
Abstract
Perovskite-type oxynitrides hold great potential for optical applications due to their excellent visible light absorption properties. However, only a limited number of such oxynitrides with modulated physical properties are available to date and therefore alternative fabrication strategies are needed to be developed. Here, we introduce such an alternative strategy involving a precursor microstructure controlled ammonolysis. This leads to the perovskite family member LaTa(IV)O2N containing unusual Ta4+ cations. The adjusted precursor microstructures as well as the ammonia concentration are the key parameters to precisely control the oxidation state and O:N ratio in LaTa(O,N)3. LaTa(IV)O2N has a bright red colour, an optical bandgap of 1.9 eV and a low (optically active) defect concentration. These unique characteristics make this material suitable for visible light-driven applications and the identified key parameters will set the terms for the targeted development of further promising perovskite family members.
Similar content being viewed by others

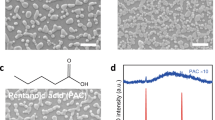

Introduction
In spite of the growing interest in perovskite materials, the number of oxynitride members is still limited. Most oxynitrides contain metal cations in d0 or d10 electronic configuration1,2,3,4,5,6,7,8, whereas alternative materials with B-site cations in e.g. d1 configuration such as Ta4+ are largely lacking. However, the latter could allow access to different electronic band structures, thus, expanding the applicability of perovskite-type oxynitrides in visible light-driven processes. The negligible number of known perovskite-type oxynitrides might be attributed to the reaction protocols normally used. Currently, the most widely used synthesis technique for this material class is the ammonolysis of oxides9,10. This procedure enables tuning of the electronic bandgap via (partial) substitution of oxygen by nitrogen1. Typically, the bandgap decreases, making the formed perovskite-type oxynitrides AB(O,N)3 interesting for visible light-driven applications1,4. An important task in order to identify perovskite-type oxynitrides is the exact determination of the O:N ratio which often causes difficulties11,12.
The precursors are often mixtures of crystalline binary oxides (e.g. La2O3 or Ta2O5)3,4,13 or ternary oxides (e.g. LaTaO4)14,15, which are either prepared by solid state reactions (SSR) or via a Pechini method. The latter allows mild reaction conditions providing excellent product homogeneity2. In the case of LaTa(V)ON2, most studies involve a high-temperature treatment of the oxide precursors prior to ammonolysis, leading to high crystallinity of the precursor13,14,16,17. The formation of several perovskite-type oxynitrides such as LaTiO2N and SrTaO2N from crystalline oxide precursors has been described by a topotactic reaction scheme18. In contrast, the reaction behaviour of amorphous/nanocrystalline oxide precursors is still unclear and the detailed ammonolysis mechanism has yet to be clarified1. A better understanding and control of essential reaction steps might help to develop new synthesis strategies. Furthermore, a target-oriented electronic configuration of the B-site cation and the precise adjustment of the O:N ratio in oxynitrides are the key factors to generate various interesting physical properties.
In this in situ and ex situ experimental study, we override the above-mentioned strong topotactic relation between oxide precursor and resulting LaTa(O,N)3 formation through a considered selection of well-characterised oxide precursors with different microstructures and an adjusted ammonia concentration. Furthermore, we demonstrate the formation of the LaTa(IV)O2N utilising nanocrystalline lanthanum tantalum oxide (n-LTO), which exhibits smaller primary particles (nm-range) and a higher specific surface area than microcrystalline LaTaO4 (m-LaTaO4). This adjusted precursor microstructure leads to a favoured Ta reduction in n-LTO. In contrast, ammonolysis of larger primary particles in the µm-range (m-LaTaO4) results in conventional LaTa(V)ON2. Hence, we expand the experimental toolbox by an additional method to access further requested perovskite-type oxynitride family members.
Results
In situ ammonolysis of lanthanum tantalum oxides
First, the reaction steps of the LaTaO2N (Fig. 1) synthesis were investigated by in situ ammonolysis (10 vol% Ar in NH3) using thermogravimetric analysis (TGA). For a better comparability, similar measurement conditions were selected for both in situ and ex situ ammonolysis (see below). Nanocrystalline lanthanum tantalum oxide (n-LTO) and microcrystalline LaTaO4 (m-LaTaO4) were synthesised as precursors in order to investigate the effect of the microstructure on the reaction behaviour (synthesis and characterisation details in Supplementary Note 1, Supplementary Fig. 1–5 and Supplementary Tables 1–4).

