
Abstract
The most commonly occurring sarcoma of the soft tissue is gastrointestinal stromal tumor (GIST). Treatment and prevention of the disease necessitate an understanding of the molecular mechanisms involved. However, the role of BRD4 in the progression of GIST is still unclear. While it is known there are abundant infiltrating tumor-associated macrophages (TAMs) in the tumor microenvironment, the exact role of these cells has yet to be studied. This work showed an upregulation of BRD4 in GIST that was associated with GIST prognosis. Through gain and loss of function studies, it was found that BRD4 promotes GIST growth and angiogenesis in vitro and in vivo. Mechanistically, BRD4 enhances CCL2 expression by activating the NF-κB signaling pathway. Furthermore, this CCL2 upregulation causes recruitment of macrophages into the tumor leading to tumor growth. A likely mechanism for interactions in the GIST microenvironment has been outlined by this work to show the role and potential use of BRD4 as a treatment target in GIST.
Similar content being viewed by others


Introduction
Among sarcomas of soft tissue, the most ubiquitous cancer is gastrointestinal stromal tumor (GIST) with gastrointestinal tract localization1,2. Previous research suggests the role of KIT and PDGFRA oncogenic mutations in interstitial cells of Cajal, which are mesenchymal pacemaker cells, in disease metastasis, proliferation, and tumorigenesis3,4. These mutations in GIST are targeted by molecular drugs, such as imatinib5,6. Despite the use of such specific drugs that alter the treatment scenario, resistance leads to recurrence in a substantial number of patients. Another challenge is a lack of effective therapy for GIST when the abovementioned genes are not mutated7,8,9. These shortcoming call for the analysis of the disease mechanisms to consider new therapeutic approaches.
There are various cells that are not malignant in the tumor microenvironment, such as the most aboundant tumor-associated macrophages (TAMs), which are ubiquitous hematopoietic cells with a migratory nature10,11,12. There are many clinical and epidemiological studies that have highlighted the poor cancer prognosis related to GIST and the number of TAMs13,14. The progression of cancer involves discrete signals of the microenvironment that influence different cells, such as macrophages15,16. The tumor microenvironment that influences macrophage orientation and differentiation is influenced by the profile of chemokines at the tumor site. An example is the upregulation and role of chemokine (C–C motif) ligand 2 (CCL2) in many cancers, including GIST17,18.
Bromodomain proteins include the mammalian bromodomain and extraterminal domain (BET) family inclusive of BRD2, 3, and 4. Research has focused on the functioning of BRD2 and 4 in elongation during transcription and regulation of the cell cycle while their possible involvement in inflammation is yet to be uncovered19,20. BRD4 has recently been shown to regulate RNA polymerase II elongation and the expression of genes involved in NF-κB-associated inflammation via activation of P-TEFb complex by CDK921,22.
In the current study, we identified BRD4, which was markedly upregulated and showed a significant association with pathology and survival in GIST patients. The expression of CCL2 was increased by BRD4 overexpression via the NF-κB signaling pathway, which led to TAM recruitment, ultimately contributing to tumor growth. These findings suggest that BRD4 is involved in GIST via TAMs.
Materials and methods
Cell culture
Jonathan Fletcher (DanaFarber Cancer Institute, Boston, MA) kindly provided the GIST882 cell line (from a patient with a K642E: KIT exon 13 homozygous missense mutation)23. Biowit Technologies (Shenzhen, China) was the source of the procured GIST-T1 cell line (from a patient with a V560Y579del: 57 nucleotide inframe mutation in KIT exon 11)24. Dulbecco’s Modified Eagle Medium (DMEM) plus 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin were used for culturing at 5% CO2 at 37 °C.
Details of patients and samples from the clinic
Patients (with written informed consent) who went through surgery at the Second Hospital of Jilin University in the period from February 2012 to March 2015 were the source of the 20 GIST samples used in this study; samples were fixed in formalin and embedded in paraffin. The samples were subjected to snap freezing in liquid nitrogen and stored at −80 °C untill assayed. HE staining was used by two pathologists to pathologically confirm the samples. The Committees for Ethical Review of Research Involving Human Subjects at the Second Hospital of Jilin University issued an approval for ethical consent.
