
Abstract
In the last two decades, studies on plant biomass-degrading fungi have remarkably increased to understand and reveal the underlying molecular mechanisms responsible for their life cycle and wood-decaying abilities. Most of the plant biomass-degrading fungi reported till date belong to basidiomycota or ascomycota phyla. Thus, very few studies were conducted on fungi belonging to other divisions. Recent sequencing studies have revealed complete genomic sequences of various fungi. Our present study is focused on understanding the plant biomass-degrading potentials, by retrieving genome-wide annotations of 56 published fungi belonging to Glomeromycota, Mucoromycota, Zoopagomycota, Blastocladiomycota, Chytridiomycota, Neocallimastigomycota, Microsporidia and Cryptomycota from JGI-MycoCosm repository. We have compared and analyzed the proteomic annotations, especially CAZy, KOG, KEGG and SM clusters by separating the proteomic annotations into lignin-, cellulose-, hemicellulose-, pectin-degrading enzymes and also highlighted the KEGG, KOG molecular mechanisms responsible for the metabolism of carbohydrates (lignocellulolytic pathways of fungi), complex organic pollutants, xenobiotic compounds, biosynthesis of secondary metabolites. However, we strongly agree that studying genome-wide distributions of fungal CAZyme does not completely corresponds to its biomass-degrading ability. Thus, our present study can be used as preliminary materials for selecting ideal fungal candidate for the degradation and conversion of plant biomass components, especially carbohydrates to bioethanol and other commercially valuable products.

Similar content being viewed by others


Background
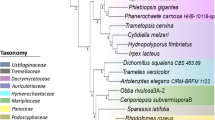
Early taxonomists classified fungi under the plant kingdom based on their lifestyle. However, with the development of advanced molecular and phylogenetic techniques fungi were given a kingdom status. Evolutionary studies have reported that fungi have evolved in the early Neoproterozoic era followed by evolution over the period of time due to the earthly impacts including oxygen, carbondioxide, solar luminosity (Baldauf and Palmer 1993; Bruns 2006) (Fig. 1a). Although early evolutionary studies have reported that fungi have evolved from a common ancestor with that of unicellular flagellated aquatic organisms. However, there is no accepted phylogenetic theory which explains about the evolution of the early fungi (James et al. 2000, 2006a; Karol et al. 2001; Tanabe et al. 2004). Previous studies have reported that chytridiomycota (flagellated cells) are group of true fungi which are phylogenetically connected to the sister groups mucoromycota, zoopagomycota, glomeromycota, ascomycota and basidiomycota which have experienced an evolutionary loss of the flagellum (James et al. 2000, 2006a; Karol et al. 2001; Tanabe et al. 2004). However, these non-flagellated fungi have evolutionarily adapted to the terrestrial habitat by developing a filamentous growth and aerially dispersed spores. Studies have reported that early fungal phylogeny and fungal tree of life remained unresolved; however, in the recent years, phylogenetic studies have questioned the phylogenetic lineage of the chytridiomycota, mucoromycota and zoopagomycota which resolved the phylogenetic aspects of these basal groups with their relationships with ascomycota and basidiomycota (James et al. 2000, 2006a; Karol et al. 2001; Tanabe et al. 2004) (Fig. 1b). Studies have classified kingdom fungi into six true phyla: chytridiomycota, zygomycota, ascomycota, basidiomycota, glomeromycota and deuteromycota (McLaughlin et al. 2001; Silar 2016). However, the recent higher level phylogenetic studies classified fungi into: Dikarya (ascomycota, basidiomycota), glomeromycota, chytridiomycota, neocallimastigomycota, blastocladiomycota, kickxellomycota, microsporidia and cryptomycota, respectively (Hibbett et al. 2007) (Fig. 1a).

a Pictorial illustration of fungal evolution since the early Neoproterozoic era and the influence of solar luminosity, carbon dioxide, oxygen, earth impacts over the geological timescale of fungal evolution (Hedges and Kumar 2009; Hedges et al. 2015), b hierarchical illustration of fungal phylogenetic classification
Advancement of molecular techniques 18srRNA and whole-genome sequencing techniques are currently being used to understand and reveal the phylogenetic relationships among the fungi. Development of advanced genome sequencing techniques and online genomic repositories have enhanced the current days knowledge of fungal lifestyle and evolution. The fungal genomic repositories such as joint genome institute MycoCosm (Grigoriev et al. 2011; Nordberg et al. 2013), 1000 fungal genome project and Hungate collection residing in the rumen microbial genomics network are continuously enhancing the genomic details of various fungi. Studies being conducted to understand the genomic potentials of different fungi belonging to different phyla were compared for their plant cell wall-degrading potentials to explore their applications in biofuel and bioremediation industries (Kameshwar and Qin 2018; King et al. 2011; Rytioja et al. 2014; Sista Kameshwar and Qin 2017; Zhao et al. 2013). Till date, there are 1054 whole-genome sequences of fungi belonging to different phyla in JGI-MycoCosm repository, out of which 444 whole-genome sequences of fungi have been published and publicly available and the remaining 610 whole-genome sequences of fungi were under study and unpublished (Grigoriev et al. 2011; Nordberg et al. 2013).
Fungi belonging to the ascomycota and basidiomycota phyla were highly studied compared to other phyla. Till date, approximately 349 basidiomycetous and 588 ascomycetous fungi whole-genome sequencing studies were reported in public repositories. During the process of evolution, fungi have developed as bio-decomposers/decayers of the organic material. In nature, fungi play various roles ecologically as saprobes, parasites, plant pathogens, symbionts and endophytes; they play a key role in wood-decay, litter decomposition and thus in maintaining the global carbon cycle (Diyarova 2016; Krishna and Mohan 2013; Wal et al. 2013). In the last two decades, various fungal strains were extensively studied for its plant cell wall (lignocellulose)-degrading abilities (Kameshwar and Qin 2016a). In our previous study, we have performed a large-scale comparative analysis of 42 wood rotting fungi representing white rot, brown rot and soft rot fungi (Sista Kameshwar and Qin 2017). This study has majorly reported that white rot fungi tentatively exhibit highest lignocellulolytic and soft rot fungi tentatively exhibit highest cellulolytic and hemicellulolytic potentials (Sista Kameshwar and Qin 2017).
The arbuscular mycorrhizal (AM) fungi belonging to the glomeromycota phyla play a crucial role both ecologically and environmentally (Morton and Benny 1990). AM fungi are asexual organisms and obligate symbiotes of vascular plants (as AM fungi penetrate the plant substrate using its mycelium). Studies have reported that plants depend on the symbiotic mycorrhizae rather than the roots for the uptake of phosphate ion from the soil (Morton and Benny 1990; Schüßler et al. 2001b; Smith and Read 1997). Thus, plants obtain inorganic micronutrients with the aid of AM fungi and in return fungi obtains carbohydrates from plant, this exchange happens through the intracellular symbiotic interfaces. The molecular evolutionary techniques such as small subunit rRNA sequences report that they share a common ancestor route with Dikarya and the present glomeromycota consists four orders: (a) diversisporales, (b) glomerales, (c) archaeosporales and (d) paraglomerales (Redecker et al. 2013; Schüßler et al. 2001a, b). Till date, studies have reported approximately 300 glomeromycota species based on their spore morphology (Chen et al. 2018).
Zygomycota are considered as true fungi and contain chitin in their cell walls. These fungi were found to be emerged from the other fungi approximately 600 to 1400 million years ago (Berbee and Taylor 2001a, b, Heckman et al. 2001). These terrestrial fungi live in decaying plants or animals and soil material, and some fungal species are parasites of plants, insects, while some species are in symbiotic relationships with the plants (Raven et al. 2005). Zygomycetes fungi are filamentous, non-flagellated, and importantly they form zygospores with in the zygosporangium formed as a result of sexual cycle. Major transition from the earliest diverging zoosporic fungi led to the phyla cryptomycota, chytridiomycota, neocallimastigomycota, blastocladiomycota and resulted towards the rise of non-flagellated filamentous multicellular Dikaryan fungi (Spatafora et al. 2016). Studies have reported that some zygomycetes fungi significantly benefit humans by producing commercially important compounds such as lycopene, fatty acids and biodiesel (Papanikolaou et al. 2007; Wang et al. 2011). Hibbett et al. (2007) have classified four divisions: entomophthoromycotina, kickxellomycotina, mucoromycotina and zoopagomycotina under the subphyla incertae sedis and a separate phylum glomeromycota. Molecular phylogenetic methods including rDNA and multigene studies have classified zygomycetes taxa into two major groups which informally include zygomycetes-I (mucoromycotina and mortierellomycotina), and few studies have also included glomeromycota phylum (Chang et al. 2015; James et al. 2006a; White et al. 2006). Phylogenetic studies conducted by Spatafora et al. (2016) have separated zygomycetes fungi into two different phylum’s mucoromycota and zoopagomycota (Spatafora et al. 2016). Mucoromycota fungi are further classified into four orders: (a) glomeromycota, (b) mucoromycotina and (c) mortierellomycotina, and zoopagomycota is further classified into (a) zoopagomycotina, (b) entomophthoromycotina and (c) kickxellomycotina. Mortierellomycotina subphylum includes common soil-inhabiting fungi, root endophytes and saprobes (Summerbell 2005). Zhao et al. (2013) have performed large-scale comparative analysis of 103 fungal proteomes representing Ascomycota, Basidiomycota, Chytridiomycota and Zygomycota. Zhao et al. (2013) have reported that fungi exhibit tremendous diversity in the number and variety of CAZymes, plant pathogenic fungi exhibit highest number of CAZymes followed by necrotrophic, and hemibiotrophic fungi and biotrophic fungi tend to exhibit fewer CAZymes (Zhao et al. 2013). This study has also reported that fungal pathogens infecting dicots exhibit more pectinases than monocot-infecting fungi, saprophytic fungi (highly active plant pathogens) also exhibited fewer CAZymes compared to plant pathogenic fungi (Zhao et al. 2013).
