
Abstract
Maturity-onset diabetes of the young (MODY) is a genetically and clinically heterogeneous group of disorders characterised by early onset, lean, non-autoimmunity mediated, non-insulin-dependent diabetes often with autosomal-dominant inheritance and specific pharmaco-genetic response. We describe two siblings with HNF1A-MODY (MODY3) due to a novel germline variant p.(His126Asp) which segregates with diabetes in the family. However, contrary to anticipated therapeutic response, blood glucose in this sib-pair did not respond to sulphonylureas (both low and high dose), dipeptidyl peptidase-4 inhibitors (DPP-4 inhibitors), and glucagon-like peptide-1 receptor agonists (GLP-1RA), also known as incretin mimetics. The unexpected limited pharmaco-therapeutic response could potentially be unique to this specific variant and/or progressive pancreatic β-cell failure associated with long-standing disease duration, higher BMI and glucose-toxicity. Therefore, we report a novel-variant MODY3 sib-pair with atypical pharmaco-therapeutic response i.e. resistant to multiple anti-diabetes agents namely sulphonylurea, DPP-4 inhibitors and GLP-1RA treatment.
Similar content being viewed by others


Introduction
Maturity-onset diabetes of the young (MODY) [OMIM: 606391] is a rare non-autoimmune forms of diabetes mellitus that share clinical similarities with classic form of type 1 and 2 diabetes mellitus (T2DM). Also known as monogenic diabetes, MODY is distinguished by causal variants within a single gene critical in pancreatic β-cell insulin secretory function. Clinically, it is characterised by young age of onset, lean, autosomal-dominant inheritance and usually non-insulin dependent, non-ketotic prone hyperglycaemia [1]. The typical onset of MODY is between 6 months and 35 years of age [2]. Fourteen genes have been associated with MODY as listed in the Online Mendelian Inheritance in Man (OMIM) database [3].
Establishing a genetic diagnosis of MODY may be clinically actionable. For instance, patients with HNF1A-MODY and HNF4A-MODY are extremely sensitive to sulfonylureas (SUs) while GCK-MODY patients do not require anti-diabetic treatment and are well-managed by diet alone [4]. Misdiagnosis of MODY as type 1 diabetes mellitus (T1DM) or young-onset T2DM is common due to their over-lapping features which consequently led to misguided pharmacological intervention (i.e. insulin therapy).
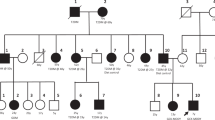
In this report, we describe a sib-pair with HNF1A-MODY associated with a novel variant. However, their glycemic response to multiple recommended non-insulin pharmaco-therapeutics was suboptimal, thereby highlighting the possible existence of MODY3 with atypical therapeutic response.
Materials (Subjects) and methods
Refer to Supplementary Material.
Results
DNA sequencing revealed a novel non-synonymous variant [NM_000545.6 (HNF1A): c.376C>G p.(His126Asp)] in both patients that was not found in population databases (Genome Aggregation Database (gnomAD), Exome Aggregation Consortium (ExAC)) (Supplementary Fig. 2). The variant is found in exon 2 of the HNF1A (hepatocyte nuclear factor 1α) gene (RefSeqGene NG_011731.2) and resides in the DNA-binding domain, which may affect transcription activity of the protein. Computational tools (SIFT, PolyPhen-2 and Mutation Taster) predicted the variant to be functionally deleterious. Taken together, the variant is interpreted to be likely pathogenic for MODY3.
Sulphonylurea therapy was initiated in both patients upon genetic diagnosis of HNF1A-MODY. Low dose glipizide (5 mg TDS) was given together with NPH insulin (isophane insulin) or insulin glargine in patient III.2 and III.3, respectively (Supplementary Fig. 3A, B). Patient III.2 showed some early response to low dose glipizide but was non-durable. Conversion to gliclazide was followed after ~4–5 months and further titrated up to supra-therapeutic dose of 280 and 400 mg/day, respectively, in both patients. Nonetheless, responsiveness to low or high dose sulphonylureas was suboptimal (HbA1c remained above 7%) in both patients. Reports have shown that incretin-based intervention, including dipeptidyl-peptidase-4 inhibitors (DPP-4 inhibitors) and glucagon-like peptide-1 receptor agonists (GLP-1RA) are effective glucose-lowering agents with lower risk of hypoglycemia in patients with MODY3 (ref. [5, 6]). Hence, both patients were tried on 50 mg of sitagliptin, an oral antihyperglycemic of the DPP-4 inhibitor class, in addition to gliclazide and basal insulin. Response to sitagliptin was short-lived (about three months) with better improvement in glucose control for Patient III.2 (close to 1% decrease in HbA1c). On the other hand, increasing dosage of sitagliptin to 100 mg OM in Patient III.3 did not improve her glycaemic control.
