was slightly higher than the overall North American population of 1.04 × 10−3, and the RU2 group had diversity equal to the overall Russian population. The isolates within the MLG groups also varied for virulence to the Thatcher near isogenic lines that carry single leaf rust resistance genes (Table 1). The NA5 group had the highest number of races with 25, that had an average virulence difference of 4.39. In contrast, the EA durum group had five isolates with the same race designation. Almost all of the groups within the regional populations had smaller average pairwise differences in virulence compared to the overall virulence difference of the regional populations. The only exception was the NA-4 group that consisted of two isolates that differed by 9.0 for virulence compared to 6.83 for the North American population. The isolates within the groups were more closely related for virulence compared to isolates in the overall regional populations.
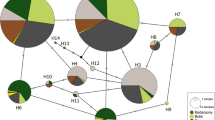
The genetic distance between the isolates was further examined using the SNP genotypes of isolates in the MLG genotype groups. In the NJ plot using Prevosti’s genetic distance the 40 groups were arranged into 15 GBS clusters (Fig. 3a). Nine of the clusters had two or more groups with bootstrap support values of >95% (Supplemental Fig 4). Six groups; ETH-EEEEE, EU1, CN3, NA1, NA2, and SA4, were not paired with any other group with high bootstrap support. Groups in cluster A were from the five groups in different regional populations that have virulence to durum wheat. Groups CN1 and CN2 were in cluster F. Seven other (E, G, H, I, J, K, M) clusters had groups highly related for genotype from different regional populations. Isolates in clusters G, I, and J, were from Europe, Central Asia, Russia, the Middle East, and Pakistan. Isolates in cluster H were from North America and South America; isolates in cluster K were from the Middle East, South America, and North America; and isolates in cluster M were from North America, South America, East Africa, Pakistan, and Europe. Isolates in EU5 were clustered with isolates in the SAF and NZ groups at 95% confidence (Supplemental Fig. 4), although these are shown as clustering separately in Fig. 4. Isolates in the ETH-EEEEE group were highly distinct from all other isolate groups from either durum wheat or common wheat. The percentages of isolates in the 15 clusters in the 11 regional populations are shown in Fig. 4.

NA North America; SA South America; EA East Africa; ME Middle East; NZ New Zealand; SAF South Africa; EU Europe; PK Pakistan; CN China; CA Central Asia; RU Russia. a Plot of Prevosti’s genetic distance of isolates in MLG groups based on 6745 SNP markers. Indicated clusters (A-O) of isolate groups have support values over 95% based on 1000 bootstrap samples. All bootstrap support values are indicated in supplemental Fig. 4. b Plot of Prevosti’s genetic distance of virulence to 20 Thatcher wheat lines near-isogenic for leaf rust resistance genes of isolates in 40 MLG genotype groups. Numbers of isolates in each region and MLG group are in Table 1.

Numbers of isolates in each region are in Table 1.
Some of the groups that were highly related for genotype also had very similar virulence to the differential wheat lines. Groups in the A (ETH Durum, NA Durum, SA Durum, EU Durum, ME Durum), F (CN1, CN2), G (RU1, CA2, EU6), I (CA1, EU2), J (CA3, PK1, EU7; EU4, CA4), K (NA5, SA2), and M (SA3, NA3, EU8) clusters were closely grouped in the NJ plot based on virulence to the Thatcher near-isogenic lines (Fig. 3b). Of the clusters with more than one group, only groups in the E cluster (SAF, NZ, and EU5) were not closely related for virulence. The frequencies of virulence to the differential lines of the isolate groups are listed in Supplemental Table 3. Excluding the groups from durum wheat in clusters A and B, the groups varied for virulence from 0 to 90% to genes Lr1, Lr2a, Lr3a, Lr3ka, Lr14a, Lr20, Lr26, Lr28, and Lr30. Virulence to genes Lr9, Lr11, Lr16, Lr18, and Lr24 were at lower frequency in all groups, and virulence to LrB, Lr10, and Lr14b were at high frequency in all groups. All groups had intermediate to high virulence to genes Lr3bg, Lr2c, and Lr17.