Earlier studies have classified blastocladiomycota, neocallimastigomycota under the phylum chytridiomycota, and with the advancement in molecular phylogenetic techniques, these orders were given the status of phylum. Chytridiomycota is considered as true fungi as it reproduces through the zoospores (motile spores) by having a posterior flagellum (Barr 1992; Bartnicki-Garcia 1970; James et al. 2006b). Together chytridiomycota, blastocladiomycota and neocallimastigomycota fungi can be grouped as true zoosporic fungi (Alexopoulous et al. 1996). Chytrids are characterized by their specific biochemical properties including alpha-aminoadipic acid, lysine synthetic pathway, chitin cell walls and ability to store glycogen (Alexopoulous et al. 1996; Barr 1992; Bartnicki-Garcia 1970; James et al. 2006b; Kendrick 2000). Blastocladiomycota is one of the recently added phyla in the kingdom fungi which was previously included as blastocladiales in the phylum chytridiomycota (Hibbett et al. 2007; James et al. 2006b). Neocallimastigomycota includes anaerobic fungi which are majorly present as symbionts in the digestive tracts of herbivores. These organisms were first discovered in the gut of ruminating animals. Recent high-throughput rumen microbiome sequencing studies have revealed the complete genome sequences of five anaerobic fungi. With the availability of genomic data, further studies were being conducted to understand the growth, development and functional roles of these anaerobic fungi in the rumen (Kameshwar and Qin 2018; Orpin 1975). In our previous study, we have extensively studied Neocallimastigomycota fungi to understand and reveal its lignin-, cellulose-, hemicellulose-, pectin-degrading potentials by extensively analyzing the genome-wide proteomic annotations, especially CAZy, InterPro, KOG, KEGG, SM clusters and MEROPS (Kameshwar and Qin 2018).
Microsporidia are typical spore-forming unicellular parasites which were primarily considered as protists or protozoans. However, recent genomic studies have classified microsporidia as a separate phylum under fungi (Hibbett et al. 2007). Microsporidian fungi were highly studied for their disease-causing properties in animals and humans. In humans, microsporidian fungi cause microsporidiosis, and apart from humans, microsporidian fungi infect various hosts including parasites which infect higher animals such as flatworms (Hoffman 1999). The term “Cryptomycota” was coined to suggest its signature characteristics and cryptic nature, once these fungi were interpreted as intermediates between ancestral protists and fungi (Lara et al. 2010). However, the present knowledge on Cryptomycota phylum is very limited compared to other phyla, and advanced genomic and molecular phylogeny studies must be conducted to understand and reveal about the growth and development of these fungi.
In this systematic review study, we have performed systematic analysis of genome-wide annotations of published fungi belonging to glomeromycota, mucoromycota, zoopagomycota, blastocladiomycota, chytridiomycota, neocallimastigomycota, microsporidia and cryptomycota to understand and reveal the evolutionary loss of genes encoding for plant biomass-degrading enzymes, complete eukaryotic orthologous groups, secondary metabolite clusters, metabolic and regulatory pathways of the selected fungi.
Review and analysis of lower-fungal genomes
In our present study, we have selected and retrieved the annotated proteomic data (including CAZy—carbohydrate active enzymes, KOG—eukaryotic orthologous groups, SM—secondary metabolite clusters, KEGG—Kyoto Encyclopedia of Genes and Genomes) of 56 fungi from https://genome.jgi.doe.gov/programs/fungi/index.jsf JGI (Joint Genome Institute) MycoCosm database.
Glomeromycota: Rhizophagus irregularis DAOM 181602 v1.0 (Tisserant et al. 2013), Rhizophagus irregularis A1 v1.0 (Chen et al. 2018), Rhizophagus irregularis A4 v1.0 (Chen et al. 2018), Rhizophagus irregularis A5 v1.0 (Chen et al. 2018), Rhizophagus irregularis B3 v1.0 (Chen et al. 2018), Rhizophagus irregularis C2 v1.0 (Chen et al. 2018) and Rhizophagus irregularis DAOM 197198 v2.0 (Chen et al. 2018), Gigaspora rosea v1.0 (Morin et al. 2019), Rhizophagus cerebriforme DAOM 227022 v1.0 (Morin et al. 2019), Rhizophagus diaphanus v1.0 (Morin et al. 2019). Mortierellomycotina (Lobosporangium transversale NRRL 3116 v1.0 (Mondo et al. 2017), Mortierella elongata AG-77 v2.0 (Uehling et al. 2017). Mucoromycotina (Absidia repens NRRL 1336 v1.0 (Mondo et al. 2017), Hesseltinella vesiculosa NRRL3301 v2.0 (Mondo et al. 2017), Lichtheimia corymbifera JMRC:FSU:9682 (Schwartze et al. 2014), Mucor circinelloides CBS277.49 v2.0 (Corrochano et al. 2016), Phycomyces blakesleeanus NRRL1555 v2.0 (Corrochano et al. 2016), Rhizopus delemar 99-880 from Broad (Ma et al. 2009), Rhizopus microsporus ATCC11559 v1.0 (Lastovetsky et al. 2016), Rhizopus microsporus var. chinensis CCTCC M201021 (Wang et al. 2013), Rhizopus microsporus var. microsporus ATCC52814 (Lastovetsky et al. 2016), Syncephalastrum racemosum NRRL 2496 v1.0 (Mondo et al. 2017), Endogone sp. FLAS 59071(Chang et al. 2019), Jimgerdemannia flammicorona AD002 (Chang et al. 2019), Jimgerdemannia flammicorona GMNB39 (Chang et al. 2019). Jimgerdemannia lactiflua OSC166217 (Chang et al. 2019). Zoopagomycotina Piptocephalis cylindrospora RSA 2659 single-cell v3.0 (Ahrendt et al. 2018), Syncephalis pseudoplumigaleata Benny S71-1 single-cell v1.0 (Ahrendt et al. 2018), Thamnocephalis sphaerospora RSA 1356 single-cell v1.0 (Ahrendt et al. 2018). Entomophthoromycotina (Conidiobolus coronatus NRRL28638 v1.0 (Chang et al. 2015). Kickxellomycotina (Coemansia reversa NRRL 1564 v1.0 (Chang et al. 2015), Linderina pennispora ATCC 12442 v1.0 (Mondo et al. 2017). Blastocladiomycota (Catenaria anguillulae PL171 v2.0 (Mondo et al. 2017). Chytridiomycota (Gonapodya prolifera v1.0 (Chang et al. 2015), Rhizoclosmatium globosum JEL800 v1.0 (Mondo et al. 2017), Spizellomyces punctatus DAOM BR117 (Russ et al. 2016). Neocallimastigomycota (Anaeromyces robustus v1.0 (Haitjema et al. 2017), Neocallimastix californiae G1 v1.0 (Haitjema et al. 2017), Orpinomyces sp. (Youssef et al. 2013), Piromyces finnis v3.0 (Haitjema et al. 2017), Piromyces sp. E2 v1.0 (Haitjema et al. 2017). Microsporidia (Antonospora locustae HM-2013 (Slamovits et al. 2004), Encephalitozoon cuniculi GB-M1 (Peyretaillade et al. 2009), Encephalitozoon hellem ATCC 50504 (Pombert et al. 2012), Encephalitozoon intestinalis ATCC 50506 (Corradi et al. 2010), Encephalitozoon romaleae SJ-2008 (Pombert et al. 2012), Enterocytozoon bieneusi H348 (Akiyoshi et al. 2009), Nematocida parisii ERTm1 (Cuomo et al. 2012), Nosema ceranae BRL01 (Cornman et al. 2009). Cryptomycota (Rozella allomycis CSF55 (James et al. 2013), Rozella allomycis CSF55 single-cell v1.0 (Ahrendt et al. 2018). We have also reported a tentative average of cellulolytic, hemicellulolytic, ligninolytic and pectinolytic potentials exhibited by the fungi belonging to the selected phyla, by considering the total number of genes encoding for the lignocellulolytic enzymes.
