
Abstract
Reactive distillation (RD) is a great process intensification concept taking advantage of the synergy created when combining (catalyzed) reaction and separation into a single unit, which allows the concurrent production and removal of products. This feat improves the productivity and selectivity, reduces the energy usage, eliminates the need for solvents, and leads to highly-efficient systems with improved sustainability metrics (e.g. less waste and emissions). This paper provides an overview of the key features of RD processes, with emphasis on novel catalytic/reactive distillation processes that can make a difference at large scale and pave the way for a more sustainable chemical process industry that is more profitable, safer and less polluting. These examples include the production of: acrylic and methacrylic monomers, unsaturated polyesters resins, di-alkyl ethers, fatty esters, as well as other short alkyl esters (e.g. by enzymatic reactive distillation). The main drivers for such new RD applications are: economical (large reduction of costs and energy use), environmental (lower CO2 emissions, no or reduced waste) and social (improved safety and health due to lower reactive content, reduced footprint and run away sensitivity). Hence RD technology strongly contributes to all three pillars of sustainability in the chemical process industry. Nonetheless, the potential of RD technology has not been fully tapped yet, and there is still undergoing research to improve it further by various means: e.g. ultrasound or microwave assisted RD, use of high-gravity fields (HiGee), internally heat integration, cyclic operation, or coupling RD with other operations such as membrane separations.
Similar content being viewed by others

1 Introduction
Although distillation is the oldest and most commonly used separation technique at industrial scale, there are still major developments to improve it further [1, 2]. Among them, reactive distillation (RD) is one of the greatest process intensification (PI) technique profiting from the synergy generated when combining catalyzed reactions and separation into a single unit. RD (also known as distillation with reaction) greatly improves the conversion and selectivity, reduces the energy usage, eliminates the need for solvents, and leads to eco-efficient sustainable production systems. When solid catalysts are used, RD is also referred to as catalytic distillation. RD is considered nowadays an established industrial unit operation, being the front-runner in the PI field [3, 4]. In general, RD is applicable to equilibrium limited reaction systems. While the catalyst helps increasing the reaction rate and shortening the time to reach the chemical equilibrium, it does not actually change the attainable equilibrium conversion. In RD, the equilibrium is shifted to full conversion by the continuous removal of products. Thus RD can use just the stoichiometric reactants ratio (neat operation) to pull the equilibrium to high conversions, in contrast to the usual practice of using an excess of reactant to push the equilibrium toward the desired products, with the extra cost of recovery and recycle of unreacted raw materials [2].
Figure 1 illustrates the dependence of the equilibrium conversion on the equilibrium constant (Keq) and the dramatic positive effect of increasing the reactants ratio in case of a quaternary system in which two reactants are converted to two products. Notably, in case of RD a similar effect is obtained by removing the products instead of using an excess of one of the reactants.

Equilibrium conversion vs equilibrium constant, at various reactants ratio
The literature covers very well various topics related to RD, such as: thermodynamics [5], feasibility and applicability [6, 7], conceptual design [8, 9], modeling and process simulations [10, 11], optimal design [12], design and control [13], process configurations [14], process scale-up [15], green engineering aspects [16], industrial perspectives and applications [17,18,19], applications in biorefineries [20], and the status and future directions of RD technology [21]. RD technology has been applied industrially in many processes with production capacities of 100–3000 ktpy, e.g. production of methyl acetate as well as fuel additives or oxygenates (MTBE, ETBE, TAME) [3]. However, further applications remained somewhat limited.
This paper aims to provide some insights into the key features of RD processes, along with a focused presentation of novel catalytic/reactive distillation (industrial) processes reported in the past decade, such as the production of acrylic and methacrylic monomers, polyesters, alkyl ethers (dimethyl ether, di-n-pentyl ether), fatty esters, and other short chain alkyl esters.
2 Reactive Distillation Technology
Along the way from taking any brilliant idea into practical process implementation, there are several essential R&D steps that combine computation and process simulations with lab- and pilot-scale experimental work—as conveniently illustrated in Fig. 2. These steps have the role to provide (preferably in the shortest time) valuable insights that increase the knowledge about the process and reduce the uncertainty to low levels that allow a confident design and construction of the plant. This is also the case for reactive distillation processes, where the simulations are used to guide the conceptual process design and the experimental work, while the experiments decide upon the validity of the simulated model. The validated model can be used then for process scale-up, sensitivity analysis studies, process control and optimization.