In the previous study with SSR markers, a large number of isolates in different regional populations had identical MLG. We wished to determine if these isolates also had closely related SNP genotypes. A total of 132 isolates amongst 18 MLG across different regions were grouped in a NJ plot based on their SNP genotypes (Supplemental Fig. 5), in addition to 10 other isolates in eight MLG. The genetic distance between MLG is related to the number designation, such that MLG closer in number are more related for SSR genotype compared to MLG that are further apart. Isolates with identical or closely related MLG that were found in different regions were clustered into groups of highly related SNP genotypes. Isolates with MLG 6, 25, 65, 59, 77, and 99 were in discrete groups based on their SNP genotypes. Isolates with MLG 41, 42, 46, 49, 52 were clustered in one group, as were isolates in MLG 7 and 9, isolates in MLG 33, 35, and 36, isolates from durum wheat in MLG 72 and 75, isolates in MLG 79 and 82, and isolates in MLG 83 and 84. The number of isolates within MLG groups for each MLG are listed in Supplemental Table 4.
Divergence of host-associated groups of P. triticina
The SNP genotypes distinguished three different types of P. triticina isolates on wheat: isolates from common wheat found worldwide, isolates from durum wheat found worldwide; and isolates from durum wheat that were found only in Ethiopia. The SNP genotypes of the two isolates from A. speltoides were also included to determine relative divergence times of the four P. triticina groups using a molecular clock based on a genome-wide mutation rate. Divergence times in relative generations or cell divisions, were estimated using the nucleotide diversities of the four groups, nucleotide divergence between the groups, and the genome-wide mutation rate as determined from R. toruloides (Table 2). Divergence times were also estimated by dividing branch lengths of the NJ tree (Fig. 5) by 2μ. The ETH-EEEEE isolates were estimated to be the oldest type found on wheat, having diverged from the ancestral A. speltoides type ≈ 14.9–20.1 × 106 cell divisions ago (Table 2, Fig. 5). The isolates found on durum wheat worldwide diverged from the ETH-EEEEE types ≈ 1.6–4.8 × 106 divisions ago, and the isolates virulent to common wheat diverged from the durum isolates ≈9.5 × 105–4.6 × 106 divisions ago. The two A. speltoides isolates showed the least relative divergence from the common wheat samples likely due to reference sequence bias, since the reference genome is a common wheat isolate.

Divergence times between groups were based on branch lengths scaled by 2x the genome-wide mutation rate. AEG isolates collected from Aegilops speltoides; NA North America; SA South America; EA East Africa; ME Middle East; NZ New Zealand; SAF South Africa; EU Europe; PK Pakistan; CN China; CA Central Asia; RU Russia.
Discussion
In this study, isolates of P. triticina with highly related SNP genotypes were widely distributed across geographically distant wheat growing regions, which provided strong evidence for recent and historical migration of this important wheat pathogen. Regional differentiation was also found, as there were endemic genotype groups that were not closely related to any groups from other regions. The distribution of genetic variation in the worldwide populations has been influenced by host selection and the wind-blown dispersal of asexual urediniospores within and across geographically distant populations.