Immunohistochemistry
Sectioning of the samples fixed in formalin and embedded in paraffin into 4-μm sections was performed. Individually, BRD4, CD31, and CD68 antibody staining was performed with selected slides that were then analyzed by two experienced pathologists. After normalization to the staining of the nucleus and the cytoplasm, the staining intensity scores were based on the following scoring system: 0 = no staining; 1 = weak staining; 2 = moderate staining; and 3 = strong staining. The combination of these scores with the positive cell percentage yielded the final immunohistochemistry (IHC) score.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed as previously described25,26. Briefly, TRIzol Reagent (Invitrogen) was used to extract total RNA, followed by reverse transcription with SuperScript II Reverse Transcriptase (Invitrogen) in accordance with the protocols of the manufacturer. An ABI 7300 real-time PCR system (Applied Biosystems) was used for the analysis with primers specific for BRD4 and GAPDH. The 2−ΔΔCT method was employed for assessing the changes in the expression of BRD4, with the control being GAPDH mRNA levels.
Western blotting
Western blotting was performed as previously described27,28. RIPA buffer (Pierce, Rockford, IL, USA) plus a protease inhibitor cocktail (Roche, Pleasanton, CA, USA) was utilized for lysing cells. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was utilized for resolving proteins, followed by the transfer of proteins to polyvinylidene difluoride membranes. Overnight incubation with primary antibodies was performed at 4 °C, followed by exposure to the corresponding secondary antibodies conjugated to horseradish peroxidase (HRP). Then, blots were incubated with the corresponding HRP-conjugated secondary antibodies. ECL regents (Thermo Scientific) were used to observe the resulting bands. The antibodies used in this study were as follows: BRD4 (ab128874), β-actin (ab8226), MMP9 (ab38898), LOX (ab174316), VEGFA (ab52917), p65 (ab16502), p-p65 (ab86299), Lamin A/C (ab108595), and CCL2 (ab9851; Abcam).
Assay for cell viability
Cells were seeded into 96-well plates, followed by an exchange of the media to one with CCK8 (100 μl for 10 μl of the latter) for 1 h after overnight culture. The optical density (OD) was measured using absorbance at 450 nm at various time points: 24, 48, and 72 h.
BrdU/PI assay
This protocol was in accordance with that used in a previous study29. To summarize, six-well plates were used for overnight culture of cells, and cells were subsequently incubated with 10 μg/ml BrdU for 20 min. This was followed by a double phosphate-buffered saline (PBS) wash and overnight fixation utilizing 70% ethanol at −20 °C. This was followed by a 45 min denaturation in 2 N HCL and FITC secondary antibody staining at room temperature for 1 h. Then, 40 g/ml RNase A and 200 g/ml PI were added for 30 min, followed by flow cytometry analysis.
Isolation and differentiation of cells
The Buffy coats from healthy voluteers were used to isolate human monocytes. Ficoll-Hypaque (Pharmacia Corporation) was utilized to isolate peripheral blood monocytes followed by density gradient centrifugation for 50 min at 400 g. Cells were seeded into 24-well plates at a density of 2 × 106 cells/ml in RPMI 1640 medium plus 10% heat-inactivated human AB serum, 50 U penicillin and streptomycin/ml, 2 mM L-glutamine, and 100 ng/ml human macrophage colony-stimulating factor, which was aimed at stimulating the differentiation of macrophages. Warm medium was used to wash away the cells that did not adhere gently and repeatedly after 6 days of culture. CD14+ macrophages constituted over 95% of the adherent cells using this approach. For the activation of M1 macrophages in vitro, 2 × 106 cells/l of the isolated cells (as described above) were treated for 1 day with 25 μg/ml LPS (lipopolysaccharide; Sigma) to the above-isolated cells, while for the activation of M2-polarized macrophages, 45 ng/ml recombinant human interleukin-4 (IL-4, R&D) was used. Flow cytometry was conducted to check the differentiated macrophages from monocytes. Conditioned media for the next round of in vitro experiments was obtained from PBS washing of cells followed by incubation in medium minus supplements for another 24 h.
Assay for endothelial cell tube formation
Endothelial cell tube formation was performed as previously described30. Incubation of the GIST cell lines in DMEM minus serum was done as indicated overnight. Ninety-six-well plates with Matrigel were seeded with 20,000 HUVECs per well along with the above-described conditioned medium. After incubation, imaging of tube formation by microscopy and quantification the branch number in the formed HUVEC tubes were performed.
Enzyme-linked immunosorbent assay
After treatment in DMEM minus serum overnight as indicated, conditioned medium was collected from the cells for VEGFA-enzyme-linked immunosorbent assay (ELISA) analysis in accordance with the prescribed protocols of the manufacturer. The absorbance at 470 nm was recorded for the OD values.