The KOG, SM clusters and KEGG annotations
The eukaryotic cluster of orthologous group classifies protein sequences from a completed genome sequence of eukaryotes based on their orthology (Tatusov et al. 2000). The KOG database of JGI-MycoCosm database classifies the protein sequences of fungal whole-genome sequences among four major groups as (a) cellular signaling and processing (CSP), (b) information storage and processing (ISP), (c) metabolism and (d) poorly characterized. We have retrieved and compared the genome-wide KOG annotations of all the selected fungi and tentatively calculated the average of all the KOG annotations and compared among the divisions (Fig. 2a). Results obtained from our systematic review potentially propose that fungi belonging to microsporidia and cryptomycota divisions might have experienced serious evolutionary loss of several genes classified among KOG groups. Previous studies have reported that compared to other fungal divisions microsporidia fungi have smallest genomes and these fungi have experienced a significant loss of mitochondrial, ribosomal RNAs and Golgi complex genes (Corradi and Selman 2013), whereas the fungi belonging to chytridiomycota, neocallimastigomycota and glomeromycota exhibited higher number of genes encoding for KOG groups (Fig. 2). Understanding the molecular mechanisms involved in degradation of plant cell wall components will significantly benefit in developing recombinant strains with extrinsic degrading potentials. Thus, we have specifically compared the KOG groups corresponding to the fungal defense mechanisms (V), carbohydrate transport and metabolism (G), secondary metabolite biosynthesis, transport and catabolism (Q) and energy production and conversion (C). Results obtained from the comparative analysis have shown that the selected glomeromycota fungi exhibit highest number of genes encoding for secondary metabolite biosynthesis, transport and catabolism (Q) KOG group. The selected neocallimastigomycota fungi have shown highest number of genes encoding for carbohydrate transport and metabolism (G). Similarly, genes encoding for defense mechanisms (V) was found to be highest among the selected neocallimastigomycota and chytridiomycota fungi. Interestingly, the genes encoding for energy production and conversion (C) group was highly observed among most of the selected fungi. Contrastingly, all the selected fungi belonging to microsporidia and cryptomycota exhibited lower number of genes encoding for all the selected KOG groups.

Pictorial illustration of the selected non-Dikarya fungi in descending orders based on the distribution of eukaryotic orthologous groups (KOG) and secondary metabolite clusters. NRPS non-ribosomal peptide synthases, PKS polyketide synthases, NRPS–PKS hybrid, DMATSs prenyl-transferases, TCs terpene cyclases
Fungal growth is seriously challenged by various biotic and abiotic stressors ranging from nutrient limitations, environmental conditions such as pH and temperature and other microorganisms competing for nutrients (Macheleidt et al. 2016). As an immediate physiological response, fungi produce a wide range of secondary metabolites (Macheleidt et al. 2016). Fungi are rich sources of various commercially important secondary metabolites including pharmaceutical compounds, antibiotics, etc. Interestingly, genes responsible for the transcriptional regulation, biosynthesis and export of these secondary metabolites were found in adjoining gene clusters. We have retrieved and compared the secondary metabolite clusters (SM clusters) of all the selected fungi from the JGI-MycoCosm database. The secondary metabolite gene clusters are currently divided into non-ribosomal peptide synthases (NRPS), polyketide synthases (PKS), hybrid NRPS–PKS enzymes, prenyl-transferases (DMATSs) and terpene cyclases (TCs) (Khaldi et al. 2010). Interestingly, the systematic comparison of secondary metabolite gene cluster annotations has shown that the selected non-Dikarya except neocallimastigomycota fungi contain single copies of NRPS and PKS gene clusters and completely lacks prenyl-transferases (DMATSs) and hybrid gene clusters, whereas neocallimastigomycota fungi exhibited highest number of NRPS, NRPS-like, PKS and PKS-like gene clusters (Fig. 2).
The JGI-MycoCosm predicted genes are majorly divided into KEGG metabolic and KEGG regulatory pathways which are further divided into reference pathways. The KEGG metabolic and regulatory pathways are divided into 12 reference pathways: (a) amino acid metabolism (AAM), (b) biosynthesis of polyketides and non-ribosomal peptides (BpNp), (c) biosynthesis of secondary metabolites (BSM), (d) carbohydrate metabolism (CM), (e) energy metabolism (EM), (f) glycan biosynthesis and metabolism (GBM), (g) lipid metabolism (LM), (h) metabolism of cofactors and vitamins (MCV), (i) metabolism of other amino acids (MAA), (j) nucleotide metabolism (NM), (k) overview (O) and (l) xenobiotics biodegradation and metabolism (XBM). The results obtained from our systematic comparison have shown that the selected microsporidia, cryptomycota, zoopagomycotina and neocallimastigomycota fungal genomes contained lowest number of genes distributed among KEGG metabolic and regulatory pathways. Interestingly, the selected glomeromycota, mucoromycota, kickxellomycotina, fungal genomes exhibited higher number of genes encoding for KEGG metabolic and regulatory pathways (Fig. 3). We have also compared the important KEGG pathways involved in degradation of plant cell wall components, fungal metabolism and regulatory pathways. These KEGG pathways are: BpNp—biosynthesis of polyketides and non-ribosomal peptides, BSM—biosynthesis of secondary metabolites, CM—carbohydrate metabolism, EM—energy metabolism, and XBM—xenobiotic biodegradation metabolism pathways. Results obtained from this comparative analysis have shown that the selected glomeromycota fungi have exhibited higher number of genes encoding xenobiotic biodegradation metabolism (XBM), biosynthesis of secondary metabolites (BSM) and biosynthesis of polyketides and non-ribosomal peptides (BpNp). In particular, Neocallimastix californiae (Neocallimastigomycota), Gonapodya prolifera (Chytridiomycota), Rhizopus microsporus var. chinensis (Mucoromycotina) and Mortierella elongata (Mortierellomycotina) encode higher number of genes distributed among the pathways except XBM and BpNp pathways. The selected microsporidia and cryptomycota fungi exhibited lower number of genes for all the selected pathways (Table 1).

Pictorial illustration of the selected non-Dikarya fungi in descending order based on the distribution of KEGG pathway classes encoding genes. AAM amino acid metabolism, BpNp biosynthesis of polyketides and non-ribosomal peptides, BSM biosynthesis of secondary metabolites, CM carbohydrate metabolism, EM energy metabolism, GBM glycan biosynthesis and metabolism, LM lipid metabolism, MCV metabolism of cofactors and vitamins, MAA metabolism of other amino acids, NM nucleotide metabolism, O overview, XBM xenobiotics biodegradation and metabolism
Carbohydrate active enzymes (CAZymes)
Fungi secrete a variety of carbohydrate active enzymes for the infection and degradation of the plant cell wall components. The carbohydrates released during the process of degradation are further used for fungal growth and development (Zhao et al. 2013). Several basidiomycetous fungi have been extensively studied for its lignocellulose-degrading abilities. White and brown rot fungi especially Phanerochaete chrysosporium and Postia placenta were highly studied for their extrinsic lignocellulose-degrading ability (Zhao et al. 2013). For most of the fungal pathogens, it is highly important to access plant cytoplasm and reach across the plant tissues. Previous studies have reported that several plant cell wall-degrading fungal enzymes such as xylanases and pectinases play a crucial role in imparting virulence and pathogenicity toward its substrate (Douaiher et al. 2007; Ferrari et al. 2008; Kikot et al. 2009). Till date, 445 fungal genomes have been completely sequenced and extensively studied for their lignocellulose-degrading abilities. Complete genome sequencing studies of Saccharomycetes and Schizosaccharomycetes revealed the functional role of various cell wall-degrading enzymes in plant infection and mainly fungal growth and development (Zhao et al. 2013). Recent genome sequencing studies have also reported about the evolutionary loss of CAZyme-encoding genes in few fungal divisions (Skamnioti et al. 2008). Comparative metadata analysis of fungal genome-wide annotations especially CAZymes has been studied and reported (Kameshwar and Qin 2018; Sista Kameshwar and Qin 2017; Zhao et al. 2013). However, previous studies have majorly focused on higher fungi belonging to Ascomycota and Basidiomycota divisions. The genome-wide annotations of Glomeromycota, Chytridiomycota, Blastocladiomycota, Neocallimastigomycota and Microsporidia phyla were not highly studied compared to its counter parts. The systematic review of 40 fungal genomes belonging to the Glomeromycota, Chytridiomycota, Blastocladiomycota, Neocallimastigomycota and Microsporidia phyla has shown that genes encoding for carbohydrate active enzymes was found to decline during its evolution from Glomeromycota to Microsporidia. The selected fungi belonging to microsporidia and cryptomycota phyla fungi have experienced a serious evolutionary loss of CAZyme-encoding genes. Contrastingly, the analyzed neocallimastigomycota phyla fungi were found to contain higher number of CAZyme-encoding genes compared to other selected fungi (Kameshwar and Qin 2018). The neocallimastigomycota fungi encode highest number of CAZymes followed by mucoromycotina, chytridiomycota, glomeromycota, monoblepharidomycetes, mortierellomycotina, entomophthoromycotina, kickxellomycotina, blastocladiomycota, zoopagomycotina, cryptomycota and microsporidia (Table 7).