Geographic proximity affected the relative differentiation of the regional populations. The high similarity of the Russian and Central Asia populations suggests a stepwise progression of wind disseminated urediniospores across wheat fields in these two regions. Both groups from Russia were closely related to groups from Central Asia. The high similarity of isolates from Central Asia, Russia, the Middle East, Europe, and Pakistan in the GBS clusters G (CA2,RU1, EU6), I (CA1, EU2, PK3, RU2), and J (EU7, ME2, EA2, PK1, CA3, CA4, EU4) indicated the movement of P. triticina genotypes across Europe and Asia. Movement of wheat rust fungi across continental land masses has been regularly documented. Races of wheat stem rust caused by P. graminis f. sp. tritici with virulence to genes Sr31 and Sr38 (Wanyera et al. 2006; Jin et al. 2009; Jin et al. 2008) have spread across East Africa into parts of the Middle East and southwest Asia. Races of P. triticina found in the southern Great Plains of the U.S. are wind dispersed by southerly winds to northern wheat growing regions. The same races of P. triticina can be found in Texas, Minnesota and Manitoba in the same year (Kolmer and Hughes 2014; McCallum et al. 2018). Races of the stripe rust fungus, P. striiformis f. sp. tritici, have spread across Asia into western Europe (Hovmøller et al. 2015). The relatedness of the NA and SA populations is likely due to the introduction of P. triticina from common sources in Europe during the establishment of wheat cultivation. Groups NA3, SA3; NA5, SA2; and NA4 and SA1 were highly related. Since the mid-1990s P. triticina races with identical SSR genotypes and virulence in the NA3 and SA3 groups have spread across both continents (Ordoñez et al. 2010).
Genotype groups that were not closely related to other groups contributed to the differentiation of the regional populations. These groups may be endemic in their respective regions. The CN population in particular was strongly differentiated from other populations, as the three groups from China were unrelated to groups from other regions. Similarly, groups in North America (NA1, NA2), South America (SA4), and Europe (EU1, EU5) that were unrelated to other groups contributed to the differentiation of these regional populations. Host selection may account for these unique groups. Wheat cultivars with certain combinations of leaf rust resistance genes may have selected and maintained unique P. triticina races. Within the U.S., different leaf rust resistance genes are present in the soft red winter wheat cultivars grown in the southern and eastern states; the hard red winter wheat cultivars of the southern and mid-Great Plains, and the hard red spring wheat cultivars of the northern Great Plains (Kolmer and Hughes 2018). The different host cultivars and their respective leaf rust resistance genes have selected different leaf rust races in these three regions. The unique genotype groups in this study may also be a result of genetic drift due to sampling. Additional collections in additional years across all populations may have resulted in fewer unrelated groups. To reduce the effects of drift due to sampling, all populations ideally would have been sampled in an identical manner within a time period of a few years with approximately an equal number of collections and derived isolates.
Groups from different regions were highly related, which had the effect of reducing the overall association between geographic distance and genotype. The pan-global distribution of these groups indicates long-distance movement of wind-blown urediniospores between continental regions. Isolates of P. triticina with virulence to durum wheat have been found in durum growing regions across North America (Singh et al. 2004), South America (Ordoñez et al. 2010), Europe (Goyeau et al. 2011), the Middle East (Kolmer et al. 2011), and East Africa (Kolmer and Acevedo 2016). These isolates in MLG 72 and 75 (Supplemental Fig. 5) are highly related for SNP genotype, and likely have a common origin. Races of P. triticina with virulence to Lr72 in durum wheat have spread to most durum growing regions of the world since the early 2000s (Soleiman et al. 2016). Isolates in NA3, SA3, EA1, PK2, and EU8 are highly related and most have virulence to Lr17 and Lr3bg. Isolates in these groups were first found in the United States (Long et al. 2000) and Canada (Kolmer 1998) in the mid-1990s, Uruguay and Argentina in 1999 (German et al. 2007), and in France in 2000 and 2001 (Goyeau et al. 2006). Isolates with this virulence are in MLG 33, 35, and 36 in Supplemental Fig. 5, and have nearly identical SNP genotypes. The initial appearance of isolates in PK2 and EA1 is unknown since regular surveys of P. triticina in these regions have not been conducted. Isolates in SA3, NA3, and EU8; and EA1 and PK2 were also highly similar for virulence. Given the high molecular and virulence similarity, isolates in these groups likely have a common origin. The New Zealand and South Africa populations were also similar for SNP genotype, however only a small number of isolates from both of these populations were collected in the mid-1990s and may not be representative of the current populations. Foreign incursions of P. triticina races were previously noted in Australia and New Zealand (Park et al. 2002; Park et al. 1995).
