Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Cells
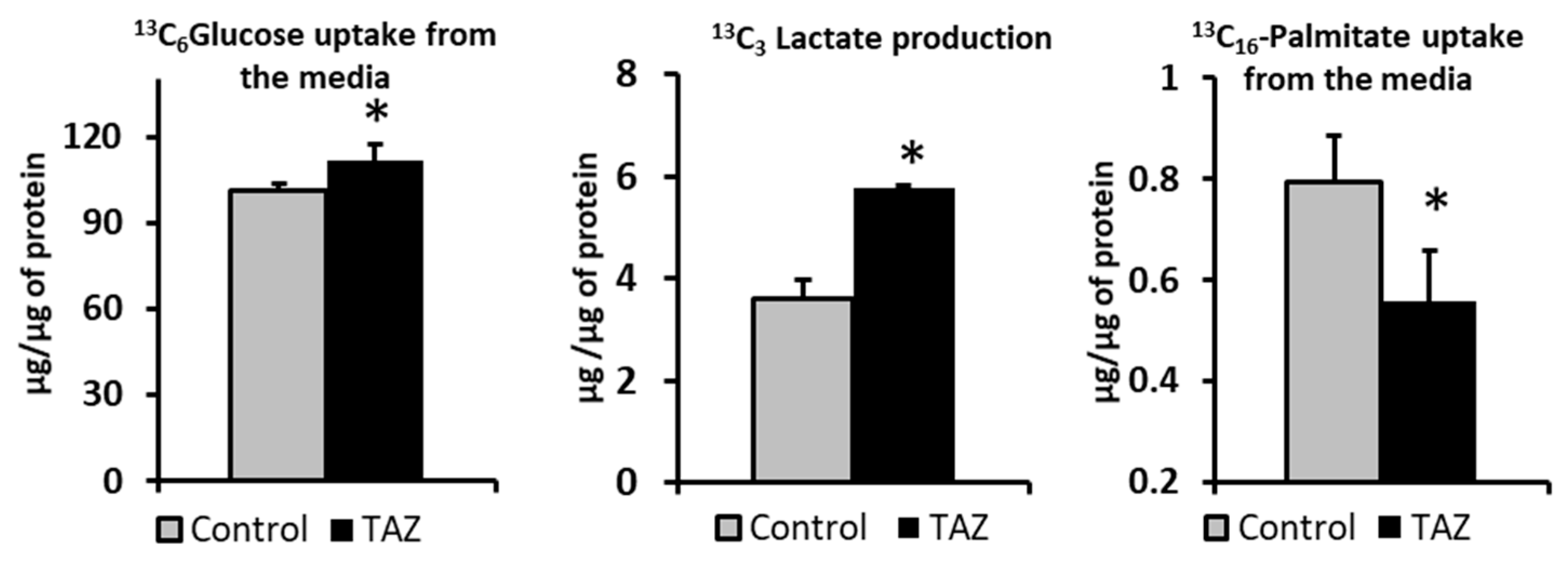
2.2. Carbon Substrates Preferences
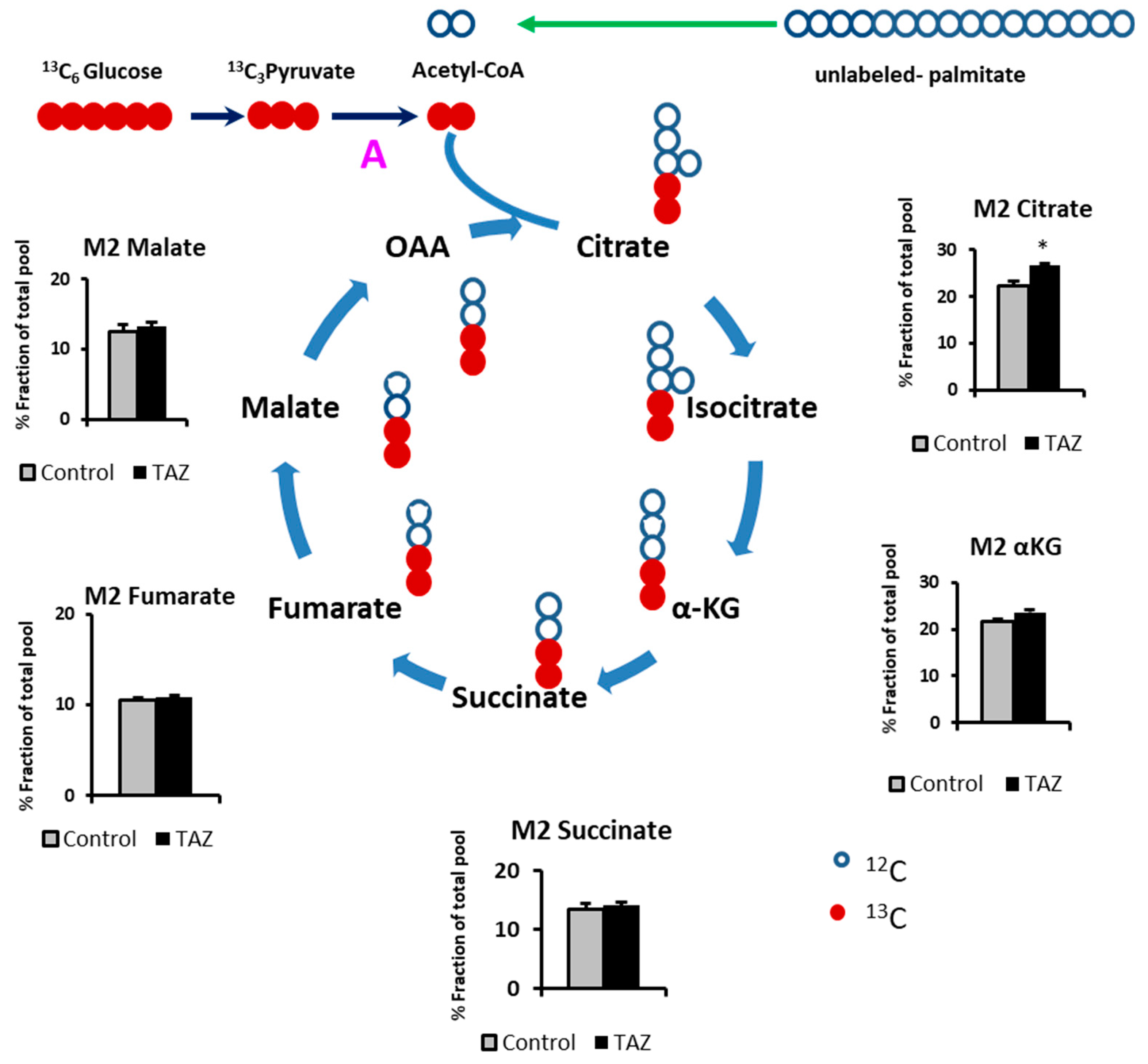
2.3. Glucose Carbons Incorporation into Krebs Cylcle Intermediates
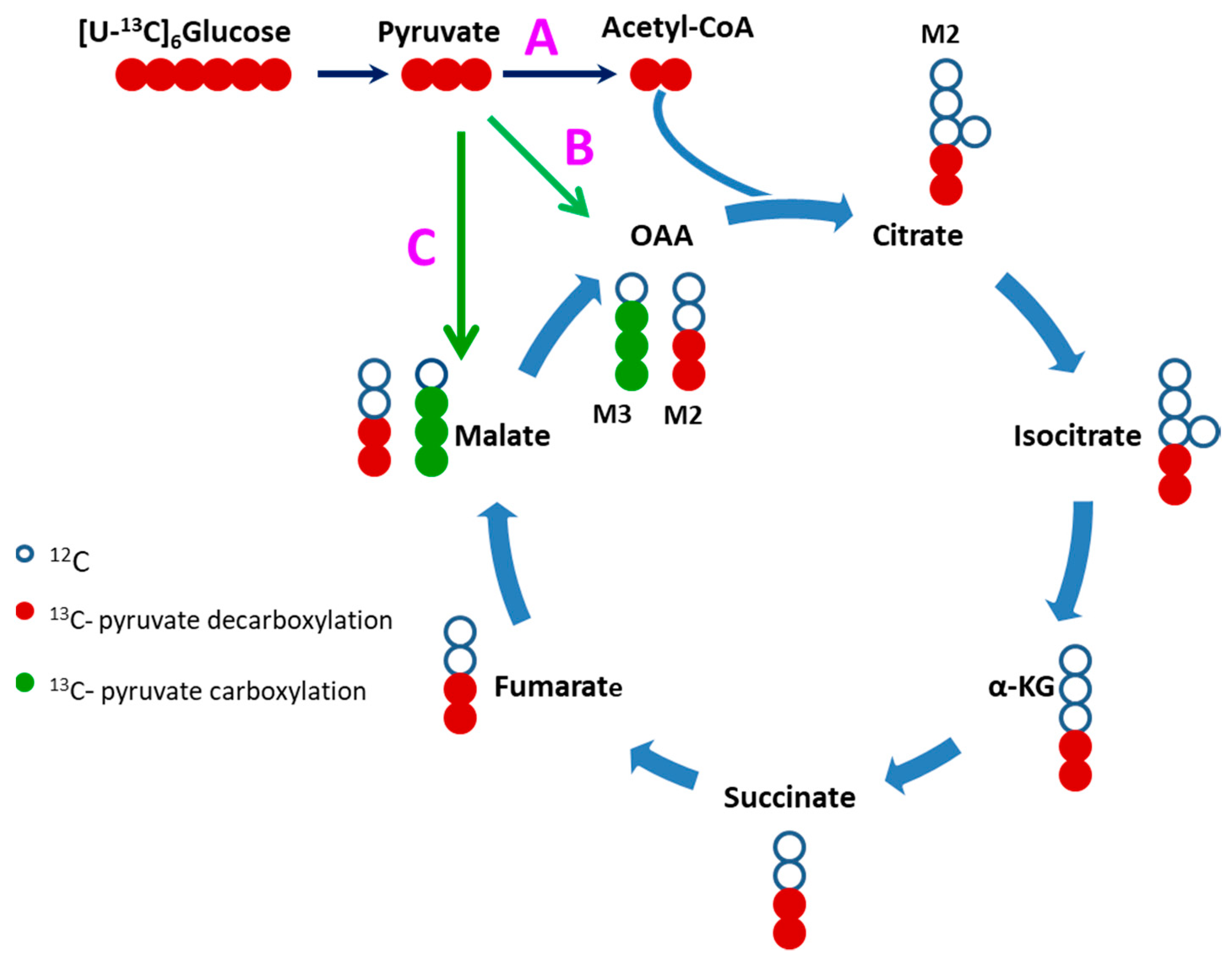
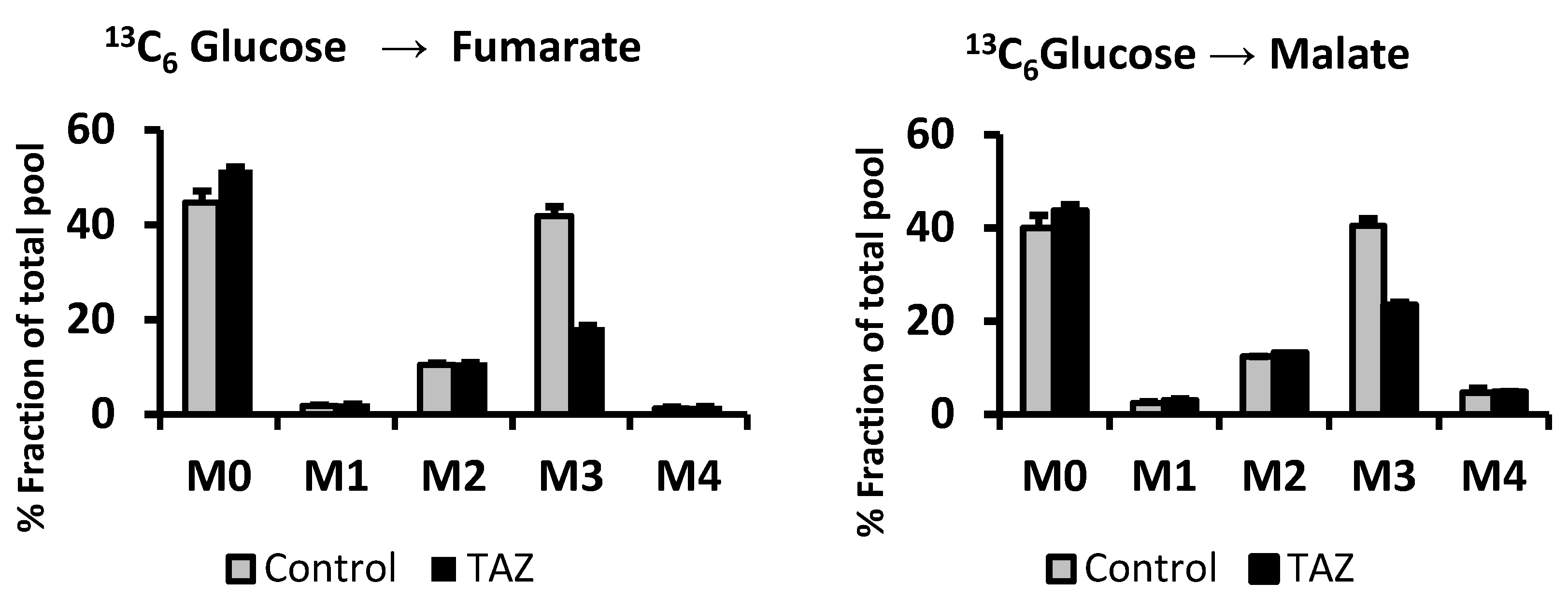
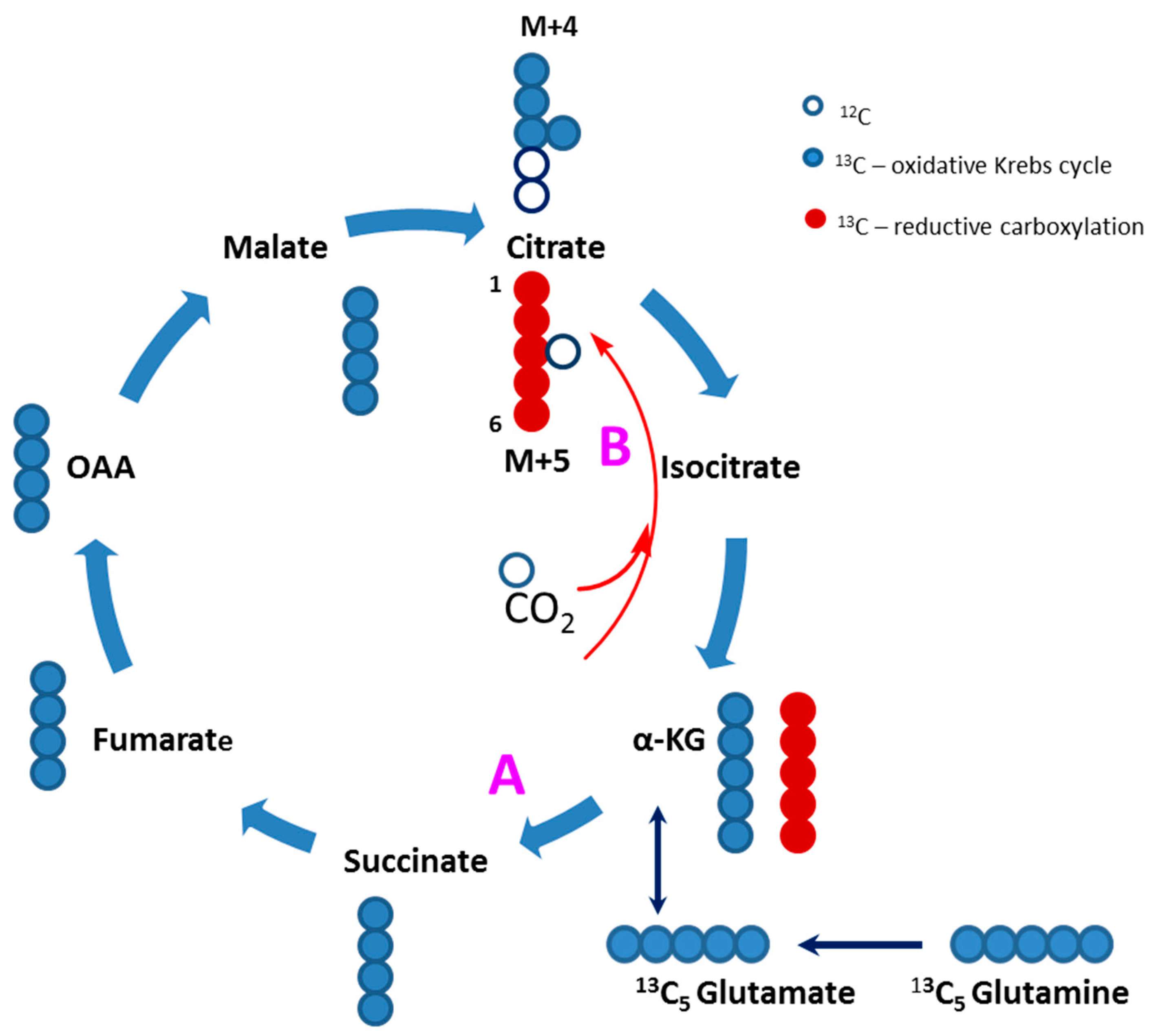
2.4. Evidence of Attenuated Pyruvate Anaplerosis
2.5. Alteration in Levels of Krebs Cycle Intermediates
2.6. Alteration in Amino Acids Levels
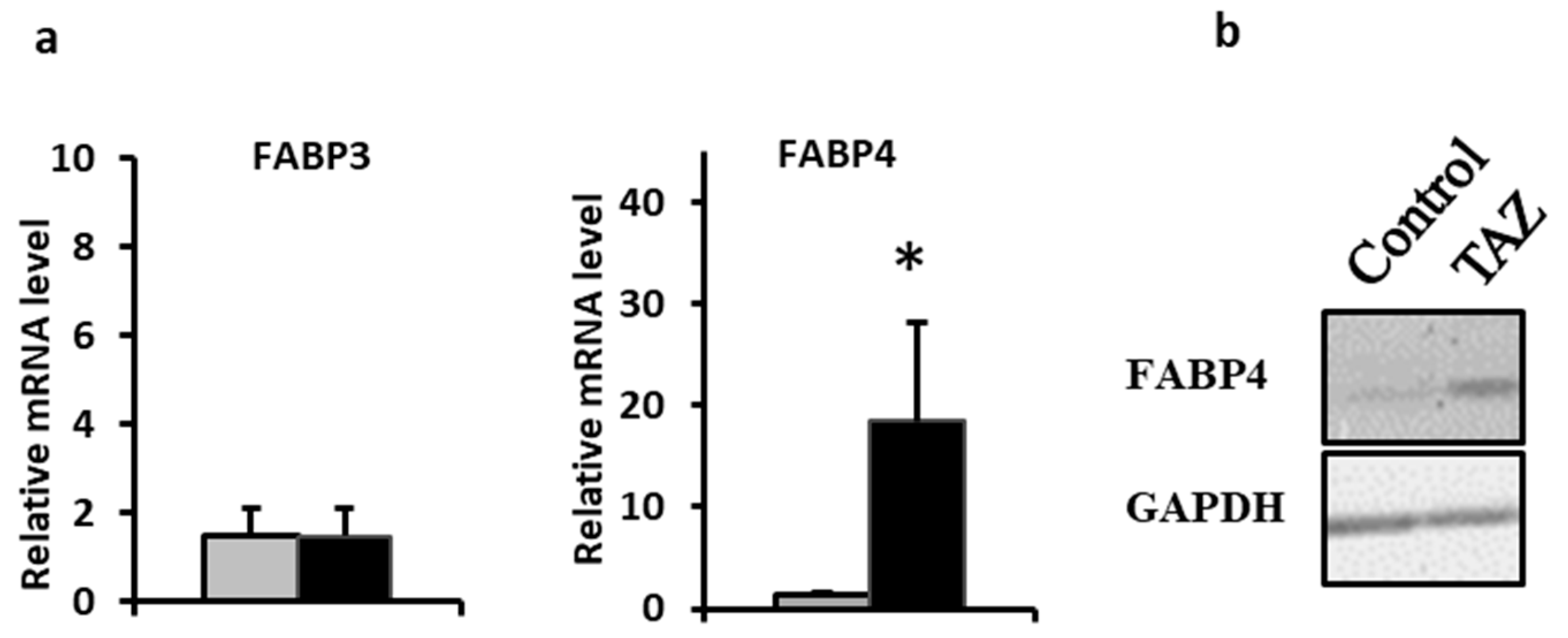
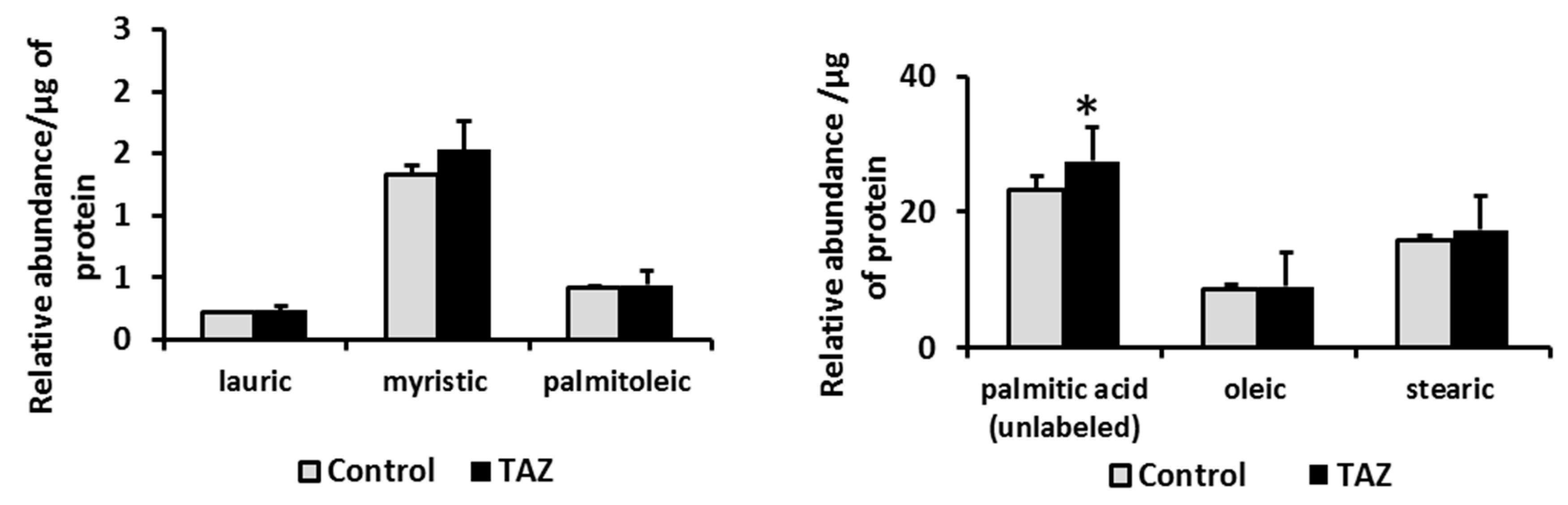
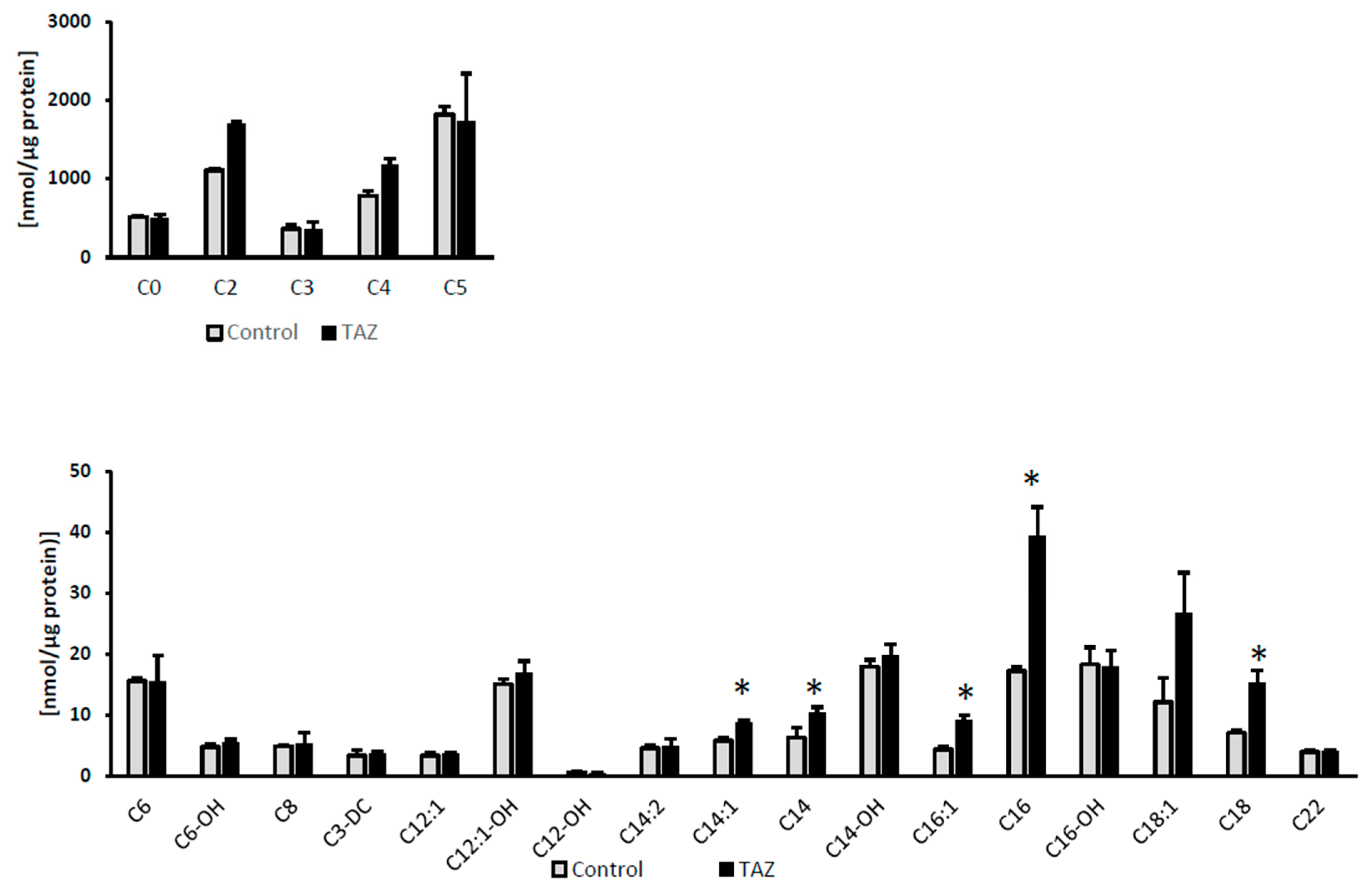
2.7. Disturbances in Fatty Acids Metabolism
2.8. Glucose Independent Glutamine Metabolism
3. Discussion
3.1. Lactate Production
3.2. Alteration in Substrate Preferences
3.3. Disturbances in Krebs Cycle Intermediates
3.4. Defective Pyruvate Anaplerosis
3.5. Alteration in Proline and Arginine Level
3.6. Glutamine as a Carbon Source
3.7. Alterations in Fatty Acids Metabolism
4. Materials and Methods
4.1. iPS Cells
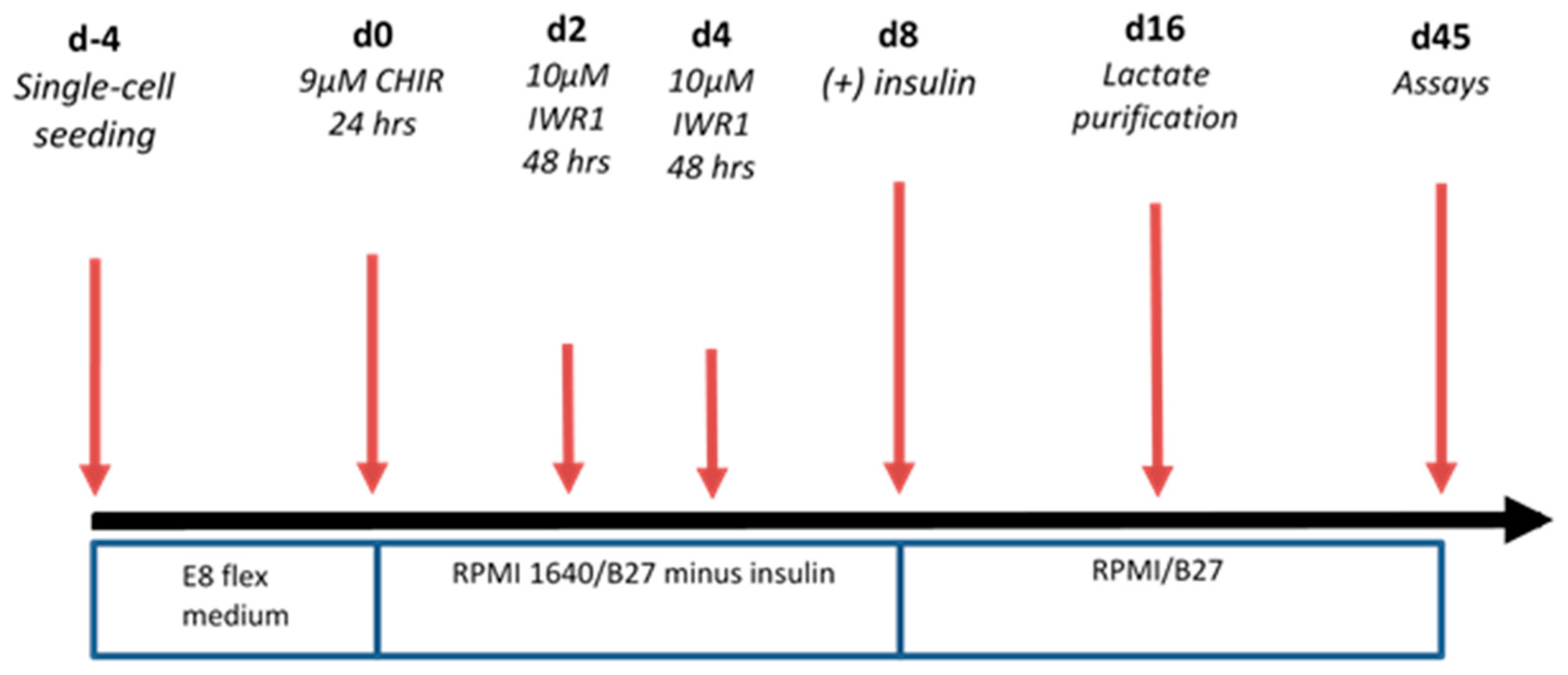
4.2. Differentiation to Cardiomyocytes
4.3. Glucose Uptake and Lactate Production
4.3.1. Glucose Uptake
4.3.2. Lactate Production
4.4. Palmitate Uptake
4.5. Metabolic Analysis
4.5.1. Krebs Cycle Intermediates
4.5.2. Acylcarnitine Analysis
4.5.3. Amino Acids Analysis
4.5.4. Free Fatty Acids Analysis
4.6. Gene Expression Analysis
4.7. Calculations
4.7.1. Metabolites Level
4.7.2. Statistical Analysis
4.7.3. Mass Isotopomer Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.T.; Bryant, R.M.; Day, J.; Gonzalez, I.L.; Colan, S.D.; Thompson, W.R.; Berthy, J.; Redfearn, S.P.; Byrne, B.J. Cardiac and Clinical Phenotype in Barth Syndrome. Pediatrics 2006, 118, e337–e346. [Google Scholar] [CrossRef]
- Jefferies, J.L. Barth syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2013, 163, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Woiewodski, L.; Ezon, D.; Cooper, J.; Feingold, B. Barth Syndrome with Late-Onset Cardiomyopathy: A Missed Opportunity for Diagnosis. J. Pediatr. 2017, 183, 196–198. [Google Scholar] [CrossRef]
- D’Adamo, P.; Fassone, L.; Gedeon, A.; Janssen, E.A.M.; Bione, S.; Bolhuis, P.A.; Barth, P.G.; Wilson, M.; Haan, E.; Örstavik, K.H.; et al. The X-Linked Gene G4.5 Is Responsible for Different Infantile Dilated Cardiomyopathies. Am. J. Hum. Genet. 1997, 61, 862–867. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M.; Ren, M.; Xu, Y.; Greenberg, M.L.; Haller, I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 2005, 138, 38–49. [Google Scholar] [CrossRef]