Partly motivated for the desire for weight control, dulaglutide (1.5 mg weekly) was initiated in Patient III.3 (Supplementary Fig. 3C) with very limited glycemic and weight benefit. Therefore, liraglutide (1.2 mg/day) was substituted but glycaemic control remained suboptimal (HbA1c above 8%). Consequently, both patients were converted back to insulin hormone replacement therapy in additional to oral hypoglycaemia agents.
Discussion
We present a pair of siblings with MODY3 due to a novel p.(His126Asp) variant in HNF1A which co-segregates in the family. HNF1A encodes a transcription factor that is essential for β-cell differentiation and function. Specifically, it regulates insulin secretion and glucose homoeostasis, pancreas development in the liver and kidney as well as oxidative stress among other functions [7, 8]. Therefore, affected cases have progressive β-cell defects. Genetic alterations in HNF1A may produce the MODY phenotype via a reduced function of the protein (missense variants), haploinsufficiency due to nonsense-mediated RNA decay (nonsense variants) or a dominant-negative mutational mechanism [9]. Variant in the DNA-binding site as in p.(His126Asp), could result in a reduced function of the protein, producing the MODY phenotype. There have been evidences of micro- and macro-vascular complications in MODY3 patients, highlighting the need to control blood glucose levels in these patients [10].
Existing literature suggested that patients with HNF1A-MODY are highly sensitive to the hypoglycaemia effects of sulphonylurea although the mechanism is unclear [11, 12]. Unexpectedly, the two patients presented here were found to be unresponsive to low or high doses of sulphonylurea. An extensive literature search suggested that our experience is not unique. For instance, Demol et al. described a similar case whereby a family with a novel [NM_000545.3 (HNF1A):c.618G>A p.(Trp206Ter)] termination variant are also non-responsive to glibenclamide treatment, whose severe phenotype is due to the loss of the DNA-binding domain as the variant is predicted to cause early truncation of the protein, rendering it non-functional [8]. Separately, a recent landmark study assessed the pharmacological response to sulphonylurea in a group of Mexican individuals carrying the low-frequency ethnic-specific HNF1A variant [NM_000545.6 (HNF1A):c.1522G>A p.(Glu508Lys)] and found that carriers of the variant have a reduced insulin response rather than increased sensitivity to sulphonylureas compared to non-carriers [13]. The variant, located in the transactivation domain, is rare or absent in non-Native American populations and was reported to be associated with a fivefold increased risk for development of diabetes. Carriers of the variant were expected to respond to sulphonylurea due to the partial loss of transcriptional activity conferred by the missense variant, although not as severe as other MODY3-causing variants. The unexpected non-responsiveness to sulphonylurea in carriers with diabetes compared with non-carriers may be due to a more aggressive disease progression that accelerates β-cell failure when compared with common type 2 diabetes, rendering sulphonylurea less effective in variant carriers. Another recent large observational study described that HNF1A-MODY patients, who achieved good glycaemic control upon change of treatment to sulphonylurea, had shorter diabetes duration, lower BMI and lower HbA1c at genetic diagnosis compared with those who did not achieve good control [14]. In concordant, existing literature suggested that about 50% of MODY3 patients with progressive disease require insulin therapy [15].
Despite reports of successful treatment with incretin-based agents (DPP-4 inhibitors and GLP-1RA) in MODY3 patients presenting with loss-of-function variants in HNF1A, both siblings failed to achieve optimal glycaemic control with adjunct therapy of sitagliptin, dulaglutide and liraglutide. Consequently, exogenous insulin replacement is still necessary to manage the hyperglycaemia. We acknowledge that similar therapeutic evaluations in two other family members (II.2 and III.1) who also carry the same variant will provide additional insights in understanding the physiological responses in carriers of HNF1A p.(His126Asp). However, information on the clinical management of the two family members is not available as they are under the care of a separate institution. Moreover, through the probands, we have been informed that these two family members had significant diabetic kidney disease, so much so that one of them has been receiving long-term renal replacement therapy (RRT). RRT is an absolute (or relative) contraindication for anti-diabetic therapy like GLP-1RA and certain DPP-4 inhibitors (e.g. Sitagliptin). Therefore, it will not be feasible to conduct similar glycemic pharmaco-genetic evaluation in the other two family members receiving care from the other institution. Taken together, systemic-studies on different variant types and pharmacogenomic evaluations over a long period are required to understand the therapeutic responses in patients carrying HNF1A variants.