Schematic synthesis path of LaTa(IV)O2N. Initially, a black xerogel is obtained by a Pechini method. A subsequent low temperature calcination leads to white, nanocrystalline lanthanum tantalum oxide (n-LTO). Finally, ammonolysis of the n-LTO under flowing ammonia at higher temperatures yields the desired bright red LaTa(IV)O2N. Scalebar of the SEM images: 2 µm, Scalebar of the inset: 100 nm.
The TGA curve of n-LTO during in situ ammonolysis (Fig. 2) reveals several mass changes, the origin of which were determined by termination experiments. The initial mass change of −0.7% in the range of 25 °C to 588 °C (region I) can be assigned to the desorption of residual water or organic contaminants, which is why the precursor remains white (Fig. 2).

In situ TGA ammonolysis of nanocrystalline lanthanum tantalum oxide (n-LTO). In situ TGA of the ammonolysis (10 vol% Ar in NH3) of n-LTO, including respective powder X-ray diffraction (PXRD) patterns and anionic compositions determined via hot gas extraction (HGE) after selected termination experiments. The illustrative coloured powders at specific temperatures complement the analysis. The reduction of tantalum at 844 °C is highlighted with a red frame. Additionally, the first four mass changes (region I–IV) during in situ ammonolysis of n-LTO are magnified in the inset. The in situ ammonolysis results of the microcrystalline LaTaO4 (cyan curve (micro) in the same inset) can be found below.
The subsequent larger mass change of −2.9% (region II) is accompanied by a local mass minimum at 844 °C. At this temperature, the powder is black and the powder X-ray diffraction (PXRD) pattern shows the onset of crystallisation. In addition, hot gas extraction (HGE) reveals a composition of LaTaO3.44(4)N0.41(2)□0.14(6) (product of the termination experiment at 844 °C) with an assumed ratio of La:Ta:O = 1:1:4 (HGE results are listed in Supplementary Table 5). Therefore, region II (between 588 °C and 844 °C) is characterised by an oxygen vacancy formation with simultaneous nitrogen incorporation. According to literature19, LaTaO4 crystallises in space group A21am up to around 800 °C and exhibits octahedron chains with corresponding interspace (Fig. 1). Therefore, we assume that vacancy formation and nitrogen incorporation takes place in this interspatial region.
At the same time, the colour-change of the powder from white to black indicates a reduction of the contained Ta. As can be shown by X-ray photoelectron spectroscopy (XPS), heating to 844 °C in ammonia atmosphere causes the two existing Ta(V)–O binding characters in n-LTO (Fig. 3a, n-LTO) to change their character. The evaluation of the Ta(V)–O binding characters in n-LTO and m-LaTaO4 is described in Supplementary Note 4. The two new binding energies of the Ta 4f7/2 orbitals in LaTaO3.44(4)N0.41(2)□0.14(6) of EB(Ta 4f7/2) = 25.2 eV and EB(Ta 4f7/2) = 24.4 eV, respectively, can be derived from the applied peak fittings (Fig. 3a, n-LTO, NH3/ 844 °C).