- Bowron, A.; Frost, R.; Powers, V.E.C.; Thomas, P.H.; Heales, S.J.R.; Steward, C.G. Diagnosis of Barth syndrome using a novel LC-MS/MS method for leukocyte cardiolipin analysis. J. Inherit. Metab. Dis. 2013, 36, 741–746. [Google Scholar] [CrossRef]
- Acehan, D.; Vaz, F.; Houtkooper, R.H.; James, J.; Moore, V.; Tokunaga, C.; Kulik, W.; Wansapura, J.; Toth, M.J.; Strauss, A.; et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 2011, 286, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Acehan, D.; Xu, Y.; Stokes, D.L.; Schlame, M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Investig. 2007, 87, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Kelley, R.I.; Cheatham, J.P.; Clark, B.J.; Nigro, M.A.; Powell, B.R.; Sherwood, G.W.; Sladky, J.T.; Swisher, W.P. X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J. Pediatr. 1991, 119, 738–747. [Google Scholar] [CrossRef]
- Vernon, H.J.; Sandlers, Y.; McClellan, R.; Kelley, R.I. Clinical laboratory studies in barth syndrome. Mol. Genet. Metab. 2014, 112. [Google Scholar] [CrossRef]
- Dudek, J.; Maack, C. Barth syndrome cardiomyopathy. Cardiovasc. Res. 2017, 113, 399–410. [Google Scholar] [CrossRef]
- Steward, C.G.; Newbury-Ecob, R.A.; Hastings, R.; Smithson, S.F.; Tsai-Goodman, B.; Quarrell, O.W.; Kulik, W.; Wanders, R.; Pennock, M.; Williams, M.; et al. Barth syndrome: An X-linked cause of fetal cardiomyopathy and stillbirth. Prenat. Diagn. 2010, 30, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.T.; Byrne, B.J.; Gewitz, M.H.; Wechsler, S.B.; Kao, A.C.; Gerstenfeld, E.P.; Merliss, A.D.; Carboni, M.P.; Bryant, R.M. Ventricular Arrhythmia in the X-linked Cardiomyopathy Barth Syndrome. Pediatr. Cardiol. 2005, 26, 632–637. [Google Scholar] [CrossRef]
- Chanana, A.M.; Rhee, J.-W.; Wu, J.C. Human-induced pluripotent stem cell approaches to model inborn and acquired metabolic heart diseases. Curr. Opin. Cardiol. 2016, 31, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Buikema, J.W.; Wu, S.M. Untangling the Biology of Genetic Cardiomyopathies with Pluripotent Stem Cell Disease Models. Curr. Cardiol. Rep. 2017, 19, 30. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra47. [Google Scholar] [CrossRef] [Green Version]
- Gintant, G.; Fermini, B.; Stockbridge, N.; Strauss, D. The Evolving Roles of Human iPSC-Derived Cardiomyocytes in Drug Safety and Discovery. Cell Stem Cell 2017, 21, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Burridge, P.W.; Kropp, E.M.; Chuppa, S.L.; Kwok, W.-M.; Wu, J.C.; Boheler, K.R.; Gundry, R.L. High Efficiency Differentiation of Human Pluripotent Stem Cells to Cardiomyocytes and Characterization by Flow Cytometry. J. Vis. Exp. 2014, 52010. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; van der Meer, P.; Serra, M.; et al. Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes by Inhibition of HIF1α and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13 C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef]