In conclusion, we report a sib-pair genetically diagnosed with MODY3 caused by a novel HNF1A variant p.(His126Asp) but are non-responsive to sulphonylurea, DPP-4 inhibitors and GLP-1RA therapy, possibly due to their progressive pancreatic β-cell failure associated with long-standing disease duration (>20 years), higher BMI and glucose-toxicity. Our report suggests that variant-type and clinical features (i.e. duration of diabetes, glucose levels and BMI) should be taken into consideration when counselling glucose-lowering therapy for patients with MODY3.
References
Jason F, Stefan J, Pal RN. Common and rare forms of diabetes mellitus: towards a continuum of diabetes subtypes. Nat Rev Endo 2016;12:394–406.
Molven A, Njolstad PR. Role of molecular genetics in transforming diagnosis of diabetes mellitus. Expert Rev Mol Diagn. 2011;11:313–20.
Shields BM, Hicks S, Shephard MH, Colclough K, Hattersley AT, Ellard S, et al. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504–8.
Owen KR, Thanabalasingham G, James TJ, Karpe F, Farmer AJ, McCarthy MI, et al. Assessment of high-sensitivity C-reactive protein levels as diagnostic discriminator of maturity-onset diabetes of the young due to HNF1A mutations. Diabetes Care. 2010;33:1919–24.
Østoft SH. Incretin hormones and maturity onset diabetes of the young-pathophysiological implications and anti-diabetic treatment potential. Dan Med J. 2015;62:B4860.
Østoff SH, Bagger JI, Hansen T, Pedersen O, Holst JJ, Knop FK, et al. Incretin effect and glucagon responses to oral and intravenous glucose in patients with maturity-onset diabetes of the young-type 2 and type 3. Diabetes. 2014;63:2838–44.
Juszczak A, Owen KR. Identifying subtypes of monogenic diabetes. Diabetes Manag. 2014;4:49–61.
Demol S, Lebenthal Y, Bar-Meisels M, Phillip M, Gat-Yablonski G, Gozlan Y. A family with a novel termination mutation in hepatic nuclear factor 1α in maturity-onset diabetes of the young type 3 which is unresponsive to sulphonylurea therapy. Horm Res Paediatr. 2014;81:280–4.
Harries LW, Hattersley AT, Ellard S, Messenger RNA. Transcripts of the hepatocyte nuclear factor-1alpha gene containing premature termination codons are subject to nonsense-mediated decay. Diabetes. 2004;53:500–4.
Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER, et al. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med. 2010;27:157–61.
Lachanse CH. Practical aspects of monogenic diabetes: a clinical point of view. Can J Diabetes. 2016;40:368–75.
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362:1275–81.
Martagon AJ, Bello-Chavolla OY, Arellano-Campos O, Alemda-Valdes P, Walford GA, Cruz-Bautista I, et al. Mexican carriers of the HNF1A p.E508K variant do not experience an enhanced response to sulphonylureas. Diabetes Care. 2018;41:1726–31.
Shephard MH, Shields BM, Hudson M, Pearson ER, Hyde C, Ellard S, et al. A UK nationwide prospective study of treatment change in MODY: genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia. 2018;61:2520–7.
Wobser H, Bonner C, Nolan JJ, Byrne MM, Prehn JH. Downregulation of protein kinase B/Akt-1 mediates INS-1 insulinoma cell apoptosis induced by dominant negative suppression of hepatocyte nuclear factor-1alpha function. Diabetologia. 2006;49:519–26.
Acknowledgements
We would like to thank the patients for giving their consent to take part in our study and members of the KTPH Diabetes Centre for their continuing support. The work was supported by NHG-KTPH Small Innovative Grant II/14001 and KTPH STAR17201.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tan, C.S.H., Ang, S.F. & Lim, S.C. Response to multiple glucose-lowering agents in a sib-pair with a novel HNF1α (MODY3) variant. Eur J Hum Genet 28, 518–520 (2020). https://doi.org/10.1038/s41431-019-0561-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0561-8
This article is cited by
-
HNF1A binds and regulates the expression of SLC51B to facilitate the uptake of estrone sulfate in human renal proximal tubule epithelial cells
Cell Death & Disease (2023)
-
Diagnosis of hepatic nuclear factor 1A monogenic diabetes mellitus (HNF1A-MODY) impacts antihyperglycemic treatment
Wiener klinische Wochenschrift (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}