Main research and development steps to go from an idea to process implementation
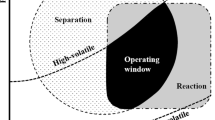
When deciding whether RD is the right choice or not, one must keep in mind both the benefits and limitations of reactive distillation—as summarized in Table 1 [4]. Since both operations take place simultaneously in the same unit, there must be a good match between the temperatures and pressures required for reaction and separation. The operating window is typically limited by the thermodynamic properties (e.g. boiling points) of the components involved. But also the windows in which the (catalytic) reaction delivers acceptable yields and selectivity has usually a limited overlap with the separation window. This overlap may be reduced further by the feasibility window concerning the equipment design. This leads usually to a very restricted area in which reactive separation is feasible [4]. The combination of R&D is not possible if there is no major overlapping of the operating conditions of reaction and separation (e.g. a high pressure reaction can not be combined with a vacuum distillation).
Figure 3 shows the most common RD configuration alternatives: conventional RD column, heterogeneous azeotropic RD column, and RD column combined with pre-reactor and/or side reactors [1]. Note that distillation columns are a suitable solution only for reactions that are sufficiently fast in order to reach high conversions within the residence time range of such columns. Previous research [22] distinguished two main operating conditions: (1) distillation controlled range (conversion is influenced by the concentration of the components to be separated), and (2) kinetics controlled range (conversion is influenced mainly by the residence time and the reaction constant). The industrial design of RD should aim at operating conditions within these two ranges, just the sufficient residence time and only the necessary expenditure for the distillation. Figure 4 presents the resulting principles for the choice of equipment for homo- and heterogeneous catalysis [1, 22]. In case of heterogeneous catalysis additional, separate reaction volumes are necessary to retain the catalyst inside the column. These volumes can be arranged either within the equipment or in a side reactor unit coupled by recycle streams. As catalysis plays an important role in RD processes, several situations are possible in practice:

Reactive distillation process configurations

Equipment choice for reactive distillation processes in case of homogeneous catalysis (left) and heterogeneous catalysis (right)
-
Non catalyzed reaction: the reaction takes place almost anywhere in the RD column (where reactants are present), and no catalyst separation or recovery is required.
-
Homogeneous catalyst: the reaction takes place almost anywhere in the RD column. The catalyst may be left in one of the product streams (if acceptable according to the specifications), or it can be neutralized and separated as salt waste stream (cheap acid/base catalysts), or it has to be recovered and recycled (expensive catalysts).
-
Heterogeneous catalyst: the reaction zone is well defined (i.e. where the solid catalyst is present), the catalyst does not leave the RD column hence no recovery is needed. This particular case is also called catalytic distillation.
Notably, the choice of internals is much more limited for RD columns as compared to classic distillation. Figure 5 presents some examples of catalytic packing [1, 10, 23]. The most important type of internals includes the following:

Various internals for catalytic distillation: ‘tea-bag’ configurations for trays, gutters (top), and structured catalyst-sandwiches for packed columns (bottom)
-
Catalyst bales, formed by wrapped wire sheets filled with catalyst. The steel mesh creates voids that allow V–L contact and vapor traffic. Bales are piled on top of each other to provide the required height to achieve the desired extent of reaction. This is used by Chemical Research & Licensing in their RD technology.
-
Catalytic Raschig rings (random packing) with surface coated catalyst. Alternatively, the solid catalyst itself can be shaped as packing.
-
Catalytic structured packing, with elements shaped as sandwiches manufactured from corrugated wire gauze sheets hosting catalyst bags (assembled as cylinders or rectangular boxes). The packing structure consists of alternating catalyst bags and open channel spaces. To ensure higher efficiency of combined reaction and diffusion the catalyst particle should have a diameter of about 0.8–1 mm. Commercial offers include KATAPAK-S® manufactured by Sulzer ChemTech, MultiPak® supplied by Julius Montz, and KATAMAX® from Koch-Glitsch.
3 Novel Reactive Distillation Processes
This section describes novel catalytic/reactive distillation processes of industrial importance, reported in the past decade or so, with major advantages and improved sustainability metrics.
3.1 Acrylic and Methacrylic Monomers
Acrylic and methacrylic monomers are essential chemicals largely used in the coatings industry. Coatings are primarily waterborne emulsions, which combine methacrylates as a hard momoner and a softer acrylate monomer to improve flexibility. A strong industrial drive aims to move away from the existing batch processes to multi-product continuous plants that allow increased capacities, constant product quality, less waste, and lower energy usage. As shown in this section, RD technology can play an important role in this transition.
The (meth)acrylic monomers are usually obtained by esterification reactions that make use of acid catalysts, either homogeneous (e.g. H2SO4, pTSA, MSA, or triflic acid) or heterogeneous catalysts (e.g. Amberlyst, Nafion, sulfated Zr/Ti, metal oxides):
A key challenge that must be solved when using RD is to avoid polymerization in the column (e.g. by working at low pressure and temperature). In addition, one or more polymerization inhibitors are also included to prevent unintended polymerization of the acrylic acid (AA), methacrylic acid (MA) or their corresponding esters (monomers). Suitable inhibitors include: phenothiazine, hydroquinone, hydroquinone methyl ether, or other hydroquinone derivatives [24]. The inhibitors are added in the usual amounts (ranging 200–2000 ppm), and can be included in one of the feed streams or it can be added separately at the top of the RD column. Many known polymerization inhibitors are more efficient in the presence of oxygen, hence it is preferred that the RD process is carried out in the presence of air.
Figure 6(left) illustrates a reactive distillation column for the synthesis of short chain acrylic monomers. Such an experimental setup has been successfully proven at pilot-scale, for the production of n-butyl acrylate [25]. Sulzer Katapak SP-11 was employed, including Amberlyst 131 as solid acid catalyst, while phenothiazine was used as inhibitor. A non-equilibrium model was used and validated later based on the experimental data. The packing characteristics were adapted to an industrial scale column to allow a reliable process optimization. Coupling the RD with a decanter was shown to be the most economic process. This is mainly because using RD to overcome the equilibrium limitations, the recycle of reactants is minimized and the difficult separation of acrylic acid (AA) and n-butyl acrylate (n-BA) is avoided.