- Cade, W.T.; Spencer, C.T.; Reeds, D.N.; Waggoner, A.D.; O’Connor, R.; Maisenbacher, M.; Crowley, J.R.; Byrne, B.J.; Peterson, L.R. Substrate metabolism during basal and hyperinsulinemic conditions in adolescents and young-adults with Barth syndrome. J. Inherit. Metab. Dis. 2013, 36, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Sandlers, Y.; Mercier, K.; Pathmasiri, W.; Carlson, J.; McRitchie, S.; Sumner, S.; Vernon, H.J. Metabolomics Reveals New Mechanisms for Pathogenesis in Barth Syndrome and Introduces Novel Roles for Cardiolipin in Cellular Function. PLoS ONE 2016, 11, e0151802. [Google Scholar] [CrossRef]
- Rigaud, C.; Lebre, A.-S.; Touraine, R.; Beaupain, B.; Ottolenghi, C.; Chabli, A.; Ansquer, H.; Ozsahin, H.; Filippo, S. Di; Lonlay, P. De; et al. Natural history of Barth syndrome: A national cohort study of 22 patients. Orphanet J. Rare Dis. 2013, 8, 70. [Google Scholar] [CrossRef]
- Rodríguez-Calvo, R.; Girona, J.; Alegret, J.M.; Bosquet, A.; Ibarretxe, D.; Masana, L. Role of the fatty acid-binding protein 4 in heart failure and cardiovascular disease. J. Endocrinol. 2017, 233, R173–R184. [Google Scholar] [CrossRef]
- Baessler, A.; Lamounier-Zepter, V.; Fenk, S.; Strack, C.; Lahmann, C.; Loew, T.; Schmitz, G.; Blüher, M.; Bornstein, S.R.; Fischer, M. Adipocyte fatty acid-binding protein levels are associated with left ventricular diastolic dysfunction in morbidly obese subjects. Nutr. Diabetes 2014, 4, e106. [Google Scholar] [CrossRef]
- Takagi, W.; Miyoshi, T.; Doi, M.; Okawa, K.; Nosaka, K.; Nishibe, T.; Matsuo, N.; Hirohata, S.; Ito, H. Circulating adipocyte fatty acid-binding protein is a predictor of cardiovascular events in patients with stable angina undergoing percutaneous coronary intervention. BMC Cardiovasc. Disord. 2017, 17, 258. [Google Scholar] [CrossRef] [Green Version]