Assays for wound healing and invasion
The assay for wound healing involved six-well plates used for seeding GIST cells. A sterile 200-μm plastic pipette tip was employed to softly scratch the monolayer once cells were 95% confluent, followed by documentation of the wound with photographs. The Transwell invasion assay involved seeding of 4 × 104 cells suspended in medium lacking serum in the membranes of the upper chamber without/with Matrigel (BD Biosciences) coating. The lower chamber received 600 μl medium plus 10% FBS. After 24 h, the membrane bottom was subjected to 30-min fixation and 0.1% crystal violet staining. Following the use of a cotton swab to wipe the interior of the membrane, microscopy was utilized to quantify the cells.
Colony formation
Six-well plates were used to culture 500–1000 cells/well that has been exposed to the aforementioned chemicals and combinations. Renewal of the medium was performed every third day untill the 14th day, after which cells were fixed and stained with crystal violet. Cells that had been exposed to drugs were compared to control cells exposed to DMSO, and assays were completed in triplicate.
Studies using mouse xenograft tumors
The Animal Care and Use Committee of the Second Hospital of Jilin University issued approval for the following experiments. Balb/c nude mice (famale, 4 weeks in age) were randomly divided into two groups (n = 6). The animals received subcutaneous administration of GIST-882 cells that expressed the vector or BRD4 resuspended in PBS with Matrigel (1:1). The tumor volumes were computed with the following formula expression once a visible tumor mass had developed in the animals: (length × width2)/2. Animals were sacrificed 19 days after injection.
Statistical analysis
Prism 5.0 (GraphPad software) was employed for analyses. The mean±s.d. was utilized to calculate the data of a minimum of three independent experiments. An unpaired two-tailed Student’s t-test was used for analyzing two data sets, and one-way analysis of variance was used for more sets. Significance was set at p < 0.05.
Results
The expression of BRD4 is elevated in GIST
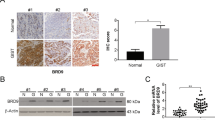
As described clinical samples were examined for their levels of BRD4 to examine the functioning of BRD4 in GIST. The samples were observed to have increased expression of BRD4 mRNA and protein compared with matched healthy tissue controls (Fig. 1a, b). The levels of BRD4 detected by IHC were also in line with this observation (Fig. 1c, d). The increased levels showed a correlation with poor overall survival (OS) and disease-free survival in the patients (P < 0.01; Fig. 1e). These data indicate the potential involvement of BRD4 in the progression of GIST.

a BRD4 expression was examined in normal and GIST tissues by Western blotting. b BRD4 mRNA expression was detected in normal and GIST tissues. c and d BRD4 protein expression in normal and GIST tissue was analyzed by IHC. Scale bar: 25 μm. e Kaplan–Meier curves for OS of GIST patients with low vs. high expression of BRD4; **P < 0.01.
Stimulation of the proliferation, migration, and invasion of GIST cells by BRD4
Once the increased expression was observed, the possible role of BRD4 in disease advancement was studied next. The role of BRD4 was assessed by the construction ofcontrol pcDNA-3.1(+) and pcDNA-3.1(+)-BRD4 vectors that were transfected into GIST-882 and GIST-T1 cell lines. Exposure to G418 caused expression of the vector or BRD4. The transfection of the pcDNA-3.1(+)-BRD4 vector led to increased BRD4 in the GIST-882 and GIST-T1 cell lines compared with the transfection of pcDNA-3.1(+) alone (Fig. 2a). The CCK8 assay showed that the cells expressing BRD4 showed higher growth patterns than those expressing control vector (Fig. 2b). In line with these observations, the BrdU/PI assay results displayed a higher BrdU-incorporated cell number in the lines expressing BRD4 than that seen in control vector-transfected cells (Fig. 2c), which is indicative of increased DNA synthesis due to BRD4. The colony formation assay showed higher numbers and sizes of colonies in the cells, expressing BRD4 than those in control vector-transfected cells (Fig. 2d). These observations are indicative of BRD4 stimulating the proliferation of GIST cells.

a GIST882 and GIST-T1 cells stably expressing vector or BRD4 were harvested. BRD4 protein expression was examined by western blotting. b The viability of GIST882 and GIST-T1 cells stably expressing vector or BRD4 was detected by CCK8 assay. c DNA synthesis was examined in cells stably expressing vector and cells stably expressing BRD4 using BrdU/PI assays. d GIST882 and GIST-T1 cells stably expressing vector or BRD4 were seeded on six-well plates for colony formation assays. e and f Both GIST882 and GIST-T1 cells stably expressing vector or BRD4 were plated in the upper chamber with/without Matrigel for 24 h. Then, cell migration and invasion were assessed by observing the cells that migrated to the underside of the transwell insert. Scale bar: 25 μm. g Wound healing assays were used to examine cell migration in GIST882 cells stably expressing vector or BRD4. h Wound healing assays were used to examine cell migration in GIST-T1 cells stably expressing vector or BRD4; *P < 0.05; **P < 0.01.