Reactive distillation column for synthesis of short chain acrylic monomers (left) and long chain (meth)acrylic monomers (right)
A later study focused on the design, control and economic evaluation of a RD-based process [26]. As n-butanol is not completely converted in a single pass, a decanter-flash system was used to recover and recycle the alcohol, as well as obtain a high purity water stream (by-product). The top stage pressure is 13 kPa, while the reboiler pressure is 27.5 kPa hence the maximum temperature in the column is in the reboiler (104.5 °C) and below the safety limits to avoid the polymerization. Sulzer Katapak SP-12 was employed, which consists of catalyst granules immobilized in wire gauze layers, combined with layers of MellapakPlus structured packing. The number of catalyst to MellapakPlus layers is 1:2, thus the amount of catalyst is about 30% volume. The separation efficiency is about 2.5 theoretical stages per meter (HETP = 0.4 m). As the packing (column) diameter is 1.22 m, and the catalyst density is 740 kg/m3, this leads to a maximum allowed amount of catalyst of 104 kg per tray. About 2000 kg of catalyst is needed in total in the RD column, for an annual capacity of 20.6 ktpy n-BA. The economic analysis shows that a cost of 762 €/t of acrylate can be achieved, which is 43.5% lower than those of classic batch processes (1350 €/t).
Figure 6(right) illustrates a more generic RD configuration for the synthesis of long chain (meth)acrylic monomers, as described in a recent patent [24]. This RD process allows high throughput and short residence times of the polymerizable monomers (to avoid undesirable waste of high boiling residue), reduced capital expenditure and energy usage, and lower operating costs. Just as in the previous case, a catalyst (pTSA) and an inhibitor are used here. Since C5+ alcohols have a higher boiling point that of the (meth)acrylic acid, the alcohol is fed on top of the reactive zone in the RD column. Typical alcohols used include: 2-ethylhexyl alcohol, cyclohexanol, isoamyl alcohol, and abietyl alcohol. The pressure limit can be well correlated with the boiling temperature of the alcohol at ambient pressure [24]:
where Tb(ROH) is the boiling point in °C of the mono-alcohol at atmospheric pressure. For example, in the synthesis of 2-ethyl hexyl methacrylate (EHMA) a pressure of 0.15 bar is maintained in operation, leading to a maximum temperature of 145 °C in the reboiler. The top product is water (> 99.95 wt%) while the bottom product is the methacrylate (> 99.3 wt%) with minor amounts of alcohol, inhibitor and catalyst (which are usually allowed in the product). A higher purity product can be obtained if the monomer is removed as vapor product from the last stage of the RD column, or alternatively from the vapor return line from the reboiler, but this require an additional heat exchanger to condense the vapor stream of monomers.
3.2 Unsaturated Polyesters
Unsaturated polyester resins are important chemicals, used in many applications such as sheet or bulk moulding compound, printer toners, fiberglass reinforced plastics, or cured-in-place pipe applications. These unsaturated polyesters are formed by the reaction of diols/polyols (e.g. ethylene glycol) with saturated or unsaturated di-acids (e.g. maleic or phthalic acid). The reaction scheme is rather complex and includes several types of reactions: ring opening, poly-esterification, isomerisation and saturation [27].
Several known catalyst for polyester synthesis can be used, such as titanium-, antimony-, and tin-based catalysts, in their various forms such as alkoxylates or as carboxylates [28]. The common practice is that the small amount of catalyst used is practically left in the polyester product, hence the separation and recovery of catalyst is best avoided.
The problem is that at industrial scale, polyesters are still produced using batch reactors and distillation columns, with a total production time of about 12 h or even longer. Such processes are plagued by batch-to-batch inconsistencies and high capital and operating costs.
In a series of papers by Shah et al. [27, 29, 30] a novel RD process was proposed for the synthesis of polyesters. This is an enhanced process alternative able to reduce the production time and to achieve a better quality of the product at lower costs. Also, complete conversion can be achieved in a short time, as the water by-product is continuously removed from the system. Reliable kinetic and thermodynamic models were developed, and subsequently used in rigorous simulations testing various process alternatives—as shown in Fig. 7 [27]. Moreover, various internals configurations (packing and trays) were evaluated and the experimental validation of the process was successfully carried out at pilot scale using two configurations: a single RD column, as well as a RD column coupled with a pre-reactor. The product specification of the polyester produced in the continuous RD column was compared with the polyester product manufactured at industrial scale. Remarkable, it was found that the product specifications of the polyester produced in the RD column is well comparable to that of the industrial product manufactured in a classic batch process.