- Fuseya, T.; Furuhashi, M.; Yuda, S.; Muranaka, A.; Kawamukai, M.; Mita, T.; Ishimura, S.; Watanabe, Y.; Hoshina, K.; Tanaka, M.; et al. Elevation of circulating fatty acid-binding protein 4 is independently associated with left ventricular diastolic dysfunction in a general population. Cardiovasc. Diabetol. 2014, 13, 126. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Bruntz, R.C.; Lane, A.N.; Higashi, R.M.; Fan, T.W. Exploring cancer metabolism using Stable isotope-resolved metabolomics (SIRM). J. Biol. Chem. 2017, 292, 11601–11609. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Valianpour, F.; Chen, S.; Vaz, F.M.; Hakkaart, G.A.; Wanders, R.J.A.; Greenberg, M.L. Aberrant cardiolipin metabolism in the yeast taz1 mutant: A model for Barth syndrome. Mol. Microbiol. 2003, 51, 149–158. [Google Scholar] [CrossRef]
- Whited, K.; Baile, M.G.; Currier, P.; Claypool, S.M. Seven functional classes of Barth syndrome mutation. Hum. Mol. Genet. 2013, 22, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Valianpour, F.; Wanders, R.J.A.; Overmars, H.; Vaz, F.M.; Barth, P.G.; van Gennip, A.H. Linoleic acid supplemention of Barth syndrome fibroblasts restores cardiolipin levels. J. Lipid Res. 2003, 44, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sutachan, J.J.; Plesken, H.; Kelley, R.I.; Schlame, M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab. Investig. 2005, 85, 823–830. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Maianski, N.A.; Tool, A.T.J.; Becker, K.; Plecko, B.; Valianpour, F.; Wanders, R.J.A.; Pereira, R.; Van Hove, J.; Verhoeven, A.J.; et al. Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood 2004, 103, 3915–3923. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Wang, M.; Harris, N.; Han, X. Tafazzin knockdown interrupts cell cycle progression in cultured neonatal ventricular fibroblasts. Am. J. Physiol. Circ. Physiol. 2013, 305, H1332–H1343. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Harris, N.; Ren, J.; Han, X. Mitochondria-targeted antioxidant prevents cardiac dysfunction induced by tafazzin gene knockdown in cardiac myocytes. Oxid. Med. Cell. Longev. 2014, 2014, 654198. [Google Scholar] [CrossRef] [Green Version]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Miller, P.C.; Schlame, M.; Phoon, C.K.L. A Critical appraisal of the tafazzin knockdown mouse model of Barth syndrome: what have we learned about pathogenesis and potential treatments. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1183–H1193. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Powers, C.; Moore, V.; Schafer, C.; Ren, M.; Phoon, C.K.L.; James, J.F.; Glukhov, A.V.; Javadov, S.; Vaz, F.M.; et al. The PPAR pan-agonist bezafibrate ameliorates cardiomyopathy in a mouse model of Barth syndrome. Orphanet J. Rare Dis. 2017, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, L.; Donati, M.A.; Funghini, S.; Malvagia, S.; Catarzi, S.; Lugli, L.; Ragni, L.; Bertini, E.; Vaz, F.M.; Cooper, D.N.; et al. New clinical and molecular insights on Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, V.; Reynolds, C.A.; Greenberg, M.L. Barth syndrome: A life-threatening disorder caused by abnormal cardiolipin remodeling. J. Rare Dis. Res. Treat. 2017, 2, 58–62. [Google Scholar] [PubMed]