As GIST therapy is hindered by metastasis at a clinical level, we assessed the role of BRD4 in metastasis. BRD4 overexpression resulted in increased migration of cells, as observed in the transwell assays (Fig. 2e, f) as well as an increased ability to migrate, as observed in the wound-healing assays (Fig. 2g, h). Thus, these results suggest that BRD4 plays a key role in migration and invasion in GIST.
The emerging role of BRD4 in angiogenesis in GIST
The progression of tumors involves the vital formation of new capillaries from existing ones, which is referred to as angiogenesis. This process involves angiogenic factors, such as bFGF, PDGFB, and VEGF secreted by the cells of the tumor. This understanding led us to next assess the effect of BRD4 on angiogenesis in GIST. Higher levels of VEGFA, LOX, and MMP9 were displayed by the cell lines expressing BRD4 than by cells transfected with the control vector (Fig. 3a). In line with these observations, there was a lower level of VEGFA in the shBRD4 cells than in the shNC cells (Fig. 3c). ELISA corroborated the observations of higher VEGFA with forced BRD4 expression (Fig. 3b), while knocking out BRD4 showed the reverse observations (Fig. 3d).

a GIST882 and GIST-T1 cells stably expressing vector or BRD4 were harvested, and expression of the indicated proteins was examined by Western blotting. b GIST882 and GIST-T1 cells stably expressing vector or BRD4 were cultured with DMEM without serum overnight. Conditioned media were collected for ELISA assays to assess VEGFA secretion. c GIST882 and GIST-T1 cells stably expressing shcon or shBRD4 were harvested and expression of the indicated protein was examined by Western blotting. d GIST882 and GIST-T1 cells stably expressing shcon or shBRD4 were cultured with DMEM without serum overnight. Conditioned media were collected for ELISA assays to assess VEGFA secretion. e Conditioned media from GIST882 cells stably expressing vector or BRD4 were incubated with HUVECs cells in 96-well plates. The tube formation was examined by the branching points per filed. f Conditioned media from GIST-T1 cells stably expressing shcon or shBRD4 were incubated with HUVECs cells in 96-well plates. Tube formation was examined based on the number of branching points per field. Scale bar: 100 μm; **P < 0.01; ***P < 0.001.
The conditioned media from cells expressing vector, BRD4, shBRD4, or shNC were individually incubated with endothelial cells. Increased formation of tubes was seen when medium from BRD4-expressing cells was compared with the level seen in cells expressing vector alone and knockdown cells showed the opposite observations of shNC cells (Fig. 3e, f). Overall, our findings indicated that BRD4 is likely to be involved in angiogenesis in GIST.
Overexpression of BRD4 stimulates growth and angiogenesis in vivo
The abovementioned effects were assessed in the animal models as described in the materials section. The growth of cells, as well as the weight and size of tumors, was distinctly greater in cells expressing BRD4 than in cells expressing vector alone (Fig. 4a, b). The density of microvessels in tumors was shown to be increased in cells expressing BRD4, as suggested by the elevated levels of CD31 determined by IHC (Fig. 4C). This finding was also corroborated by the higher levels of VEGFA, as measured by Western blotting, seen in animals with BRD4-expressing tumors than seen in animals with vector-only tumors (Fig. 4d).

a GIST882 control vector cells or BRD4 cells were subcutaneously injected into Balb/c nude mice. Tumor volume growth curves from day 1 to 19 of treatment are presented. b After 19 days, mice were sacrificed, and tumor weights were examined in the two groups. c Representative IHC staining of BRD4 and CD31 in the two groups is presented. Scale bar: 25 μm. d The level of MMP9, LOX, and VEGFA expression in xenograft tumors was analyzed by Western blotting; *P < 0.05.
BRD4 exerts its tumorigenic effect via CCL2
Next, we analyzed the potential target genes of BRD4. Our qRT-PCR results showed a decrease in CCL2 in BRD4 knockdown cells compared with shcontrol cells (Fig. 5a). The downregulation of CCL2 was further confirmed by ELISA (Fig. 5b). In addition, overexpression of BRD4 increased CCL2 expression (Fig. 5c). This is suggestive of the induction of CCL2 by BRD4 in GIST.