Reactive distillation processes for synthesis of unsaturated polyesters: reactive stripping column (left) and reactive distillation with pre-reactor (right)
The reported results show that the total production time in a RD system is only 1.8–2 h, which is actually 6–8 times lower as compared to the batch process (12 h)—as illustrated in Fig. 8 [27, 29, 30]. In case of using a RD column coupled with a pre-reactor the improvements are clearly especially promising: 90% lower reaction volume, 93% shorter production time, and 66% energy savings (and lower CO2 emissions) as compared to the traditional batch process.

Key performance indicators for various RD configurations relative to a conventional batch reactor
3.3 Fatty Esters
The production of fatty esters (biodiesel) by reactive separations received significant attention during the past decades [31]. Being a mixture of fatty acid methyl esters (FAME), biodiesel is a renewable fuel used complementary to petro-diesel fuel. Its main synthesis routes are by either trans-esterification of tri-alkyl glycerides (TAG) or esterification of free fatty acids (FFA). Both routes use catalysts (homogeneous or solid acid/base catalysts) that can be applied in a number of industrial processes for biodiesel manufacturing: e.g. batch, continuous, supercritical, enzymatic, multi-step, or reactive separations [31].
The trans-esterification reaction is typically catalyzed by bases, while the esterification is catalyzed by acids. Alternative acid/base catalysts could be used but at prohibitive reaction rates. For example, Fig. 9 illustrates the reaction profiles (conversion vs time) for the synthesis of fatty esters using various solid acid catalysts, such as: tungsto-phosphoric acid, ion exchange resins (Amberlyst and Nafion), mixed metal oxides (sulfated zirconia, sulfated titania, or tin oxide), and sulfated carbon fibers among others [32]. Sulfuric acid is shown only as a benchmark for comparison against homogeneous catalysts.

Reaction profiles (conversion vs time) for the synthesis of fatty esters (2-ethyl hexyl dodecanoate) using various (solid) acid catalysts
The reaction time can be shortened by increasing the liquid–liquid interfacial area by various PI techniques (e.g. static mixers, micro-channels reactors, microwaves/ultrasound assisted reactors, rotating/spinning tube reactors and centrifugal contactors) or by integrating the reaction and separations steps: e.g. reactive distillation/absorption/extraction, reactive membrane separators, and centrifugal contactors [33]. After the FAME synthesis stage, there are several costly downstream processing steps required for catalyst neutralization and salt removal, excess alcohol recovery and recycle, as well as glycerol and biodiesel purification. Among the process intensification alternatives investigated, RD is the most promising option. Pilot scale experiments have been reported [34] for a RD process for fatty esters synthesis using a strong acidic ion-exchange resin (Amberlyst 15) hosted in catalytic packing (Katapak-S/-SP). Another process reported for fatty acids esterification [31] makes use of sulfated zirconia as solid catalyst in a heat integrated RD process (Fig. 10, top). A simpler version similar to a reactive absorption (RA) uses a reactive column without reboiler and condenser [35]. Another possible alternative consists of a dual RD process using two alcohol feeds: a light and a heavy alcohol [36]. Table 2 provides a comparison of the process parameters for RD and RA processes, without and with heat integration [35].