- Chabowski, A.; Górski, J.; Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Protein-mediated Fatty Acid Uptake in the Heart. Curr. Cardiol. Rev. 2008, 4, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Stanley, W.C. Myocardial energy metabolism in dilated cardiomyopathy. Heart Metab. 2011, 49, 5–8. [Google Scholar]
- Dávila-Román, V.G.; Vedala, G.; Herrero, P.; de las Fuentes, L.; Rogers, J.G.; Kelly, D.P.; Gropler, R.J. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar]
- Mejia, E.M.; Zinko, J.C.; Hauff, K.D.; Xu, F.Y.; Ravandi, A.; Hatch, G.M. Glucose Uptake and Triacylglycerol Synthesis Are Increased in Barth Syndrome Lymphoblasts. Lipids 2017, 52, 161–165. [Google Scholar] [CrossRef]
- Fournier, N.C.; Richard, M.A. Fatty acid-binding protein, a potential regulator of energy production in the heart. Investigation of mechanisms by electron spin resonance. J. Biol. Chem. 1988, 263, 14471–14479. [Google Scholar] [PubMed]
- Cade, W.T.; Bohnert, K.L.; Peterson, L.R.; Patterson, B.W.; Bittel, A.J.; Okunade, A.L.; de las Fuentes, L.; Steger-May, K.; Bashir, A.; Schweitzer, G.G.; et al. Blunted fat oxidation upon submaximal exercise is partially compensated by enhanced glucose metabolism in children, adolescents, and young adults with Barth syndrome. J. Inherit. Metab. Dis. 2019, 42, 12094. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Powers, C.; Madala, S.K.; Greis, K.D.; Haffey, W.D.; Towbin, J.A.; Purevjav, E.; Javadov, S.; Strauss, A.W.; Khuchua, Z. Cardiac Metabolic Pathways Affected in the Mouse Model of Barth Syndrome. PLoS ONE 2015, 10, e0128561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Lou, W.; Raja, V.; Denis, S.; Yu, W.; Schmidtke, M.W.; Reynolds, C.A.; Schlame, M.; Houtkooper, R.H.; Greenberg, M.L. Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J. Biol. Chem. 2019, 294, 11568–11578. [Google Scholar] [CrossRef] [PubMed]
- Chatzispyrou, I.A.; Guerrero-Castillo, S.; Held, N.M.; Ruiter, J.P.N.; Denis, S.W.; IJlst, L.; Wanders, R.J.; van Weeghel, M.; Ferdinandusse, S.; Vaz, F.M.; et al. Barth syndrome cells display widespread remodeling of mitochondrial complexes without affecting metabolic flux distribution. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 3650–3658. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Stanley, W.C.; Lopaschuk, G.D.; Hall, J.L.; McCormack, J.G. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc. Res. 1997, 33, 243–257. [Google Scholar] [CrossRef]