a The indicated cytokines expression in GIST882 cells stably expressing shcon or shBRD4 were analyzed by RT-PCR. b ELISA of CCL2 expression in the GIST882 and GIST-T1 cells stably expressing shcon or shBRD4. c ELISA of CCL2 expression in the GIST882 and GIST-T1 cells stably expressing vector or BRD4. d GIST882 control vector cells or BRD4 cells were subcutaneously injected into Balb/c nude mice. Mice were treated with IgG or αCCL2 antibody. Tumor volume growth curves from day 1 to 19 of treatment are presented. e GIST882 cells stably expressing shcon or shCCL2 with BRD4 overexpression were subcutaneously injected into Balb/c nude mice. Mice were treated with IgG or αCCL2 antibody. Tumor volume growth curves from day 1 to 19 of treatment are presented. *P < 0.05; **P < 0.01.
These experiments were followed by the use of an antibody to inhibit chemokine signaling, which was assessed in vivo. The growth of tumors was distinctly lowered by the CCL2-neutralizing antibody (Fig. 5d). Moreover, BRD4-promoted tumor growth was blocked by CCL2 downregulation (Fig. 5e). This is suggestive of the role of CCL2 produced by GIST cells in metastasis due to BRD4.
NF-κB is involved in the CCL2 expression induced by BRD4
The next analysis was to examine a potential mechanism behind the abovementioned results. The regulation of the NF-κB pathway by BRD4 has been shown in previous work. The NF-κB circuit involves p65 phosphorylation and nuclear translocation, which influence gene activation. S536 phosphorylation (the key site of regulation) was observed on p65 in proportion to BRD4 expression in GIST-882 and GIST-T1 cells (Fig. 6a). The phosphorylation of p65 was downregulated in BRD4-kockdown cells (Fig. 6a). p65 knockdown using siRNA abolished the induction of CCL2 in GIST-882 cells (Fig. 6b). Western blotting showed p65 translocation to the nucleus with overexpression of BRD4 (Fig. 6c). A further attempt to decipher the role of NF-κB was to pretreat cells with an inhibitor of NF-κB, BAY 11–7082, that prevents translocation to the nucleus (Fig. 6c). This treatment abrogated the expression of CCL2 as well as the phosphorylation of p65 via BRD4 overexpression (Fig. 6d). These results indicate a role for p65 in the activation of CCL2 by BRD4.

a The phosphorylation of p65 was analyzed in the indicated cells by Western blotting. b CCL2 expression in GIST882 cells stably expressing shcon or shp65 was analyzed by Western blotting. c GIST882 cells stably expressing vector or BRD4 were treated with 10 μmol/l BAY11-7082 for 1 h. Nuclear fractions were isolated from cells and analyzed for p65 expression by Western blotting. Lamin A/C and β-actin, which are expressed in nucleus and cytoplasm, respectively, were used as controls for loading and fractionation. d GIST882 cells stably expressing vector or BRD4 were treated with 10 μmol/l BAY11-7082 for 1 h. The levels of p-p65 (S536) and BRD4 were analyzed by Western blotting.
Macrophages activated by CCL2 stimulate tumor progression
CD68 (a macrophage marker) was detected at high levels in GIST intratumoral and peritumoral regions, in line with the observations from the mouse model that showed the association of macrophage infiltration with the level of BRD4 (Fig. 7a). This association is indicative of a link between BRD4 and TAMs. The next set of experiments included analyzing the effect of BRD4 overexpression on TAMs. This involved the culturing of human monocytes as described in the materials section. The conditioned medium from the cells transfected with BRD4 caused the monocytes to be activated into macrophages, as seen by the CD206-high/HLA-DR-low phenotype, which was not observed in cells treated with the conditioned medium from controls (Fig. 7b). Flow cytometry of M1- and M2-specific markers was performed to confirm the expression. The expression of CD206 and CD163 (M2 markers) was increased in the medium from cells transfected with BRD4, while that of HLA-DR and CD86 (M1 markers) was lower in the medium fom cells transfected with BRD4 than that in the controls (Fig. 7b, c). These findings provide proof that increasing levels of BRD4 activate TAMs. Exposure to a CCL2-neutralizing antibody caused distinct in vivo inhibition of these macrophages, indicating the vital role of CCL2 in TAM activation by BRD4 (Fig. 7d).