Heat-integrated reactive distillation process (top) and reactive dividing-wall column (bottom) for synthesis of fatty acid methyl esters
Note that when the FFA content of the feed is variable, a slight excess of methanol can be used (up to 15%) but this requires a reactive dividing-wall column—as illustrated in Fig. 10(bottom) [31]. In this case the excess methanol is recovered as top distillate product and recycled, while water by-product is removed as side-stream and treated similarly as in the RD process.
Table 3 compares the energy requirements for a classic two-step process based on pre-treatment of FFAs and trans-esterification of glycerides vs previously reported reactive separation processes based on esterification of waste oils with high FFA content [33]. These values are worth noting, especially considering the on-going quest on increasing the eco-efficiency of biodiesel production. The specific energy use in reactive separation processes is drastically lower than e.g. the FAME purification step alone in the conventional process. On top of the energy savings (and lower CO2 emissions) of about 90%, the reactive separation processes benefit from lower investment costs and reduced plant footprint due to less equipment being used.
3.4 Di-alkyl Ethers
Dimethyl ether (DME) is of much interest due to its use as clean fuel for diesel engines or in combustion cells, as a precursor to organic compounds, or green aerosol propellant. Currently, DME is produced by conversion of feedstock such as natural gas, coal, oil residues and bio-mass into syngas (CO and H2), followed by a two-step process: methanol synthesis, and then dehydration. Syngas is converted to methanol over a copper-based catalyst (Cu/Zn, Cu/Zn/Al, Cu/Zn/Co), which is then dehydrated over a solid catalyst to produce DME. The dehydration step takes place at 250-400 °C and 10–20 bars. As solid catalyst, γ-alumina is preferred due to its thermal stability, mechanical resistance, high surface area and catalytic properties [37]. The current process involves a gas-phase reactor (70–80% conversion of methanol), followed by a direct sequence of distillation columns that deliver high-purity DME (over 99.99 wt%). Figure 11 shows the ternary diagram of the DME process, including the mass balance lines for reaction (Rx), first and second distillation column (DC1 and DC2). The equilibrium limited dehydration reaction is carried out in a fixed-bed catalytic reactor:

Ternary diagram illustrating the main steps in the DME process (top), and reactive dividing-wall column for DME synthesis (bottom)
The outlet of the reactor (consisting of DME, water and unreacted methanol) is cooled and then distilled in the first tower to yield DME, while methanol is separated from water in a second column and recycled back to the reactor. A major problem of this process is the large equipment cost and high energy requirements (in spite of using various heat integration options).
Intensified DME technologies (e.g. coupled and dual type reactors, micro-reactors, membrane reactors, and spherical reactors) are well described in the review paper of Azizi et al. [38]. Among them, RD is the most promising. An improved alternative combining all functions into one unit is based on a reactive dividing-wall column (R-DWC), as shown in Fig. 11 [39]. The reaction takes place in liquid phase and the catalyst is an acidic ion-exchange resin. Methanol is fed on top of the reactive zone where the solid catalyst is located, while DME is produced as top distillate, water as bottom product, and the remaining methanol (due to a conversion of 50%) as side stream product that is recycled. The R-DWC unit has 35 stages, with a reactive zone (located on stages 8–31 on the feed side), a common stripping (stages 32–35) and rectifying section (stages 1–7). Methanol is fed on stage 8 (at the top of the reactive zone). The feed side of the DWC acts as the RD zone where the solid acid catalyst is present. High purity (99.99 wt%) DME is obtained as distillate, while high-purity water is removed as bottom product. The results reported show that the process using an R-DWC has better performances as compared to the classic process or RD combined with distillation: energy savings (and lower CO2 emissions) of up to 60%, and about 30% lower capital investment costs.
A classic RD configuration can be used when the dehydration of methanol in liquid phase is catalyzed by more active and thermally stable resins, such as Amberlyst-35 which ensures high activity and selectivity at temperatures up to 150 °C. Figure 12(top) illustrates such a RD process, including the mass balance and the key operating parameters [40]. The RD column for DME synthesis has the following key features:

Reactive distillation process for DME production (top), novel catalytic cyclic distillation column and specific internals—Maleta trays with catalyst (bottom)
-
When the distillate rate is set to half of the feed (based on reaction stoichiometry), methanol (as mid-boiling component) is equally distributed between the distillate and the product streams. Achieving 99.99 wt% distillate purity (DME) by manipulating the reflux ratio also ensures that the purity of the bottoms stream (water) exceeds 99.95 wt% spec.
-
OpEx represents more than 75% of the total annual cost (TAC). The main contributions to CapEx are the condenser, reboiler, column shell and structured packing, while the catalyst cost is less than 3%. Increasing the amount of catalyst per stage invariably results in a lower TAC, as the decrease of other costs clearly outweighs the cost of catalyst. However, the amount of catalyst is limited to 20% of the available volume [41].
-
Increasing the operating pressure leads to higher temperature along the RD column, hence faster reaction rates and lower OpEx (due to the reduced effort needed for separation). The optimal pressure leads then to the highest allowable temperature in the reactive section.
Table 4 provides the optimal design parameters of the RD column for DME production [40]. The operating pressure (11.36 bar) leads to temperatures in the range of 130–150 °C on the reactive stages. The amount of catalyst (193.2 kg/stage) occupies 20% of the stage volume, being in line with the recommendations for KATAPAK packing [41]. The reflux ratio (6.174 kmol/kmol) ensures the required purity. Along the reactive stages, the liquid-phase methanol mole fraction exceeds 0.8, leading to a large reaction rate. For an RD process of 100 ktpy capacity, the CapEx is only 2395 k$, the OpEx is 2604 k$/year, and the energy requirement is 2.43 MJ/kg (672.5 kWh/t) DME. It is worth mentioning that the company Oberon Fuels (http://www.oberonfuels.com) has launched the first commercial catalytic distillation column producing 9700 ton/year of fuel grade DME (98.5 wt%), in Brawley (California, US).
Figure 12(bottom) illustrates a novel PI approach integrating chemical reactions with cyclic distillation, known as catalytic cyclic distillation (CCD) which uses specific internals that can ensure an increased residence time [42]. The periodic operation mode leads to key benefits, such as: increased column throughput (without flooding), lower energy use (at the same number of stages) and much higher separation performance (over 200% equivalent Murphree tray efficiency). The synthesis of DME by catalytic cyclic distillation is a great PI alternative of industrial interest, considering the potential benefits [43].
Di-n-pentyl ether (DNPE) is another good candidate for diesel fuel formulations due to its blending cetane number, good cold flow properties and efficiency in reducing diesel exhaust emissions, particulates and smokes. Novel processes are needed to drive the production costs down and to increase the efficiency at industrial scale. The dehydration of 1-pentanol to yield DNPE is catalyzed by thermally stable resins, such as Amberlyst 70 which has high activity and selectivity at temperatures up to 190 °C.
Figure 13 shows the RD process developed for the synthesis of di-n-pentyl ether (DNPE), and the operating windows for reaction and distillation and the limiting overlapping area for RD [44]. The proposed RD process was designed and optimized in terms of total annual costs (523 k$/year for a plant capacity of 26.5 ktpy DNPE). The specific energy use is only 0.92 MJ/kg (256 kW h/ton) of DNPE product. The good controllability of the process was confirmed by dynamic simulations performed in Aspen Dynamics. Compared with other PI alternatives (membrane reactor) reported earlier, the new DNPE process based on RD is a better process candidate, requiring simpler units and leading to smaller investment costs.

Reactive distillation process for the synthesis of di-n-pentyl ether (top)
3.5 Esters Synthesis by Enzymatic RD
The first of its kind continuous RD column using immobilized enzymes as a solid catalyst has been reported recently—as shown in Fig. 14 [45]. Among the enzymes used industrially, the B-component lipase from the yeast Candida antarctica (CalB) is a particularly efficient and robust lipase catalyzing a large diversity of organic reactions, including transesterification. The enzymatic catalysts are immobilized either as enzyme beads sandwiched in structured packing, or as enzyme containing silica-gel based coating applied onto structured packing (see Fig. 14). Both catalytic packings were used in a pilot-scale RD column for experimental validation of the process. The chemical system used for this study is the production of butyl butyrate (BuBu)—a volatile ester used in the flavor industry to create sweet fruity flavors similar to that of pineapple. This trans-esterification reaction of ethyl butyrate (EtBu) with n-butanol (BuOH) forming ethanol (EtOH) uses lipase as catalyst:

Enzymatic reactive distillation (ERD) process: a scheme of ERD column with the catalyst implementations in the enzymatic reactive section, b coated packing, c Katapak-SP® packing, d enzyme beads to be filled into Katapak-SP®
Figure 15 illustrates the temperature and pressure range where the ERD process operates [45]. The temperature and pressure are linked by the VLE of the boiling system. The upper operational limits are at 343 K (as higher temperatures would lead to instability of the enzyme catalyst) and a maximum pressure of 70 kPa. Similarly, the lower limits are 323 K (lower temperatures meaning reduced reaction rates and low conversion) and about 10 kPa (lower pressures are technically possible, but require much larger equipment size). The operating window is similar to the ones reported for chemical systems catalyzed by an ion-exchange resin catalyst [44]. The catalyzed system is not mass transfer controlled hence the rate-determining step is the kinetic step. A lumped kinetic is used that comprises mass transfer and kinetics. For the enzyme coated packing a reduced ordered bi bi kinetic approach was employed, while for the enzyme beads a simplified steady-state approximation of the Ping-Pong Bi Bi-mechanism was used [45].

Operating window of the enzymatic reactive distillation for the trans-esterification of ethyl butyrate
Table 5 lists the operating and design variables for the nominal operating point of the ERD process, while Fig. 16 shows the simulated and experimental pilot-scale column profiles (in terms of molar fractions of the components and vapor temperature) for an ERD experiment using coated packing [45]. During the operation the enzyme was stable, without any activity loss. The mass of coating comprising enzyme in the experiment was estimated at 65 g/m. The modeling results are very accurate except for slight mismatches in the amounts of EtBu and BuOH in the stripping section (only max. 0.054 mol/mol deviation for EtBu, 0.028 mol/mol deviation for BuBu and 0.004 mol/mol for EtOH and 0.032 mol/mol for BuOH). The sensitivity analysis showed that high conversion rates can be reached in various ways by changing the operating and design parameters, e.g. longer reactive section or more enzyme used per column volume. Due to the excess of EtBu over BuOH the conversion of BuOH is always higher, whereas an excess of BuOH is not favorable. Notably, for both coated packing and enzyme beads, the model is capable of reproducing the experiments with great accuracy and thus it was considered validated. Moreover, by modifying the property model and the kinetics, the ERD model can be adapted to other chemical systems and can be used to analyze the ERD process and perform design studies within different project phases. This new ERD opens new avenues for research and applications using immobilized enzymes (that do not require recovery and recycling) as green catalysts in RD processes.