- Liepinsh, E.; Makrecka-Kuka, M.; Volska, K.; Kuka, J.; Makarova, E.; Antone, U.; Sevostjanovs, E.; Vilskersts, R.; Strods, A.; Tars, K.; et al. Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem. J. 2016, 473, 1191–1202. [Google Scholar] [CrossRef]
- Saric, A.; Andreau, K.; Armand, A.-S.; Møller, I.M.; Petit, P.X. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front. Genet. 2015, 6, 359. [Google Scholar] [CrossRef] [Green Version]
- Toischer, K.; Dudek, J.; Cheng, I.-F.; Bru gger, B.; Balleininger, M.; Rehling, P.; Grunau, S.; Wozny, K.; Hasenfuss, G.; Reinhold, R.; et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol. Med. 2015, 8, 139–154. [Google Scholar]
- Des Rosiers, C.; Labarthe, F.; Lloyd, S.G.; Chatham, J.C. Cardiac anaplerosis in health and disease: food for thoughts. Cardiovasc. Res. 2011, 90, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibala, M.J.; Young, M.E.; Taegtmeyer, H. Anaplerosis of the citric acid cycle: Role in energy metabolism of heart and skeletal muscle. Acta Physiol. Scand. 2000, 168, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, K.E.; Hiltunen, J.K.; Hassinen, I.E. Pyruvate carboxylation in the rat heart. Role of biotin-dependent enzymes. Biochem. J. 1989, 257, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R.; Taegtmeyer, H.; Taegtmeyer, H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J. Clin. Invest. 1991, 87, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.R.; Taegtmeyer, H. Pyruvate carboxylation prevents the decline in contractile function of rat hearts oxidizing acetoacetate. Am. J. Physiol. Circ. Physiol. 1991, 261, H1756–H1762. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Taegtmeyer, H. Metabolic recovery of isolated working rat heart after brief global ischemia. Am. J. Physiol. 1994, 267, H462–H470. [Google Scholar] [CrossRef]
- Pandhare, J.; Donald, S.P.; Cooper, S.K.; Phang, J.M. Regulation and function of proline oxidase under nutrient stress. J. Cell. Biochem. 2009, 107, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Hancock, C.N.; Liu, W.; Alvord, W.G.; Phang, J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids 2016, 48, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, R.L.S.; Rothschild, D.E.; Quinlan, C.L.; Scott, G.K.; Benz, C.C.; Brand, M.D. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biol. 2014, 2, 901–909. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J. Mol. Biol. 2006, 361, 462–469. [Google Scholar] [CrossRef]