Ligninolytic potentials
Most of the microorganisms fail to breakdown lignin compounds; however, few microorganisms especially white rot fungi have developed efficient enzyme system for the degradation of lignin (Kameshwar and Qin 2017). The lignin-degrading enzymes are mostly distributed in auxiliary activity group. However, exact molecular mechanisms and the enzymes employed by the microorganisms for the degradation of lignin are not known till date. However, major ligninolytic enzymes include laccase, peroxidases (lignin peroxidase, versatile peroxidase, manganese peroxidase); these enzymes are also called as lignin-oxidizing enzymes. The high-oxidizing potential and non-specificity are considered as the major attributes of the lignin-oxidizing enzymes (Kameshwar and Qin 2016b). These lignin-degrading enzymes depend on supporting enzymes such as aryl-alcohol oxidase, alcohol oxidase, pyranose oxidase, vanillyl alcohol oxidase, alcohol oxidase, glyoxal oxidase, galactose oxidase, 1,4-benzoquinone reductase for the supply of hydrogen peroxide, which triggers the ligninolytic enzymes (Kameshwar and Qin 2016b). Cellobiose dehydrogenase (CDH) degrades various carbohydrates especially cellobiose, mannose to lactones, CDH transfers the electrons retrieved from the substrates to electron acceptors such as quinones, phenoxy radicals and dioxygen (Cameron and Aust 2001; Henriksson et al. 1995, 2000).The prosthetic and FAD groups of CDH make it suitable for the reduction of metals and radicals, though it exhibits high tendency toward amorphous cellulose and it helps in degradation of other plant cell wall components like lignin and xylan (Cameron and Aust 2001; Henriksson et al. 1995, 2000). Studies have also reported that both CDH and LPMO have exhibited and high-oxidative cleavage of lignin compounds (Cameron and Aust 2001; Henriksson et al. 1995, 2000). Similar to hemicellulose and pectin, lignin is also partially and completely esterified by O-acetyl and methyl groups. The acetylated lignin components of plant cell wall inhibit the activity of ligninolytic enzymes. Thus, feruloyl and glucuronoyl esterases play a most significant role in deacetylating the esterified lignin–carbohydrate complexes. Thus, in our report we have considered CDH, LPMO enzymes, lignin-oxidizing, lignin-degrading auxiliary activity enzymes, feruloyl and glucuronoyl esterases in lignin-degrading CAZymes (Table 2).
The glomeromycota, chytridiomycota, blastocladiomycota fungal genomes analyzed in this study lack several copies of ligninolytic auxiliary activity enzymes. The selected glomeromycota fungal genomes specifically lack genes encoding for AA2 (lignin, manganese and versatile peroxidases), AA4 (vanillyl alcohol oxidase), AA8 (iron reductase), AA9, AA10, AA13, AA14 (LPMO), AA12 (pyrroloquinoline quinone-dependent oxidoreductase) and AA15 (lytic cellulose monooxygenase), whereas the selected mortierellomycotina and zoopagomycotina fungal genomes lack genes encoding for AA2, AA4, AA9 (except Coere1 and Linpe1), AA10, AA13, AA14 and AA15 families. Similarly, the analyzed fungal genomes belonging to blastocladiomycota and chytridiomycota have also experienced serious loss of various genes encoding for auxiliary activity class enzymes such as AA2, AA4, AA8, AA10, AA13, AA14 and AA15. All the selected neocallimastigomycota and microsporidia fungal genomes completely lack genes encoding for the auxiliary activity class enzymes. The selected fungi belonging to the phylum’s glomeromycota, kickxellomycotina, entomophthoromycotina, mortierellomycotina, mucoromycotina have exhibited higher ligninolytic potentials followed by the fungi belonging to chytridiomycota, zoopagomycota, monoblepharidomycetes, blastocladiomycota, cryptomycota, neocallimastigomycota, microsporidia (Table 7).
Cellulolytic potentials
Breakdown and conversion of cellulose to glucose is performed by three classes of enzymes: (a) (EnG) endo-β-1-4-glucanase (EC 3.2.1.4), (b) exo-β-1-4-glucanase (EC 3.2.1.94) and (c) β-glucosidase (EC 3.2.1.21) (Silveira et al. 2014). The glycosidic linkages of cellulose are primarily cleaved by EnG on microfibrils surface resulting in long chains with reducing and non-reducing ends making them accessible for ExG and BG enzymes. The ExG acts on the obtained degraded products and further breaks it down to cellobiose and oligosaccharide chains which are further degraded to glucose by BG (Silveira et al. 2014). Apart from these three major cellulolytic hydrolases, microorganisms also secrete other cellulolytic hydrolase called cellodextrinase, which are found to act on the soluble cello-oligosaccharides resulting in cellobiose and shorter oligosaccharide chains (Ferreira et al. 1991; Huang and Forsberg 1987). Studies have reported that cellodextrinases are highly active on soluble cello-oligosaccharides but were totally inactive against insoluble cellulose (Ferreira et al. 1991; Huang and Forsberg 1987). Generally, cellulases are composed of distinct catalytic domain (CD), a linker and a cellulose-binding module (CBM) (Linder and Teeri 1997). The CBM recognizes and binds to the surface of cellulose and acts upon it by cleaving single cellodextrin chain and feeding it in the active site of the enzyme, where the catalytic domains hydrolyze it to cellobiose (Zhao et al. 2008). Fungal CBMs belonging to the carbohydrate-binding domain family 1 exhibit a small wedge-shaped fold composed of three aromatic amino acid residues which features cellulose-binding surface (Kraulis et al. 1989; Mattinen et al. 1997). Previous studies have reported that the aromatic residues present in the active site of these hydrolases play a crucial role in binding of the CBM to the surface of cellulose (Lehtiö et al. 2003). Simulation studies conducted by Nimlos et al. (2012) have reported that the CBM sites are active in binding on the hydrophilic region of the cellulose than on the hydrophobic surface of substrate (Nimlos et al. 2012). These studies have also reported that the CBM can also diffuse from hydrophilic to the hydrophobic regions of the substrates surface, but the opposite is not possible from these simulation experiments (Nimlos et al. 2012). As CBMs play a crucial role in cellulose hydrolysis, it will be incomplete if we do not consider CBM encoding genes in determining the genomic cellulolytic ability. The cellulose-binding modules are distributed among 20 CBM classes.
Degradation of cellulose involves not only glycoside hydrolases but also strong oxidases such as lytic polysaccharide monooxygenases (LPMO). Discovery of LPMO and its involvement in cellulose degradation was considered as a breakthrough in the field of biofuel production, as LPMO cleaves the glycosidic bonds of cellulose and makes it highly susceptible for other cellulolytic hydrolases to act upon it (Harris et al. 2014; Hemsworth et al. 2015; Johansen 2016). The cellulolytic activity of LPMO is triggered by a reducing agent which activates the oxygen present on copper active site positioned on the surface of the enzyme. Electron donors such as ascorbate, gallic acid/pyrogallol, sulfur-containing compounds and other small molecule reductants triggers the activity of various systems including LPMOs, cellobiose dehydrogenase and GMC (glucose–methanol–choline) oxidoreductases. However, recent studies have also reported the involvement of complex enzymes such as LPMO, cellobiose dehydrogenase (CDH), other glycoside hydrolases and enzymes involved in Fenton’s mechanism was found to play a role in conversion and degradation of cellulose (Beeson et al. 2015; Phillips et al. 2011). Studies have also reported that fungal or plant-derived phenols and plant pigments like chlorophyll can also trigger LPMO activity (Garajova et al. 2016; Kracher et al. 2016; Westereng et al. 2015). The cellulolytic LPMOs are classified among the classes AA9, AA10 and AA15, with cellobiose dehydrogenases and GMC-oxidoreductases classified in AA3 class. The oxidative cleavage of cellulose by LPMO results in aldonic acids (glucose units oxidized on C-1 positions) and gemdiols (4-ketoaldoses, if oxidized on C-4 position) (Villares et al. 2017) (Table 3).