Simulated and experimental column profiles with molar fractions of the components (left) and vapor temperature (right) for the ERD with coated packing
4 Conclusions
Despite the relatively complex design, control and equipment, RD remains one of the best process intensification technologies that fulfills all the principles of green engineering: design for separation, maximize efficiency, use renewable vs depleting sources, prevent instead of treat, meet need and minimize excess, integrate local material and energy flows, output-pulled vs input-pushed, and design for a commercial afterlife. RD offers key unique features such as: low number of processing units, enhanced overall rates, overcome unfavorable equilibrium, avoid difficult separations, improved selectivity, reduced energy use, less CO2 emissions, low or no solvent use.
Considering the outstanding progress in equipment development, modeling and simulation, design and control strategies, real time optimization, and the rapid pace of exploring new applications, RD remains an important PI technique capable to bring green chemistry and sustainable engineering into the chemical process industry. The examples presented in this study clearly illustrate that catalyzed RD processes can drastically improve the production of important chemicals such as: acrylic and methacrylic monomers, unsaturated polyesters resins, di-alkyl ethers, and (fatty) alkyl esters.
References
Kiss AA (2013) Advanced distillation technologies—design, control and applications. Wiley, Chichester
Kiss AA (2014) Distillation technology - Still young and full of breakthrough opportunities. J Chem Technol Biot 89:479–498
Harmsen GJ (2007) Reactive distillation: The front-runner of industrial process intensification: A full review of commercial applications, research, scale-up, design and operation. Chem Eng Process 46:774–780
Kiss AA (2017) Process intensification for reactive distillation. In: Rong B-G (ed) Process synthesis and process intensification: Methodological approaches. de Gruyter, Berlin
Frey T, Stichlmair J (1999) Thermodynamic fundamentals of reactive distillation. Chem Eng Technol 22:11–18
Shah M, Kiss AA, Zondervan E, de Haan AB (2012) A systematic framework for the feasibility and technical evaluation of reactive distillation processes. Chem Eng Process 60:55–64
Muthia R, Reijneveld AGT, van der Ham AGJ, Bargeman G, ten Kate AJB, Kersten SRA, Kiss AA (2018) Novel method for mapping the applicability of reactive distillation. Chem Eng Process 128:263–275
Doherty MF, Malone MF (2001) Conceptual design of distillation systems. McGraw-Hill, New York
Almeida-Rivera CP, Swinkels PLJ, Grievink J (2004) Designing reactive distillation processes: present and future. Comput Chem Eng 28:1997–2020
Taylor R, Krishna R (2000) Modelling reactive distillation. Chem Eng Sci 55:5183–5229
Noeres C, Kenig EY, Gorak A (2003) Modelling of reactive separation processes: reactive absorption and reactive distillation. Chem Eng Process 42:157–178
Segovia-Hernández JG, Hernández S, Bonilla Petriciolet A (2015) Reactive distillation: a review of optimal design using deterministic and stochastic techniques. Chem Eng Process 97:134–143
Luyben WL, Yu CC (2008) Reactive distillation design and control. Wiley, Hoboken
Baur R, Krishna R (2004) Distillation column with reactive pump arounds: an alternative to reactive distillation. Chem Eng Process 43:435–445
Hoffman A, Noeres C, Gorak A (2004) Scale-up of reactive distillation columns with catalytic packings. Chem Eng Process 43:383–395
Malone MF, Huss RS, Doherty MF (2003) Green chemical engineering aspects of reactive distillation. Environ Sci Technol 37:5325–5329
Tuchlenski A, Beckmann A, Reusch D, Dussel R, Weidlich U, Janowsky R (2001) Reactive distillation - Industrial applications, process design & scale-up. Chem Eng Sci 56:387–394
Stankiewicz A (2003) Reactive separations for process intensification: an industrial perspective. Chem Eng Process 42:137–144
Hiwale RS, Bhate NV, Mahajan YS, Mahajani SM (2004) Industrial applications of reactive distillation: Recent trends. Int J Chem React Eng 2:R1
Kiss AA, Lange JP, Schuur B, Brilman DWF, van der Ham AGJ, Kersten SRA (2016) Separation technology - Making a difference in biorefineries. Biomass Bioenergy 95:296–309