- Kühl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 2017, 6, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Mitochondrial Medicine Society’s Committee on Diagnosis; Haas, R.H.; Parikh, S.; Falk, M.J.; Saneto, R.P.; Wolf, N.I.; Darin, N.; Wong, L.-J.; Cohen, B.H.; Naviaux, R.K. The in-depth evaluation of suspected mitochondrial disease. Mol. Genet. Metab. 2008, 94, 16–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, W.-J.; Zeng, X.-W.; Deng, A.; Zhao, S.-J.; Luo, D.-Z.; Ma, G.-Z.; Wang, H.; Liu, Q. Circulating FABP4 (Fatty Acid-Binding Protein 4) Is a Novel Prognostic Biomarker in Patients with Acute Ischemic Stroke. Stroke 2017, 48, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Zoair, A.; Mawlana, W.; Abo-Elenin, A.; Korrat, M. Serum Level of Heart-Type Fatty Acid Binding Protein (H-FABP) Before and After Treatment of Congestive Heart Failure in Children. Pediatr. Cardiol. 2015, 36, 1722–1727. [Google Scholar] [CrossRef]
- Gan, L.; Liu, Z.; Cao, W.; Zhang, Z.; Sun, C. FABP4 reversed the regulation of leptin on mitochondrial fatty acid oxidation in mice adipocytes. Sci. Rep. 2015, 5, 13588. [Google Scholar] [CrossRef] [Green Version]
- Lamounier-Zepter, V.; Look, C.; Alvarez, J.; Christ, T.; Ravens, U.; Schunck, W.-H.; Ehrhart-Bornstein, M.; Bornstein, S.R.; Morano, I. Adipocyte Fatty Acid-Binding Protein Suppresses Cardiomyocyte Contraction: A New Link Between Obesity and Heart Disease. Circ. Res. 2009, 105, 326–334. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 2009, 1788, 2080–2083. [Google Scholar] [CrossRef] [Green Version]
- Noël, H.; Pande, S.V. An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity. Lipid requirement of carnitine acylcarnitine translocase. Eur. J. Biochem. 1986, 155, 99–102. [Google Scholar] [CrossRef]
- Kalim, S.; Clish, C.B.; Wenger, J.; Elmariah, S.; Yeh, R.W.; Deferio, J.J.; Pierce, K.; Deik, A.; Gerszten, R.E.; Thadhani, R.; et al. A Plasma Long-Chain Acylcarnitine Predicts Cardiovascular Mortality in Incident Dialysis Patients. J. Am. Heart Assoc. 2013, 2, e000542. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.A.; Kanter, E.M.; Newatia, A. Long-chain acylcarnitine induces Ca2+ efflux from the sarcoplasmic reticulum. J. Cardiovasc. Pharmacol. 2000, 36, 14–21. [Google Scholar] [CrossRef]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferro, F.; Ouillé, A.; Tran, T.-A.; Fontanaud, P.; Bois, P.; Babuty, D.; Labarthe, F.; Le Guennec, J.