Results obtained from the systematic review have shown that the selected glomeromycota fungal genomes lack genes encoding for cellulolytic glycoside hydrolase families GH1, GH2, GH3, GH6, GH7, GH8, GH12, GH38, GH45, GH48, GH74 and GH124. The selected mortierellomycotina and zoopagomycotina fungal genomes have also experienced the loss of glycoside hydrolase families GH1, GH2, GH6, GH7, GH12, GH48, GH74 and GH124. Similarly, the selected chytridiomycota and blastocladiomycota fungal genomes lack genes encoding for GH6, GH7, GH8, GH12, GH48, GH74 and GH124 families. Contrastingly, the analyzed microsporidia and cryptomycota fungal genomes have experienced complete loss of cellulolytic glycoside hydrolase families. Interestingly, the selected neocallimastigomycota phyla fungal genomes encode highest number of genes coding for cellulolytic glycoside hydrolase class enzymes compared to other selected fungi. Neocallimastigomycota phyla consist of different anaerobic fungi; these fungi were reported to develop efficient cellulosomes and hydrogenosomes aiding them in degradation of plant cell wall carbohydrate components (Kameshwar and Qin 2018). Thus, neocallimastigomycota phyla fungi stand out when compared, as they encode higher number of genes coding for carbohydrate-binding modules (CBM), dockerin proteins and glycoside hydrolases (Kameshwar and Qin 2018). The descending order of fungi based on their cellulolytic enzymes is: neocallimastigomycota > mucoromycotina > chytridiomycota > glomeromycota > monoblepharidomycetes, > mortierellomycotina > entomophthoromycotina > kickxellomycotina > blastocladiomycota > zoopagomycotina > cryptomycota > microsporidia (Table 7).
Hemicellulolytic potentials
Hemicellulose is a complex hetero-polysaccharide composed of glucomannan, xylan, glucuronoxylan, arabinoxylan and xyloglucan, In plant cell walls, hemicellulose is found in close associations with lignin, cellulose and pectin units (Scheller and Ulvskov 2010; Sista Kameshwar and Qin 2018). Thus, structurally complex hemicellulose depends on a wide range of enzymes for its degradation and conversion to simple monomers. Microorganisms secrete a wide range of hemicellulose-degrading enzymes including glycoside hydrolases, LPMO (auxiliary activity) and carbohydrate-binding modules. Studies have reported that xylan constitutes a major energy source during microbial fermentation, especially rumen microbiota (Thomson 1993). Hemicellulose is majorly composed of xylan compared to the other sugar constituents. Degradation and conversion of xylan is performed by three classes of enzymes such as endo-β-1-4-xylanase (EnX), exo-β-1-4-xylanase (ExX) and β-1-4-d-xylosidases (BX) (Saha and Bothast 1999; Subramaniyan and Prema 2002). The EnX randomly cleaves xylan backbone from inside resulting in long chains of xylan oligomers, and later BX cleaves the above obtained xylo-oligomers to xylose monomers (Saha and Bothast 1999; Subramaniyan and Prema 2002). Contrastingly, ExX attacks directly on the reducing ends of xylan backbone resulting in short chains of xylan oligomers with degree of polymerization > 2 to 3 further releasing xyloses from the oligomer (Ganju et al. 1989; Honda and Kitaoka 2004; Juturu and Wu 2014; Kubata et al. 1994). α-l-arabinofuranosidase are second most important class of hemicellulolytic enzymes which are involved in breakdown of arabinoxylans, arabinogalactans. Studies have also reported that xylanases, acetyl xylan esterases and α-l-arabinofuranosidase exhibit a strong synergism during the degradation of xylan chains. Similarly, enzymes such as β-glucosidases, β-mannosidases, glucan β-1,3-glucosidase, mannan endo-β-1,4-mannosidase, xyloglucan-specific endo-β-1,4-glucanase, glucuronoarabinoxylan-specific endo-β-1,4-xylanase, arabinoxylan-specific endo-β-1,4-xylanase are involved in degradation of polymeric chains of glucuronoxylan, arabinoxylans, glucomannan and xyloglucans. Hemicellulose is differentially esterified by O-acetyl and methyl groups, which immediately ceases the enzymes activity toward hemicellulose (Sista Kameshwar and Qin 2018). However, fungi secrete a wide range of carbohydrate esterases for N- and O-deacetylation of hemicellulosic chains. The hemicellulose deacetylating acetyl xylan esterases are classified under the carbohydrate esterase families CE1, CE2, CE3, CE4, CE5, CE6 and CE7 (Sista Kameshwar and Qin 2018) (Table 4).
Results obtained from this systematic analysis have shown that selected glomeromycota fungi lack genes encoding for GH10, GH11, GH30, GH38, GH39, GH43, GH45, GH53 and GH115 and contain genes coding for CE4 and CE16 class enzymes. Similarly, the analyzed mortierellomycotina and zoopagomycotina fungal genomes completely lack genes encoding for GH10, GH11, GH30, GH39, GH53, GH115, CE1 and CE3 families. The analyzed fungal genomes belonging to blastocladiomycota and chytridiomycota lack genes encoding for GH11, GH39, GH43, GH45, GH115, CE1, CE2, CE3 and CE6 families. Compared to other selected fungal genomes, the analyzed microsporidia fungal genomes completely lack genes coding for hemicellulolytic glycoside hydrolases, whereas the selected cryptomycota fungi only code for GH31, GH38, GH47 and CE4 families. The selected neocallimastigomycota fungal genomes outnumbers in total number of hemicellulolytic CAZymes. However, the lowest number of hemicellulolytic glycoside hydrolase encoding genes were observed in microsporidia and cryptomycota with 1 and 13 (Table 7).
Pectinolytic potentials
Pectin is also heterogeneous in nature and are richly composed of D-galacturonic acid. In plant cell walls, it occurs as galacturonans, rhamnogalacturonans-I and II and it contains anhydrogalacturonic acid backbone which is partially esterified (by methyl groups) and acetylated (on C-2 and C-3 hydroxyl groups). Fungal degradation of pectin is performed by protopectinases, endo and exo-polygalacturonases and pectin methyl esterases. The pectinolytic enzymes are mostly distributed among the GH28, GH78, GH95, GH105, GH115 glycoside hydrolase families, CE8, CE12, CE16 carbohydrate esterases families and PL1, PL3, PL4, PL9, PL11 polysaccharide lyases families (Table 5).
Our systematic review revealed that the selected fungi belonging to glomeromycota (except for CE16 class), microsporidia and cryptomycota phyla have experienced serious loss of pectinolytic enzymes. The analyzed mortierellomycotina and zoopagomycotina fungal genomes completely lack genes encoding for PL1 (except Linpe1), PL3 (except Morel2 and Conco1), PL4, PL9, PL11, GH78, GH95 (except Lobtra1 and Morel2), GH105 (except Liccor1and Synrac1), GH115 and CE12 class enzymes. Similarly, the selected chytridiomycota and blastocladiomycota phyla fungal genomes also completely lack genes encoding for pectinolytic enzymes PL1(except Ganpr1), PL3 (except Catan2 and Ganpr1), PL4, PL9, PL11, GH28 (except Ganpr1 and Rhihy1), GH78(except Ganpr1), GH95 and GH105 (Ganpr1), GH115 and CE8 (except Ganpr1), CE12 (except Ganpr1). Compared to other selected phyla fungi, neocallimastigomycota phyla fungal genomes contain several genes encoding for all the pectinolytic enzymes distributed among glycoside hydrolases, carbohydrate esterases and polysaccharide lyases except PL9 (except Anasp1) and PL11 (except Neosp1). Importantly, the selected fungi belonging to microsporidia and cryptomycota phylum exhibited a complete loss of pectinolytic genes (Table 7).
Starch- and inulin-degrading potentials
Starch biosynthesis and depolymerizing CAZymes are distributed among glycoside hydrolases, glycosyl transferases, lytic polysaccharide monooxygenases, carbohydrate-binding modules. The amylases including α-amylases, β-amylase, iso-amylases, glucoamylases are distributed among GH13, GH14, GH57, GH119 and GH126 classes, The inulin-depolymerizing CAZymes are distributed among glycoside hydrolases (GH32 and GH91) and CBM38 class (Kelly 2008; Mensink et al. 2015; Ronkart et al. 2007) (Table 6). Results obtained from our systematic review have shown that all the selected fungal genomes lack several CAZyme families including GH4, GH14, GH57, GH119, GH122 GH126, CBM34, CBM45, CBM53, CBM69, CBM74, CBM82 and AA13 (Table 6). We have observed that among the selected fungal genomes neocallimastigomycota fungi exhibited highest number of starch- and inulin-degrading CAZymes. Contrastingly, selected microsporidia, cryptomycota, zoopagomycota fungal genomes exhibited lowest number of starch- and inulin-degrading CAZymes (Table 7).