All regional populations except for East Africa, had higher levels of observed heterozygosity compared to the expected heterozygosity in sexual populations that are in Hardy-Weinberg equilibrium. The same isolates in the earlier study (Kolmer et al. 2019) also had higher levels of heterozygosity than expected for SSR loci. The high levels of heterozygosity (Halkett et al. 2005) combined with the high linkage disequilibrium in all populations were strong indicators of clonal reproduction. Furthermore, the high similarity of isolate groups for both molecular genotype and virulence was an additional indicator of clonal reproduction. If sexual recombination were occurring to some degree, there would be little if any association between race phenotype and molecular variation. In the absence of recombination, the genes affecting virulence are effectively completely linked. New races of P. triticina develop by the stepwise accumulation of new virulence genes, in existing genetic backgrounds. A sexual population of P. triticina derived in an experimental study had low levels of linkage disequilibria and no association between virulence and molecular genotype (Liu and Kolmer 1998). Despite the occurrence of aecial infections on T. speciosissimum in southern Europe (Casulii and Siniscalco 1987) there is no evidence to indicate the sexual cycle is important in the epidemiology of P. triticina (Goyeau et al. 2007).
Distinct types of P. triticina have been described based on host adaptation. Collections of P. triticina in Israel have been made from the diploid wheat progenitor A. speltoides, which is the donor of the B genome in wheat (Yehuda et al. 2004). Coalescence analysis using sequence data from 15 loci (Liu et al. 2014) indicated that the P. triticina types adapted to tetraploid durum wheat and hexaploid common wheat diverged from the type found on A. speltoides. The nucleotide divergence of P. triticina types in the GBS data also confirmed that the collections from durum wheat and common wheat were derived from the type found on A. speltoides. In both studies, the ETH-EEEEE isolates were determined to be the earliest derived form of P. triticina that have virulence to durum wheat or common wheat. These isolates are highly virulent to durum wheat cultivars, but unlike isolates in the other durum groups, are avirulent to many susceptible common wheat cultivars such as Thatcher (Kolmer and Acevedo 2016), and have only been found in Ethiopia, most likely due to the unique host environment of landrace emmer and durum wheat cultivars (Eticha et al. 2006). These isolates may be a remnant of the P. triticina population that existed before the widespread cultivation of modern improved durum wheat and common wheat cultivars. The isolates from durum wheat that are found worldwide were the next derived form, followed by the isolates from common wheat. Both the coalescence analysis and the molecular clock approaches showed that P. triticina adapted to hosts in a process that paralleled the evolution of wheat (Feldman 2001) from the B genome of A. speltoides, to the AB genomes of emmer and durum wheat and then to the ABD genomes of common wheat. The center of origin of P. triticina is most likely the Mesopotamia region of the Fertile Crescent, where wheat progenitors (Salamini et al. 2002) and the alternate host (Tatlidil et al. 2005) are native.
Over the last 25 years different molecular markers have been used to characterize populations of P. triticina in North America (Kolmer 2001; Kolmer et al. 1995) and worldwide (Kolmer and Liu 2000). In North America distinct groups of P. triticina isolates with characteristic virulence have consistently been differentiated regardless of the marker (RAPD, AFLP, SSR, SNP) employed. With all markers, the virulence phenotype of the North American isolates could reliably be used to predict the molecular genotype group. The clonal nature of these populations has retained the virulence-molecular genotype association. The SNP genotypes of the isolates had very high correspondence to the SSR genotypes as well. The 40 MLG groups in the previous study had very similar genetic distance and relationships as the same grouping of isolates with SNP genotypes (Kolmer et al. 2019). The GBS approach generated a large number of SNP markers that allowed for higher bootstrap support for separation of geographic, genetic, and host-associated groups of P. triticina, compared to SSR markers. The GBS approach is also highly cost effective in comparison with the time and labor required to genotype populations with a large number of SSR primers.