a Representative images of GIST tissues with high or low levels of CD68-positive cells in the intratumoral tissues with different BRD4 expression levels. Scale bar: 25 μm. b Flow cytometric analysis and quantification of the expressions of CD206/HLA-DR in macrophages treated with medium collected from the indicated cells for 24 h. c Flow cytometric analysis and quantification of the expressions of CD163/CD86 in macrophages treated with medium collected from the indicated cells for 24 h. d IHC staining evaluating macrophages with anti-F4/80 as indicating in tumor of nude mice. Scale bar: 25 μm; **P < 0.01.
Discussion
Despite the initial restrained success of KIT- or PDGFRA-mutant GIST patients using targeted therapies (such as imatinib), a major challenge is the development of resistance to the drugs31,32. Another factor is the nonresponsiveness of GISTs that lack such mutations to molecular targeted therapy33,34. These issues necessitate a thorough comprehension of the mechanisms of GIST pathology35. Appropriate new approaches for treating the disease (advanced or unresectable cases) are needed. This work showed overexpression of BRD4 in clinical GIST samples compared with that in corresponding healthy samples, which is suggestive of the involvement of epigenetics and posttranslational modifications. As a modulator of the microenvironment of the tumor, BRD4 was found to function in the progression of GISTs via the NF-κB circuit, leading to the activation of CCL2. This, in turn, led to the infiltration of TAMs, which affected the tumor microenvironment. This infiltration was substantially suppressed with the use of antibodies against CCL2 to reduce the growth of tumors caused by BRD4 in vivo. Thus, new insights have been gained in this work regarding the involvement of BRD4 in GIST pathogenesis.
The BET family proteins (inclusive of BRD4 and others listed in the introduction) are targeted by small molecules targeting inflammation36,37. While the role of each member in vivo has yet to be explored, the level of BRD4 in GISTs has not been examined thus far in any work. For this reason, we evaluated the pathogenic attributes of BRD4, and BRD4 was found to boost the abilities of GIST cell lines to proliferate, invade, and migrate. Hence, a potential treatment approach is to use BRD4 as a therapeutic target in GISTs.
The involvement of VEGFA in angiogenesis has been shown by research, and angiogenesis is an intricate pathway vital to tumorigenesis as well as metastasis38,39. This work explored whether BRD4 was associated with this pathway and reported that increased levels of BRD4 led to the in vitro and in vivo formation of HUVEC tubes.
The vital involvement of TAMs in the microenvironment of cancer has received much attention with the production of several growth factors by these accumulated cells shown to influence both human tumorigenesis and angiogenesis40,41. A symbiosis between cocultured macrophages and tumor cells was shown by earlier research to boost the degradation of collagen42. This work shows the role of TAMs in tumorigenesis in an animal model to provide an understanding of the mechanisms in GIST. The role of BRD4 in increasing the expression of CCL243 (which is produced in high amounts by tumors influencing tumorigenesis as well as recruitting monocytes from the circulation to become TAMs) was shown in this work. Recent research has shown the targeting of CCL2 in solid tumors with carlumab (a monoclonal antibody) in many phase I and II clinical trials44. Such targeting of CCL2 can also be explored for therapeutically addressing GIST and predicting the prognosis of patients with GIST. This work showed stimulation of the NF-κB circuit by BRD4 to boost CCL2 levels, and these levels were found to be substantially lowered by the use of anti-CCL2 antibodies. These antibodies also boosted the survival time in the animal models with high BRD4 expression due to lowered tumorigenic potential, implying the potential of blocking CCL2 in the treatment of GISTs with high levels of BRD4. This work both uncovered a mechanism and explored a potential therapeutic approach.
The regulation of both branches of the immune system by the transcription factor NF-κB is known as is its role in modulating inflammation45,46. This factor causes the induction of many genes vital for inflammation, such as chemokines and cytokines, as well as influences immune cell and inflammatory T cell functions in terms of their activation, survival, and differentiation47,48. Hence, dysregulation of NF-κB translates into several diseases involving inflammation49,50. This work showed the involvement of this pathway in the increased expression of CCL2 induced by BRD4.
To summarize, this work outlines the involvement of BRD4 in the progression of GIST. This work is distinct proof of the function of high levels of BRD4 in GIST, and BRD4 accomplishes its role via CCL2 to alter the microenvironment of tumors; these findings are in line with clinical and functional analyses. This involvement of TAMs via CCL2 induced by BRD4 can increase uncovering further mechanisms as well as addressing the treatment of GIST.