-Y. Long-chain acylcarnitines regulate the hERG channel. PLoS ONE 2012, 7, e41686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knabb, M.T.; Saffitz, J.E.; Corr, P.B.; Sobel, B.E. The dependence of electrophysiological derangements on accumulation of endogenous long-chain acyl carnitine in hypoxic neonatal rat myocytes. Circ. Res. 1986, 58, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct Metabolic Flow Enables Large-Scale Purification of Mouse and Human Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.F. Removal of fatty acids from serum albumin by charcoal treatment. J. Biol. Chem. 1967, 242, 173–181. [Google Scholar] [PubMed]
- House, A.; Fatica, E.; Shah, R.; Stergar, J.; Pearce, R.; Sandlers, Y. A protocol for metabolic characterization of human induced pluripotent derived cardiomyocytes (iPSCM). MethodsX 2019. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iPSCM | M2 Malate | M3 Malate | M3 Fumarate | FC(M3 Malate)/ FC (M2 Malate) | M3 Succinate | (FC M3 Malate)- (FC M3 Succinate) 1 |
|---|---|---|---|---|---|---|
| Control-iPS-CM | 12.43 ± 1.01 | 40.5 ± 1.4 | 41.8 ± 1.9 | 3.25 | 4.79 ± 0.4 | 35.7 |
| TAZ517delG-iPS-CM | 13.2 ± 0.52 | 23.4 ± 0.59* | 18.4 ± 0.53* | 2.68 | 3.2 ± 0.5 | 20.2 |
| a | Arginine | Proline | ||
| Current iPS-CM study (µmol/mg protein) | ||||
| Control | TAZ | Control | TAZ517delG | |
| Non starved | 5.02 | 3.05 | 0.46 | 0.44 |
| Glucose starved | NA | NA | 0.15 | 0.45 |
| Plasma clinical studies [9,24] (µmol/L) | ||||
| Control | TAZ | Control | TAZ517delG | |
| [24] | 100 | 50 | 190 | 280 |
| [11] | 69.8 | 42.9 | 164.7 | 291.1 |
| [23] | 68 | 29 | NA | NA |
| b | Arginine | Proline | ||
| Current iPS-CM study calculated fold change (FC) | ||||
| Non starved | 0.6 | 1 | ||
| Glucose starved | NA | 3 | ||
| Plasma clinical studies calculated fold change (FC) | ||||
| [24] | 0.5 | 1.5 | ||
| [11] | 0.6 | 1.8 | ||
| [23] | 0.4 | NA | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatica, E.M.; DeLeonibus, G.A.; House, A.; Kodger, J.V.; Pearce, R.W.; Shah, R.R.; Levi, L.; Sandlers, Y. Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Metabolites 2019, 9, 306. https://doi.org/10.3390/metabo9120306
Fatica EM, DeLeonibus GA, House A, Kodger JV, Pearce RW, Shah RR, Levi L, Sandlers Y. Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Metabolites. 2019; 9(12):306. https://doi.org/10.3390/metabo9120306
Chicago/Turabian StyleFatica, Erica M., Gina A. DeLeonibus, Alisha House, Jillian V. Kodger, Ryan W. Pearce, Rohan R. Shah, Liraz Levi, and Yana Sandlers. 2019. "Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes" Metabolites 9, no. 12: 306. https://doi.org/10.3390/metabo9120306