In conclusion this study has provided a first analysis of variation between and within populations of P. triticina in several important wheat producing regions worldwide using the GBS method. The high degree of correspondence between the GBS and SSR genotypes further supports previous studies of genetic variation (Kolmer et al. 2019) that used SSR markers. World-wide, P. triticina has a complex population structure as new genotypes have spread across and between continents in the last 20 years by wind dispersed urediniospores, but yet endemic genotypes often still remain (Ordoñez and Kolmer 2009), with distinct genetic groups in most regions. Different forms of P. triticina associated with tetraploid and hexaploid wheat further add to the diversity of the pathogen. New virulent races of P. triticina arise by the sequential accumulation of virulence to host resistance genes deployed in wheat cultivars. Virulence to selective host resistance genes often arises in diverse genetic backgrounds (Kolmer and Hughes 2013; Kolmer et al. 2010) further contributing to the overall diversity of this globally widespread plant pathogen.
Data archiving
Data and scripts available from the University of Minnesota data repository: https://doi.org/10.13020/0f8c-k469.
References
Bebber DP, Holmes T, Gurr SJ (2014) The global spread of crop pests and pathogens. Glob Ecol Biogeogr 23:13989–11407
Bolton MD, Kolmer JA, Garvin DF (2008) Wheat leaf rust caused by Puccinia triticina. Mol Plant Pathol 9:563–575
Casulii F, Siniscalco A (1987) Thalictrum flavum L. as an alternate host of Puccinia recondita f. sp. tritici in Southern Italy. 7th Congress of the Mediterranean Phytopathology Union, Granada, Spain
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Eticha F, Belay G, Bekele E (2006) Species diversity in wheat landrace populations from two regions of Ethiopia. Genet Resour Crop Ev 53:387–393
Farlow D, Long H, Armoux S, Sung W, Doak TG, Nordborg M, Lynch M (2015) The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 201:737–744
Feldman M (2001) Origin of cultivated wheat. In: Bonjean AP, Angus WJ (eds) The world wheat book—a history of wheat breeding. Lavoisier Publishing, Intercept Ltd, Londres, Paris, New York, p 1–56
German SE, Kohli MM, Chaves M, Barcellos A, Campos P, de Viedma L (2007) Situation of cereal rusts in the southern cone of South America and perspectives of control. Aust J Agric Res 58:620–630
Gladieux P, Fuerty A, Hood ME, Snirc A, Clavel J, Dutech C, Roy M, Giraud T (2014) The population biology of fungal invasions. Mol Ecol 24:1969–1986
Goyeau H, Berder J, Czerepak C, Gautier A, Lanen C, Lannou C (2011) Low diversity and fast evolution in the population of Puccinia triticina causing durum wheat leaf rust in France from 1999 to 2009, as revealed by an adapted differential set. Plant Pathol 61:761–772
Goyeau H, Halkett F, Zapater MF, Carlier J, Lannou C (2007) Clonality and host selection in the wheat pathogenic fungus Puccinia triticina. Fungal Genet Biol 44:474–483
Goyeau H, Park R, Schaeffer B, Lannou C (2006) Distribution of pathotypes with regard to host cultivars and French wheat leaf rust populations. Phytopathology 96:264–273
Halkett F, Simon JC, Balloux F (2005) Tackling the population genetics of clonal and partially clonal organisms. Trends Ecol Evol 20:194–201
Hovmøller MS, Walter S, Bayles RA, Hubbard A, Flath K, Sommerfeld, Leconte M, Czembor P, Rodriguez-Algaba J, Thach T, Hansen JG, Lassen P, Justesen AF, de Vallavielle-Pope C (2015) Replacement of the European wheat yellow rust population by new races from the centre of diversity in the near-Himalayan region. Plant Pathol 65:401–411
Jin Y, Sazbo LJ, Rouse MN, Fetch T, Pretorius ZA, Wanyera R, Njau P (2009) Detection of virulence to resistance gene Sr36 within the TTKS lineage of Puccinia graminis f. sp. tritici. Plant Dis 93:367–370