References
Parab, T. M. et al. Gastrointestinal stromal tumors: a comprehensive review. J. Gastrointest. Oncol. 10, 144–154 (2019).
Benesch, M., Wardelmann, E., Ferrari, A., Brennan, B. & Verschuur, A. Gastrointestinal stromal tumors (GIST) in children and adolescents: a comprehensive review of the current literature. Pediatr. Blood Cancer 53, 1171–1179 (2009).
DES, B. et al. What is changing in the surgical treatment of gastrointestinal stromal tumors after multidisciplinary approach? A comprehensive literature's review. Minerva Chir. 72, 219–236 (2017).
Akahoshi, K., Oya, M., Koga, T. & Shiratsuchi, Y. Current clinical management of gastrointestinal stromal tumor. World J. Gastroenterol. 24, 2806–2817 (2018).
Din, O. S. & Woll, P. J. Treatment of gastrointestinal stromal tumor: focus on imatinib mesylate. Ther. Clin. Risk Manag. 4, 149–162 (2008).
Zhao, W. & Cao, H. [Targeted therapy combined with immunotherapy in gastrointestinal stromal tumor: a new era of hope and challenges]. Zhonghua Wei Chang Wai Ke Za Zhi 20, 966–971 (2017).
Tang, L. et al. MRI in predicting the response of gastrointestinal stromal tumor to targeted therapy: a patient-based multi-parameter study. BMC Cancer 18, 811 (2018).
Yan, W., Zhang, A. & Powell, M. J. Genetic alteration and mutation profiling of circulating cell-free tumor DNA (cfDNA) for diagnosis and targeted therapy of gastrointestinal stromal tumors. Chin. J. Cancer 35, 68 (2016).
Balachandran, V. P. & Dematteo, R. P. Targeted therapy for cancer: the gastrointestinal stromal tumor model. Surg. Oncol. Clin. N. Am. 22, 805–821 (2013).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Tan, X., Zhang, Z., Yao, H. & Shen, L. Tim-4 promotes the growth of colorectal cancer by activating angiogenesis and recruiting tumor-associated macrophages via the PI3K/AKT/mTOR signaling pathway. Cancer Lett. 436, 119–128 (2018).
Cavnar, M. J. & DeMatteo, R. P. Sarcoma response to targeted therapy dynamically polarizes tumor-associated macrophages. Oncoimmunology 3, e28463 (2014).
Liu, J. et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE 6, e19495 (2011).
Ye, H. et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 9, 453 (2018).
Zhou, S. L. et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 150, 1646–1658 e1617 (2016).
Lee, C. H. et al. Tumor-associated macrophages promote oral cancer progression through activation of the Axl signaling pathway. Ann. Surg. Oncol. 21, 1031–1037 (2014).
Cavnar, M. J. et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J. Exp. Med 210, 2873–2886 (2013).
Klein, K. Bromodomain protein inhibition: a novel therapeutic strategy in rheumatic diseases. RMD Open 4, e000744 (2018).
Tan, X. et al. BET inhibitors potentiate chemotherapy and killing of SPOP-mutant colon cancer cells via induction of DR5. Cancer Res. 79, 1191–1203 (2019).
Jin, X. et al. DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol. Cell 71, 592–605 e594 (2018).
Sakaguchi, T. et al. Bromodomain protein BRD4 inhibitor JQ1 regulates potential prognostic molecules in advanced renal cell carcinoma. Oncotarget 9, 23003–23017 (2018).
Heinrich, M. C. et al. Sorafenib inhibits many kinase mutations associated with drug-resistant gastrointestinal stromal tumors. Mol. Cancer Ther. 11, 1770–1780 (2012).
Taguchi, T. et al. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab. Invest. 82, 663–665 (2002).
Tong, J. et al. Mcl-1 phosphorylation without degradation mediates sensitivity to HDAC inhibitors by liberating BH3-only proteins. Cancer Res. 78, 4704–4715 (2018).
Tong, J. et al. Mcl-1 degradation is required for targeted therapeutics to eradicate colon cancer cells. Cancer Res. 77, 2512–2521 (2017).
Tong, J. et al. FBW7-dependent Mcl-1 degradation mediates the anticancer effect of Hsp90 inhibitors. Mol. Cancer Ther. 16, 1979–1988 (2017).
He, K. et al. BRAFV600E-dependent Mcl-1 stabilization leads to everolimus resistance in colon cancer cells. Oncotarget 7, 47699–47710 (2016).
Tong, J., Tan, S., Zou, F., Yu, J. & Zhang, L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 36, 787–796 (2017).