Conclusion
Several studies were continuously being conducted in the last two decades to understand and reveal the plant biomass-degrading abilities of fungi. The development of next-generation sequencing studies has also significantly helped in understanding the genomic complexities and molecular mechanisms underlying several biological processes. However, most of these sequencing studies were mainly focused on fungi belonging to Basidiomycota and Ascomycota phyla. In our present study, we have retrieved and compared the genome-wide annotations of fungi belonging to glomeromycota, zygomycota, chytridiomycota, blastocladiomycota, neocallimastigomycota, microsporidia and cryptomycota phyla. We have specifically analyzed and compared the genes encoding for plant cell wall-degrading enzymes and the molecular mechanisms involved in plant biomass degradation. Results obtained in our study show that fungi belonging to microsporidia and cryptomycota have experienced serious loss of several genes encoding for plant cell wall component-degrading enzymes. Contrastingly, the analyzed fungi belonging to neocallimastigomycota phyla have exhibited extraordinary genomic potentials to degrade plant cell wall carbohydrates. The analyzed fungi belonging to glomeromycota have exhibited higher number of genes distributed under the xenobiotic biodegradation metabolism pathways compared to all the other selected fungi. Results obtained in our study can be used for finding efficient fungal strains from these selected phyla for developing commercially valuable products. However, analyzing just the genome-wide distribution of fungal CAZymes is not enough as it does not completely correspond to the fungal lignocellulolytic abilities. Thus, understanding the genome-wide CAZyme distributions mainly highlights the lignocellulolytic potential of the selected fungi. Developing efficient recombinant microbial strains will have various industrial benefits including (a) biofuel industries (conversion of plant biomass components to commercially valuable products), (b) bioremediation industries (detoxification and biodegradation of toxic environmental pollutants).
Availability of data and materials
Not applicable
Abbreviations
- CAZy:
-
carbohydrate active enzymes
- KOG:
-
eukaryotic orthologous groups
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GH:
-
glycoside hydrolases
- GT:
-
glycosyl transferases
- CBM:
-
carbohydrate-binding modules
- PL:
-
polysaccharide lyases
- CE:
-
carbohydrate esterases
- AA:
-
auxiliary activity
- AAM:
-
amino acid metabolism
- BpNp:
-
biosynthesis of polyketides and non-ribosomal peptides
- BSM:
-
biosynthesis of secondary metabolites
- CM:
-
carbohydrate metabolism
- EM:
-
energy metabolism
- GBM:
-
glycan biosynthesis and metabolism
- LM:
-
lipid metabolism
- MCV:
-
metabolism of cofactors and vitamins
- MAA:
-
metabolism of other amino acids
- NM:
-
nucleotide metabolism
- O:
-
overview
- XBM:
-
xenobiotics biodegradation and metabolism
- V:
-
fungal defense mechanisms
- G:
-
carbohydrate transport and metabolism
- Q:
-
secondary metabolite biosynthesis, transport and catabolism
- C:
-
energy production and conversion
- SM clusters:
-
secondary metabolite clusters
- NRPS:
-
non-ribosomal peptide synthases
- PKS:
-
polyketide synthases
- TC:
-
terpene cyclases
References
Ahrendt SR, Quandt CA, Ciobanu D, Clum A, Salamov A, Andreopoulos B, Cheng J-F, Woyke T, Pelin A, Henrissat B (2018) Leveraging single-cell genomics to expand the fungal tree of life. Nat Microbiol 3:1417
Akiyoshi DE, Morrison HG, Lei S, Feng X, Zhang Q, Corradi N, Mayanja H, Tumwine JK, Keeling PJ, Weiss LM (2009) Genomic survey of the non-cultivatable opportunistic human pathogen, Enterocytozoon bieneusi. PLoS pathogens 5:e1000261
Alexopoulous C, Mims C, Blackwell M (1996) Phylum chytridiomycota introductory mycology. Wiley, New York, pp 86–126
Baldauf SL, Palmer JD (1993) Animals and fungi are each other’s closest relatives: congruent evidence from multiple proteins. Proc Natl Acad Sci 90:11558–11562
Barr DJ (1992) Evolution and kingdoms of organisms from the perspective of a mycologist. Mycologia 84:1–11
Bartnicki-Garcia S (1970) Cell wall composition and other biochemical markers in fungal phylogeny. Phytochemical phylogeny. Academic Press, London
Beeson WT, Vu VV, Span EA, Phillips CM, Marletta MA (2015) Cellulose degradation by polysaccharide monooxygenases. Annu Rev Biochem 84:923–946
Berbee M, Taylor J (2001a) Systematics and evolution. Springer, Berlin, pp 229–245
Berbee ML, Taylor JW (2001b) Fungal molecular evolution: gene trees and geologic time. Systematics and evolution. Springer, Berlin, pp 229–245
Bruns T (2006) Evolutionary biology: a kingdom revised. Nature 443:758
Cameron MD, Aust SD (2001) Cellobiose dehydrogenase–an extracellular fungal flavocytochrome. Enzyme Microbial Technol 28:129–138
Chang Y, Wang S, Sekimoto S, Aerts AL, Choi C, Clum A, LaButti KM, Lindquist EA, Yee Ngan C, Ohm RA (2015) Phylogenomic analyses indicate that early fungi evolved digesting cell walls of algal ancestors of land plants. Genome Biol Evol 7:1590–1601
Chang Y, Desirò A, Na H, Sandor L, Lipzen A, Clum A, Barry K, Grigoriev IV, Martin FM, Stajich JE (2019) Phylogenomics of Endogonaceae and evolution of mycorrhizas within Mucoromycota. New Phytol 222:511–525
Chen EC, Morin E, Beaudet D, Noel J, Yildirir G, Ndikumana S, Charron P, St-Onge C, Giorgi J, Krüger M (2018) High intraspecific genome diversity in the model arbuscular mycorrhizal symbiont Rhizophagus irregularis. New Phytologist 220(4):1161–1171
Cornman RS, Chen YP, Schatz MC, Street C, Zhao Y, Desany B, Egholm M, Hutchison S, Pettis JS, Lipkin WI (2009) Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathog 5:e1000466
Corradi N, Selman M (2013) Latest progress in microsporidian genome research. J Eukaryot Microbiol 60:309–312
Corradi N, Pombert J-F, Farinelli L, Didier ES, Keeling PJ (2010) The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat Commun 1:77
Corrochano LM, Kuo A, Marcet-Houben M, Polaino S, Salamov A, Villalobos-Escobedo JM, Grimwood J, Álvarez MI, Avalos J, Bauer D (2016) Expansion of signal transduction pathways in fungi by extensive genome duplication. Curr Biol 26:1577–1584
Cuomo CA, Desjardins CA, Bakowski MA, Goldberg J, Ma AT, Becnel JJ, Didier ES, Fan L, Heiman DI, Levin JZ (2012) Microsporidian genome analysis reveals evolutionary strategies for obligate intracellular growth. Genome Res 22:2478–2488
Diyarova DK (2016) The role of wood-decaying fungi in the carbon cycle of forest ecosystems and the main ecological factors. Eur Sci J, ESJ, p 12
Douaiher MN, Nowak E, Durand R, Halama P, Reignault P (2007) Correlative analysis of Mycosphaerella graminicola pathogenicity and cell wall-degrading enzymes produced in vitro: the importance of xylanase and polygalacturonase. Plant Pathol 56:79–86
Ferrari S, Galletti R, Pontiggia D, Manfredini C, Lionetti V, Bellincampi D, Cervone F, De Lorenzo G (2008) Transgenic expression of a fungal endo-polygalacturonase increases plant resistance to pathogens and reduces auxin sensitivity. Plant Physiol 146:669–681
Ferreira L, Hazlewood GP, Barker PJ, Gilbert HJ (1991) The cellodextrinase from Pseudomonas fluorescens subsp. cellulosa consists of multiple functional domains. Biochem J 279:793–799
Ganju RK, Vithayathil PJ, Murthy S (1989) Purification and characterization of two xylanases from Chaetomium thermophile var. coprophile. Can J Microbiol 35:836–842
Garajova S, Mathieu Y, Beccia MR, Bennati-Granier C, Biaso F, Fanuel M, Ropartz D, Guigliarelli B, Record E, Rogniaux H (2016) Single-domain flavoenzymes trigger lytic polysaccharide monooxygenases for oxidative degradation of cellulose. Sci Rep 6:28276
Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA (2011) The genome portal of the department of energy joint genome institute. Nucleic Acids Res 40:D26–D32
Haitjema CH, Gilmore SP, Henske JK, Solomon KV, de Groot R, Kuo A, Mondo SJ, Salamov AA, LaButti K, Zhao Z (2017) A parts list for fungal cellulosomes revealed by comparative genomics. Nat Microbiol 2:17087
Harris PV, Xu F, Kreel NE, Kang C, Fukuyama S (2014) New enzyme insights drive advances in commercial ethanol production. Curr Opin Chem Biol 19:162–170
Heckman DS, Geiser DM, Eidell BR, Stauffer RL, Kardos NL, Hedges SB (2001) Molecular evidence for the early colonization of land by fungi and plants. Science 293:1129–1133
Hedges SB, Kumar S (2009) The timetree of life OUP. Oxford University, Oxford
Hedges SB, Marin J, Suleski M, Paymer M, Kumar S (2015) Tree of life reveals clock-like speciation and diversification. Mol Biol Evol 32:835–845
Hemsworth GR, Johnston EM, Davies GJ, Walton PH (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol 33:747–761
Henriksson G, Ander P, Pettersson B, Pettersson G (1995) Cellobiose dehydrogenase (cellobiose oxidase) from Phanerochaete chrysosporium as a wood-degrading enzyme. Studies on cellulose, xylan and synthetic lignin. Appl Microbiol Biotechnol 42:790–796
Henriksson G, Zhang L, Li J, Ljungquist P, Reitberger T, Pettersson G, Johansson G (2000) Is cellobiose dehydrogenase from Phanerochaete chrysosporium a lignin degrading enzyme? Biochem Biophys Acta 1480:83–91
Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, Kirk PM, Lücking R (2007) A higher-level phylogenetic classification of the Fungi. Mycol Res 111:509–547
Hoffman GL (1999) Parasites of North American freshwater fishes. Cornell University Press, Ithaca
Honda Y, Kitaoka M (2004) A family 8 glycoside hydrolase from Bacillus halodurans C-125 (BH2105) is a reducing end xylose-releasing exo-oligoxylanase. J Biol Chem 279:55097–55103
Huang L, Forsberg CW (1987) Isolation of a cellodextrinase from Bacteroides succinogenes. Appl Environ Microbiol 53:1034–1041
James TY, Porter D, Leander CA, Vilgalys R, Longcore JE (2000) Molecular phylogenetics of the Chytridiomycota supports the utility of ultrastructural data in chytrid systematics. Can J Bot 78:336–350
James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ, Celio G, Gueidan C, Fraker E, Miadlikowska J (2006a) Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443:818
James TY, Letcher PM, Longcore JE, Mozley-Standridge SE, Porter D, Powell MJ, Griffith GW, Vilgalys R (2006b) A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98:860–871
James TY, Pelin A, Bonen L, Ahrendt S, Sain D, Corradi N, Stajich JE (2013) Shared signatures of parasitism and phylogenomics unite cryptomycota and microsporidia. Curr Biol 23:1548–1553
Johansen KS (2016) Discovery and industrial applications of lytic polysaccharide mono-oxygenases. Biochem Soc Trans 44:143–149
Juturu V, Wu JC (2014) Microbial exo-xylanases: a mini review. Appl Biochem Biotechnol 174:81–92
Kameshwar A, Qin W (2016a) Recent developments in using advanced sequencing technologies for the genomic studies of lignin and cellulose degrading microorganisms. Int J Biol Sci 12:156–171
Kameshwar AKS, Qin W (2016b) Lignin degrading fungal enzymes production of biofuels and chemicals from lignin. Springer, Berlin, pp 81–130
Kameshwar AKS, Qin W (2017) Gene expression metadata analysis reveals molecular mechanisms employed by Phanerochaete chrysosporium during lignin degradation and detoxification of plant extractives. Curr Genet 63:877–894
Kameshwar AKS, Qin W (2018) Genome wide analysis reveals the extrinsic cellulolytic and biohydrogen generating abilities of neocallimastigomycota fungi. J Genomics 6:74
Karol KG, McCourt RM, Cimino MT, Delwiche CF (2001) The closest living relatives of land plants. Science 294:2351–2353
Kelly G (2008) Inulin-type prebiotics—a review: part 1. Altern Med Rev 13:4
Kendrick B (2000) The fifth kingdom 3rd. City name The USA. Focus Publishing, Bemidji
Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND (2010) SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol 47:736–741
Kikot GE, Hours RA, Alconada TM (2009) Contribution of cell wall degrading enzymes to pathogenesis of Fusarium graminearum: a review. J Basic Microbiol 49:231–241
King BC, Waxman KD, Nenni NV, Walker LP, Bergstrom GC, Gibson DM (2011) Arsenal of plant cell wall degrading enzymes reflects host preference among plant pathogenic fungi. Biotechnol Biofuels 4:4
Kracher D, Scheiblbrandner S, Felice AK, Breslmayr E, Preims M, Ludwicka K, Haltrich D, Eijsink VG, Ludwig R (2016) Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 352:1098–1101
Kraulis PJ, Clore GM, Nilges M, Jones TA, Pettersson G, Knowles J, Gronenborn AM (1989) Determination of the three-dimensional solution structure of the C-terminal domain of cellobiohydrolase I from Trichoderma reesei. A study using nuclear magnetic resonance and hybrid distance geometry-dynamical simulated annealing. Biochemistry 28:7241–7257
Krishna MP, Mohan M (2017) Litter decomposition in forest ecosystems: a review. Energy Ecol Environ 2(4):236–249
Kubata BK, Suzuki T, Horitsu H, Kawai K, Takamizawa K (1994) Purification and characterization of Aeromonas caviae ME-1 xylanase V, which produces exclusively xylobiose from xylan. Appl Environ Microbiol 60:531–535
Lara E, Moreira D, López-García P (2010) The environmental clade LKM11 and Rozella form the deepest branching clade of fungi. Protist 161:116–121
Lastovetsky OA, Gaspar ML, Mondo SJ, LaButti KM, Sandor L, Grigoriev IV, Henry SA, Pawlowska TE (2016) Lipid metabolic changes in an early divergent fungus govern the establishment of a mutualistic symbiosis with endobacteria. Proc Natl Acad Sci 2016:15148
Lehtiö J, Sugiyama J, Gustavsson M, Fransson L, Linder M, Teeri TT (2003) The binding specificity and affinity determinants of family 1 and family 3 cellulose binding modules. Proc Natl Acad Sci 100:484–489
Linder M, Teeri TT (1997) The roles and function of cellulose-binding domains. J Biotechnol 57:15–28
Ma L-J, Ibrahim AS, Skory C, Grabherr MG, Burger G, Butler M, Elias M, Idnurm A, Lang BF, Sone T (2009) Genomic analysis of the basal lineage fungus Rhizopus oryzae reveals a whole-genome duplication. PLoS Genet 5:e1000549
Macheleidt J, Mattern DJ, Fischer J, Netzker T, Weber J, Schroeckh V, Valiante V, Brakhage AA (2016) Regulation and role of fungal secondary metabolites. Annu Rev Genet 50:371–392
Mattinen M-L, Linder M, Teleman A, Annila A (1997) Interaction between cellohexaose and cellulose binding domains from Trichoderma reesei cellulases. FEBS Lett 407:291–296
McLaughlin D, McLaughlin E, Lemke P (2001) The Mycota. VII. Systematics and evolution, part B. The mycota. Springer, Berlin
Mensink MA, Frijlink HW, van der Voort Maarschalk K, Hinrichs WL (2015) Inulin, a flexible oligosaccharide I: review of its physicochemical characteristics. Carbohyd Polym 130:405–419
Mondo SJ, Dannebaum RO, Kuo RC, Louie KB, Bewick AJ, LaButti K, Haridas S, Kuo A, Salamov A, Ahrendt SR (2017) Widespread adenine N6-methylation of active genes in fungi. Nat Genet 49:964
Morin E, Miyauchi S, San Clemente H, Chen EC, Pelin A, de la Providencia I, Ndikumana S, Beaudet D, Hainaut M, Drula E (2019) Comparative genomics of Rhizophagus irregularis, R. cerebriforme, R. diaphanus and Gigaspora rosea highlights specific genetic features in Glomeromycotina. New Phytol 222:1584–1598
Morton JB, Benny GL (1990) Revised classification of arbuscular mycorrhizal fungi (Zygomycetes): a new order, Glomales, two new suborders, Glomineae and Gigasporineae, and two new families, Acaulosporaceae and Gigasporaceae, with an emendation of Glomaceae. Mycotaxon 37:471–491
Nimlos MR, Beckham GT, Matthews JF, Bu L, Himmel ME, Crowley MF (2012) Binding preferences, surface attachment, diffusivity, and orientation of a family 1 carbohydrate-binding module on cellulose. J Biol Chem 287:20603–20612
Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I, Smirnova T, Grigoriev IV, Dubchak I (2013) The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res 42:D26–D31
Orpin C (1975) Studies on the rumen flagellate Neocallimastix frontalis. Microbiology 91:249–262
Papanikolaou S, Galiotou-Panayotou M, Fakas S, Komaitis M, Aggelis G (2007) Lipid production by oleaginous Mucorales cultivated on renewable carbon sources. Eur J Lipid Sci Technol 109:1060–1070
Peyretaillade E, Gonçalves O, Terrat S, Dugat-Bony E, Wincker P, Cornman RS, Evans JD, Delbac F, Peyret P (2009) Identification of transcriptional signals in Encephalitozoon cuniculi widespread among Microsporidia phylum: support for accurate structural genome annotation. BMC Genom 10:607
Phillips CM, Beeson WT IV, Cate JH, Marletta MA (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem Biol 6:1399–1406
Pombert J-F, Selman M, Burki F, Bardell FT, Farinelli L, Solter LF, Whitman DW, Weiss LM, Corradi N, Keeling PJ (2012) Gain and loss of multiple functionally related, horizontally transferred genes in the reduced genomes of two microsporidian parasites. Proc Natl Acad Sci 109:12638–12643
Raven P, Evert R, Eichhorn S (2005) Biology of plants, 7th edn. WH Freeman and Co., New York
Redecker D, Schüßler A, Stockinger H, Stürmer SL, Morton JB, Walker C (2013) An evidence-based consensus for the classification of arbuscular mycorrhizal fungi (Glomeromycota). Mycorrhiza 23:515–531
Ronkart SN, Blecker CS, Fourmanoir H, Fougnies C, Deroanne C, Van Herck J-C, Paquot M (2007) Isolation and identification of inulooligosaccharides resulting from inulin hydrolysis. Anal Chim Acta 604:81–87
Russ C, Lang BF, Chen Z, Gujja S, Shea T, Zeng Q, Young S, Cuomo CA, Nusbaum C (2016) Genome sequence of Spizellomyces punctatus. Genome Announc 4:e00849–e00916
Rytioja J, Hildén K, Yuzon J, Hatakka A, de Vries RP, Mäkelä MR (2014) Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol Mol Biol Rev 78:614–649
Saha BC, Bothast RJ (1999) Enzymology of xylan degradation ACS symposium series. Am Chem Soc, Washington, pp 167–194
Scheller HV, Ulvskov P (2010) Hemicelluloses. Annu Rev Plant Biol 4:61
Schüßler A, Gehrig H, Schwarzott D, Walker C (2001a) Analysis of partial Glomales SSU rRNA gene sequences: implications for primer design and phylogeny. Mycol Res 105:5–15
Schüßler A, Schwarzott D, Walker C (2001b) A new fungal phylum, the Glomeromycota: phylogeny and evolution. Mycol Res 105:1413–1421
Schwartze VU, Winter S, Shelest E, Marcet-Houben M, Horn F, Wehner S, Linde J, Valiante V, Sammeth M, Riege K (2014) Gene expansion shapes genome architecture in the human pathogen Lichtheimia corymbifera: an evolutionary genomics analysis in the ancient terrestrial mucorales (Mucoromycotina). PLoS Genet 10:e1004496
Silar P (2016) Protistes Eucaryotes: origine, evolution et biologie des microbes eucaryotes. HAL Arch Ouver 53:462
Silveira MHL, Aguiar RS, Siika-aho M, Ramos LP (2014) Assessment of the enzymatic hydrolysis profile of cellulosic substrates based on reducing sugar release. Bioresour Technol 151:392–396
Sista Kameshwar AK, Qin W (2017) Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 9:1–13
Sista Kameshwar AK, Qin W (2018) Understanding the structural and functional properties of carbohydrate esterases with a special focus on hemicellulose deacetylating acetyl xylan esterases. Mycology 9:1–23
Skamnioti P, Furlong RF, Gurr SJ (2008) The fate of gene duplicates in the genomes of fungal pathogens. Commun Integr Biol 1:196–198
Slamovits CH, Fast NM, Law JS, Keeling PJ (2004) Genome compaction and stability in microsporidian intracellular parasites. Curr Biol 14:891–896
Smith S, Read D (1997) Mycorrhizal symbiosis, 2nd edn. Academic Press, Cambridge
Spatafora JW, Chang Y, Benny GL, Lazarus K, Smith ME, Berbee ML, Bonito G, Corradi N, Grigoriev I, Gryganskyi A (2016) A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108:1028–1046
Subramaniyan S, Prema P (2002) Biotechnology of microbial xylanases: enzymology, molecular biology, and application. Crit Rev Biotechnol 22:33–64
Summerbell RC (2005) Root endophyte and mycorrhizosphere fungi of black spruce, Picea mariana, in a boreal forest habitat: influence of site factors on fungal distributions. Stud Mycol 53:121–145
Tanabe Y, Saikawa M, Watanabe MM, Sugiyama J (2004) Molecular phylogeny of Zygomycota based on EF-1α and RPB1 sequences: limitations and utility of alternative markers to rDNA. Mol Phylogenet Evol 30:438–449
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Thomson JA (1993) Molecular biology of xylan degradation. FEMS Microbiol Lett 104:65–82
Tisserant E, Malbreil M, Kuo A, Kohler A, Symeonidi A, Balestrini R, Charron P, Duensing N, Frey NF, Gianinazzi-Pearson V (2013) Genome of an arbuscular mycorrhizal fungus provides insight into the oldest plant symbiosis. Proc Natl Acad Sci 110:20117–20122
Uehling J, Gryganskyi A, Hameed K, Tschaplinski T, Misztal P, Wu S, Desirò A, Vande Pol N, Du Z, Zienkiewicz A (2017) Comparative genomics of Mortierella elongata and its bacterial endosymbiont Mycoavidus cysteinexigens. Environ Microbiol 19(8):2964–2983
Villares A, Moreau C, Bennati-Granier C, Garajova S, Foucat L, Falourd X, Saake B, Berrin J-G, Cathala B (2017) Lytic polysaccharide monooxygenases disrupt the cellulose fibers structure. Sci Rep 7:40262
Wal A, Geydan TD, Kuyper TW, Boer W (2013) A thready affair: linking fungal diversity and community dynamics to terrestrial decomposition processes. FEMS Microbiol Rev 37:477–494
Wang L, Chen W, Feng Y, Ren Y, Gu Z, Chen H, Wang H, Thomas MJ, Zhang B, Berquin IM (2011) Genome characterization of the oleaginous fungus Mortierella alpina. PLoS ONE 6:e28319
Wang D, Wu R, Xu Y, Li M (2013) Draft genome sequence of Rhizopus chinensis CCTCCM201021, used for brewing traditional Chinese alcoholic beverages. Genome Announc 1:e00195–e00212
Westereng B, Cannella D, Agger JW, Jørgensen H, Andersen ML, Eijsink VG, Felby C (2015) Enzymatic cellulose oxidation is linked to lignin by long-range electron transfer. Sci Rep 5:18561
White MM, James TY, O’Donnell K, Cafaro MJ, Tanabe Y, Sugiyama J (2006) Phylogeny of the Zygomycota based on nuclear ribosomal sequence data. Mycologia 98:872–884
Youssef NH, Couger M, Struchtemeyer CG, Liggenstoffer AS, Prade RA, Najar FZ, Atiyeh HK, Wilkins MR, Elshahed MS (2013) The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Appl Environ Microbiol 79:4620–4634
Zhao X, Rignall TR, McCabe C, Adney WS, Himmel ME (2008) Molecular simulation evidence for processive motion of Trichoderma reesei Cel7A during cellulose depolymerization. Chem Phys Lett 460:284–288
Zhao Z, Liu H, Wang C, Xu J-R (2013) Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 14(1):274
Acknowledgements
Not applicable.
Funding
This work was supported by Natural Sciences and Engineering Research Council of Canada Funding (RGPIN-2017-05366) to Wensheng Qin and Ontario Trillium Scholarship (OTS) to Ayyappa Kumar Sista Kameshwar.
Author information
Authors and Affiliations
Contributions
AKSK is involved in collecting, reviewing the literature and performing the systematic review of the publicly available non-Dikaryon fungal genomes. WQ is involved in guiding the analysis, improving and revising the manuscript. Both AKSK and WQ are involved in writing the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sista Kameshwar, A.K., Qin, W. Systematic review of publicly available non-Dikarya fungal proteomes for understanding their plant biomass-degrading and bioremediation potentials. Bioresour. Bioprocess. 6, 30 (2019). https://doi.org/10.1186/s40643-019-0264-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-019-0264-6