Jin Y, Szabo LS, Pretorius ZA, Singh RP, Ward R, Fetch T (2008) Detection of virulence to resistance gene Sr24 within race TTKS of Puccinia graminis f. sp. tritici. Plant Dis 92:923–926
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281
Keenan K, McGinnity P, Cross T, Crozier WW, Prodohl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Kolmer JA (1998) Physiologic specialization of Puccinia recondita f. sp. tritici in Canada in 1996. Can J Plant Pathol 20:176–181
Kolmer JA (2001) Molecular polymorphism and virulence phenotypes of the wheat leaf rust fungus Puccinia triticina in Canada. Can J Bot 79:917–926
Kolmer JA, Acevedo MA (2016) Genetically divergent types of the wheat leaf rust fungus Puccinia triticina in Ethiopia, a center of tetraploid wheat diversity. Phytopathology 106:380–385
Kolmer JA, Hughes ME (2013) Physiological specialization of Puccinia triticina on wheat in the United States in 2011. Plant Dis 97:1103–1108
Kolmer JA, Hughes ME (2014) Physiologic specialization of Puccinia triticina on wheat in the United States in 2012. Plant Dis 98:1145–1150
Kolmer JA, Hughes ME (2018) Physiologic specialization of Puccinia triticina on wheat in the United States in 2016. Plant Dis 102:1066–1071
Kolmer JA, Liu JQ (2000) Virulence and molecular polymorphism in international collections of the wheat leaf rust fungus Puccinia triticina. Phytopathology 90:427–436
Kolmer JA, Liu JQ, Sies M (1995) Virulence and molecular polymorphism in Puccinia recondita f. sp. tritici in Canada. Phytopathology 85:276–285
Kolmer JA, Long DL, Hughes ME (2010) Physiologic specialization of Puccinia triticina on wheat in the United States in 2008. Plant Dis 94:775–780
Kolmer JA, Ordoñez ME, German SE, Morgounov A, Pretorious Z, Visser B, Goyeau H, Anikster Y, Acevedo M (2019) Multilocus genotypes of the wheat leaf rust fungus Puccinia triticina in worldwide regions indicate past and current long-distance migration. Phytopathology 109:1453–1463
Kolmer JA, Ordoñez ME, Manisterski J, Anikster Y (2011) Genetic differentiation of Puccinia triticina populations in the Middle East and genetic similarity with populations in central Asia. Phytopathology 101:870–877
Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100
Liu JQ, Kolmer JA (1998) Molecular and virulence diversity and linkage disequilibria in asexual and sexual populations of the wheat leaf rust fungus, Puccinia recondita. Genome 41:832–840
Liu M, Rodrigue N, Kolmer J (2014) Population divergence in the wheat leaf rust fungus Puccinia triticina is correlated with wheat evolution. Heredity 112:443–453
Long DL, Kolmer JA (1989) A North American system of nomenclature for Puccinia recondita f. sp. tritici. Phytopathology 79:525–529
Long DL, Leonard KJ, Hughes ME (2000) Virulence of Puccinia triticina on wheat in the United States from 1996 to 1998. Plant Dis 84:1334–1341
Long H, Behringer MG, Williams E, Te R, Lynch M (2016) Similar mutation rates but highly diverse mutation spectra in Asomycete and Baidiomycete yeasts. Genome Biol Evol 8:3815–3821
Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkami S, Hartl DL, Thomas WK (2018) A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci 105:9272–9277
Malinsky M, Trucchi E, Lawson DJ, Falush D (2018) RADpainter and fineRADstructure: population inference and RADseq data. Mol Biol Evol 35:1284–1290
McCallum B, Seto-Goh P, Foster A, Xue A (2018) Physiological specialization of Puccinia triticina, the causal agent of wheat leaf rust, in Canada in 2012. Can J Plant Pathol 40:434–441
Ordoñez ME, German SE, Kolmer JA (2010) Genetic differentiation within the Puccinia triticina population in South America and comparison with the North American population suggests common ancestry and intercontinental migration. Phytopathology 100:376–383