Zhang, Z. et al. GNA13 promotes tumor growth and angiogenesis by upregulating CXC chemokines via the NF-kappaB signaling pathway in colorectal cancer cells. Cancer Med. 7, 5611–5620 (2018).
Nishida, T. et al. Efficacy and safety profile of imatinib mesylate (ST1571) in Japanese patients with advanced gastrointestinal stromal tumors: a phase II study (STI571B1202). Int. J. Clin. Oncol. 13, 244–251 (2008).
Demetri, G. D. et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 347, 472–480 (2002).
Hung, K. D., Van, Q. L., Hoang, G. N. & Bich, P. N. T. Imatinib mesylate for patients with unresectable or recurrent gastrointestinal stromal tumors: 10-year experience from vietnam. Cancer Control 26, 1073274819863776 (2019).
Ogata, K. et al. Long-term imatinib treatment for patients with unresectable or recurrent gastrointestinal stromal tumors. Digestion 97, 20–25 (2018).
Platoff, R. M. et al. Recurrent gastrointestinal stromal tumors in the imatinib mesylate era: treatment strategies for an incurable disease. Case Rep. Oncol. Med 2017, 8349090 (2017).
Donati, B., Lorenzini, E. & Ciarrocchi, A. BRD4 and cancer: going beyond transcriptional regulation. Mol. Cancer 17, 164 (2018).
Pericole, F. V. et al. BRD4 inhibition enhances azacitidine efficacy in acute myeloid leukemia and myelodysplastic syndromes. Front Oncol. 9, 16 (2019).
Horwitz, E. et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Disco. 4, 730–743 (2014).
Waldner, M. J. et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J. Exp. Med. 207, 2855–2868 (2010).
Sengupta, M. et al. Anticancer efficacy of noble metal nanoparticles relies on reprogramming tumor-associated macrophages through redox pathways and pro-inflammatory cytokine cascades. Cell Mol. Immunol. 15, 1088–1090 (2018).
Genard, G., Lucas, S. & Michiels, C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front Immunol. 8, 828 (2017).
Yang, L. & Zhang, Y. Tumor-associated macrophages: from basic research to clinical application. J. Hematol. Oncol. 10, 58 (2017).
Sierra-Filardi, E. et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J. Immunol. 192, 3858–3867 (2014).
Zheng, X. et al. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 8, 48436–48452 (2017).
Hoesel, B. & Schmid, J. A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 12, 86 (2013).
Dorrington, M. G. & Fraser, I. D. C. NF-kappaB signaling in macrophages: dynamics, crosstalk, and signal integration. Front Immunol. 10, 705 (2019).
Tornatore, L., Thotakura, A. K., Bennett, J., Moretti, M. & Franzoso, G. The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol. 22, 557–566 (2012).
Kempe, S., Kestler, H., Lasar, A. & Wirth, T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 33, 5308–5319 (2005).
Miller, S. A. et al. Effects of consumption of whole grape powder on basal NF-kappaB signaling and inflammatory cytokine secretion in a mouse model of inflammation. J. Nutr. Intermed. Metab. 11, 1–8 (2018).
Abraham A. C., et al. Targeting the NF-kappaB signaling pathway in chronic tendon disease. Sci. Transl. Med. 11, eaav4319 (2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mu, J., Sun, P., Ma, Z. et al. BRD4 promotes tumor progression and NF-κB/CCL2-dependent tumor-associated macrophage recruitment in GIST. Cell Death Dis 10, 935 (2019). https://doi.org/10.1038/s41419-019-2170-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-2170-4
This article is cited by
-
KIT mutations and expression: current knowledge and new insights for overcoming IM resistance in GIST
Cell Communication and Signaling (2024)
-
Advances in immunology and immunotherapy for mesenchymal gastrointestinal cancers
Molecular Cancer (2023)
-
Elian granules alleviate precancerous lesions of gastric cancer in rats by suppressing M2-type polarization of tumor-associated macrophages through NF-κB signaling pathway
BMC Complementary Medicine and Therapies (2023)
-
Design, synthesis and pharmacological characterization of N-(3-ethylbenzo[d]isoxazol-5-yl) sulfonamide derivatives as BRD4 inhibitors against acute myeloid leukemia
Acta Pharmacologica Sinica (2022)
-
The KDM6A-SPARCL1 axis blocks metastasis and regulates the tumour microenvironment of gastrointestinal stromal tumours by inhibiting the nuclear translocation of p65
British Journal of Cancer (2022)