Sundmacher K, Kienle A (2003) Reactive distillation: Status and future directions. Wiley, Weinheim
Schoenmakers HG, Bessling B (2003) Reactive and catalytic distillation from an industrial perspective. Chem Eng Process 42:145–155
Krishna R (2002) Reactive separations: more ways to skin a cat. Chem Eng Sci 57:1491–1504
Kiss AA, ten Kate AJB, Conte E (2015) Continuous process for the esterification of an alpha,beta-unsaturated carboxylic acid and an alcohol, Patent No. WO/2015/018773, Publication date: 12.02.2015
Niesbach A, Fuhrmeister R, Keller T, Lutze P, Górak A (2012) Esterification of acrylic acid and n-butanol in a pilot-scale reactive distillation column—experimental investigation, model validation, and process analysis. Ind Eng Chem Res 51:16444–16456
Moraru MD, Bildea CS (2017) Process for n-butyl acrylate production using reactive distillation: design, control and economic evaluation. Chem Eng Res Des 125:130–145
Shah M, Kiss AA, Zondervan E, de Haan AB (2013) Evaluation of configuration alternatives for multi-product polyester synthesis by reactive distillation. Comput Chem Eng 52:193–203
de Haan AB, Kiss AA, Oudshoorn ML, Shah MR (2013) Manufacturing polyesters by reactive distillation, Patent No. WO/2013/048247, Publication date: 4.04.2013
Shah M, Kiss AA, Zondervan E, de Haan AB (2012) Influence of liquid back mixing on a kinetically controlled reactive distillation process. Chem Eng Sci 68:184–191
Shah M, Kiss AA, Zondervan E, de Haan AB (2012) Pilot-scale experimental validation of unsaturated polyesters synthesis by reactive distillation. Chem Eng J 213:175–185
Kiss AA (2014) Process intensification technologies for biodiesel production—reactive separation processes. Springer, Heidelberg
Kiss AA, Rothenberg G, Dimian AC, Omota F (2006) The heterogeneous advantage: biodiesel by catalytic reactive distillation. Top Catal 40:141–150
Kiss AA, Bildea CS (2012) A review on biodiesel production by integrated reactive separation technologies. J Chem Technol Biotechnol 87:861–879
Steinigeweg S, Gmehling J (2003) Esterification of a fatty acid by reactive distillation. Ind Eng Chem Res 42:3612–3619
Kiss AA, Bildea CS (2011) Integrated reactive absorption process for synthesis of fatty esters. Bioresour Technol 102:490–498
Dimian AC, Bildea CS, Omota F, Kiss AA (2009) Innovative process for fatty acid esters by dual reactive distillation. Comput Chem Eng 33:743–750
Muller M, Hubsch U (2005) Dimethyl ether. In: Ullmann’s encyclopedia industrial chemistry, 7th edn. Wiley, Weinheim
Azizi Z, Rezaeimanesh M, Tohidian T, Rahimpour MR (2014) Dimethyl ether: a review of technologies and production challenges. Chem Eng Process 82:150–172
Kiss AA, Suszwalak DJPC (2012) Innovative dimethyl ether synthesis in a reactive dividing-wall column. Comput Chem Eng 38:74–81
Bildea CS, Gyorgy R, Brunchi CC, Kiss AA (2017) Optimal design of intensified processes for DME synthesis. Comput Chem Eng 105:142–151
Goetze L, Bailer O, Moritz C, von Scala C (2001) Reactive distillation with Katapak. Catal Today 69:201–208
Patrut C, Bildea CS, Kiss AA (2014) Catalytic cyclic distillation—a novel process intensification approach in reactive separations. Chem Eng Process 81:1–12
Kiss AA, Bildea CS, Patrut C (2015) Process and installation for the production of dialkyl ethers. Patent No. WO/2015/113914, Publication date: 06.08.2015
Bildea CS, Gyorgy R, Sánchez-Ramírez E, Quiroz-Ramírez JJ, Segovia-Hernandez JG, Kiss AA (2015) Optimal design and plantwide control of novel processes for di-n-pentyl ether production. J Chem Technol Biotechnol 90:992–1001
Wierschem M, Schlimper S, Heils R, Smirnova I, Kiss AA, Skiborowski M, Lutze P (2017) Pilot-scale validation of enzymatic reactive distillation for butyl butyrate production. Chem Eng J 312:106–117
Acknowledgements
The author gratefully acknowledges the Royal Society Wolfson Research Merit Award. The author also thanks all the participants of the Rideal Conference 2018 (UK) for the useful discussions, and the reviewers for their insightful comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kiss, A.A. Novel Catalytic Reactive Distillation Processes for a Sustainable Chemical Industry. Top Catal 62, 1132–1148 (2019). https://doi.org/10.1007/s11244-018-1052-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-018-1052-9