Ordoñez ME, Kolmer JA (2009) Differentiation of molecular genotypes and virulence phenotypes of Puccinia triticina from common wheat in North America. Phytopathology 99:750–758
Paradis E, Claude J, Strimmer K (2004) APE: analysis of phylogenetics and evolution in R language. Bioinformatics 20:289–290
Park RF, Bariana HS, Wellings CR, Wallwork H (2002) Detection and occurrence of a new pathotype of Puccinia triticina with virulence for Lr24 in Australia. Aust J Agric Res 53:1069–1076
Park RF, Burdon JJ, McIntosh RA (1995) Studies in the origin, spread, and evolution of an important group of Puccinia recondita f. sp. tritici pathotypes in Australasia. Eur J Plant Pathol 101:613–622
Peakall R, Smouse PE (2012) GenAlex 6: Genetic analysis in Excel. Population genetic software for teaching and research: an update. Bioinformatics 28:2537–2539
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Gen 5:92–102
Rosen Z, Bhaskar A, Roch S, Song YS (2018) Geometry of the sample frequency spectrum and the perils of demographic inference. Genetics 201:655–682
Saari EE, Prescott JM (1985) World distribution in relation to economic losses. In: Roelfs AP, Bushnell WR (eds) The Cereal Rusts. Academic Press, Orlando, p 259–298
Saari EE, Young HC, Kernkamp MF (1968) Infection of North American Thalictrum spp. with Puccinia recondita f. sp. tritici. Phytopathology 58:939–943
Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W (2002) Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet 3:429–441
Samborski DJ (1985) Wheat leaf rust. In: Roelfs AP, Bushnell WR (eds) The Cereal Rusts. Academic Press, Orlando, Fla., p 39–59. vol 2
Savary S, Willocquet L, Pethybridge SJ, Esker P, McRoberts N, Nelson A (2019) The global burden of pathogens and pests on major food crops. NatEcol Evol 3:430–439
Singh RP, Huerta-Espino J, Pfeiffer W, Figueroa-Lopez P (2004) Occurrence and impact of a new leaf rust race on durum wheat in northwestern Mexico from 2001 to 2003. Plant Dis 88:703–708
Soleiman NH, Solis I, Soliman MH, Sillero JC, Villegas D, Alvaro F, Royo C, Serra J, Ammar K, Martinez-Moreno F (2016) Emergence of a new race of leaf rust with combined virulence to Lr14a and Lr72 genes on durum wheat. Span J Agric Res 14(3):e10SC02. https://doi.org/10.5424/sjar/2016143-9184
Tatlidil S, Bicakci A, Malyer H, Can Baser KH (2005) Pollen morphology of Thalictrum L., species (Ranunculaceae) in Turkey. Pak J Bot 37:203–212
Wanyera R, Kinyua MG, Jin Y, Singh RP (2006) The spread of stem rust caused by Puccinia graminis f. sp. tritici, with virulence on Sr31 in wheat in Eastern Africa. Plant Dis 90:113
Yehuda PB, Eilam T, Manisterski J, Shimoni A, Anikster Y (2004) Leaf rust on Aegilops speltoides caused by a new forma specialis of Puccinia triticina. Phytopathology 94:94–101
Zhu YO, Siegel ML, Hall DW, Petrov DA (2014) Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci 111:E2310–E2318
Acknowledgements
The authors thank all of the collaborators who have sent collections of Puccinia triticina to the USDA-ARS Cereal Disease Laboratory for virulence typing and genotype analysis. The authors thank Kun Xiao for excellent greenhouse and laboratory assistance. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kolmer, J.A., Herman, A., Ordoñez, M.E. et al. Endemic and panglobal genetic groups, and divergence of host-associated forms in worldwide collections of the wheat leaf rust fungus Puccinia triticina as determined by genotyping by sequencing. Heredity 124, 397–409 (2020). https://doi.org/10.1038/s41437-019-0288-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-019-0288-x
This article is cited by
-
Nuclear exchange generates population diversity in the wheat leaf rust pathogen Puccinia triticina
Nature Microbiology (2023)

{kind=link}
{kind=link}