XPS spectra in the Ta 4 f region and point charge model. a XPS spectra in the Ta 4 f region of the intermediates obtained by termination of the in situ ammonolysis at 844 °C and 891 °C, as well as of n-LTO and m-LaTaO4 (measurement data: open black circles and solid black line: overall fit). The Ta 4 f7/2 peaks are marked with their respective fitted binding energy and, in case of m-LaTaO4, additionally with the respective space group of the polymorph (cf. SI) b Applied point charge model20,21 in the Ta 4f region of LaTaO3.44(4)N0.41(2)□0.14(6) (in situ), LaTaO2N (ex situ) and LaTaON2 (ex situ). The Ta(V)–O binding character of m-LaTaO4 (P21/c) was used as reference for Ta5+. The black dots represent the reference binding energies of Ta(0)22 and Ta(V) (measured) connected by the black dashed line. The grey dashed line represents the shifted Ta5+ binding energy (from 25.3 eV to 25.0 eV because of N substitution) of LaTaON2 (ex situ). c XPS spectra in the Ta 4 f region of LaTaO2N and LaTaON2 with the respective fitted binding energies and corresponding binding characters. The open circles represent the measurement data, the solid lines show the overall fit and the dashed lines the fitting results of the respective Ta 4 f orbitals.
Calculations using the equations estimated by Nordling20 (Supplementary Note 5) suggest that substitution of oxygen with nitrogen at persisting Ta5+ causes a chemical shift of 0.3 eV to lower binding energies. The in situ observed conversion of n-LTO to LaTaO3.44(4)N0.41(2)□0.14(6) between 588 °C and 844 °C is accompanied by a Ta 4f7/2 binding energy shift from 25.5 eV to 25.2 eV for Ta(V)–O by nitrogen incorporation. Therefore, the binding energy EB(Ta 4f7/2) = 25.2 eV can be allocated to a Ta(V)–(O,N) binding character since the original binding energy lowers by the expected ∆EB = 0.3 eV. The other Ta 4f7/2 binding energy of Ta(V)–O, however, shifts from 26.7 eV in n-LTO to 24.4 eV in LaTaO3.44(4)N0.41(2)□0.14(6), the difference being much higher than ∆EB = 0.3 eV. This might be explained by a change of the Ta oxidation state in addition to nitrogen substitution. Therefore, the binding energy at EB(Ta 4f7/2) = 24.4 eV can be assigned to a Ta(IV)–(O,N) binding character. The Ta(IV)–(O,N) binding character can be determined by applying the point charge model20,21 assuming that the atomic potential of Ta remains unaffected by a change of the oxidation state. Ta(0) at EB(Ta 4f7/2) = 21.9 eV22 and Ta(V) at EB(Ta 4f7/2) = 25.3 eV in m-LaTaO4 (P21/c) (instead of EB(Ta 4f7/2) = 25.9 eV in m-LaTaO4 (Cmc21)) were selected as references (Fig. 3b). The selection of the Ta(V) binding energy was based on the similar interatomic distances in and between the [TaVO6]7- octahedron together with the amount of neighbouring ions of Ta compared to the respective oxynitride ([Ta(O,N)6]z- octahedron). Thus, due to the linear relationship between the oxidation state and the binding energy in the point charge model, a Ta oxidation state of 4 + (d1 electronic configuration) at EB(Ta 4f7/2) = 24.4 eV was determined (Fig. 3b).
A further mass change of + 1.0% occurs in the temperature range of 844 °C to 891 °C (region III). Simultaneously, the colour of the powder changes from black to ochre. The weight fractions at 891 °C determined by HGE amount to 13.7 wt% O and 6.7 wt% N (compared to 14.4 wt% O and 1.5 wt% N at 844 °C). However, the total mass increase due to the strong nitrogen enrichment accompanied by only a small oxygen loss cannot be explained by a simple refill of the previously generated oxygen vacancies in “LaTaO4”, since the total anionic weight fraction substantially exceeds the calculated maximum value of 16.7 wt% for “LaTaO4” (Supplementary Table 5). Evaluation of the respective powder pattern shows that the ochre-coloured phase is not fully crystallised (Supplementary Note 6 and Supplementary Fig. 6) suggesting the formation of a nitrogen-rich intermediate (proposed composition: LaTa(O,N,□)4:(N2)y). Such intermediates are well-known from reoxidation experiments of several other oxynitrides including LaTiO2N23. The XPS measurement (Fig. 3a, n-LTO, NH3/ 891 °C) again reveals a chemical shift of the Ta 4f7/2 binding energy from 24.4 eV (Ta(IV)–(O,N)) to 24.0 eV (Ta(IV)–N). This indicates an increased nitrogen content in the chemical environment of Ta and, hence, in the whole sample. The other Ta(V)–(O,N) binding character at EB(Ta 4f7/2) = 25.1 eV (previously 25.2 eV) remains unchanged. A further temperature increase to 950 °C (region IV) leads to an abrupt mass change of −6.9%. During the following 10 h ammonolysis prior to cooling to 25 °C the mass remains near-constant. The respective termination experiment indicates a colour change from ochre to red after the 10 h ammonolysis. HGE of the red phase reveals a composition of LaTaO1.44(1)N1.63(9). This phase is further transformed to red LaTaO1.26(9)N1.83(4) by a second heating cycle under ammonia at 1000 °C for 14 h. Such intermediate compositions during the synthesis of LaTa(O,N)3 are often reported in literature when large amounts of oxide precursors are used or the applied precursor has been crystallised at around 1000 °C before the ammonolysis24. Additionally, a large sample amount can cause inhomogeneous exposure to the reducing species during ammonolysis. The positive and negative mass changes observed during the heating and cooling steps seem to be caused by desorption and adsorption of gaseous species (e.g. H2O and/or NH3). The nitrogen content of both red phases falls short of that of LaTaON2, revealing that n-LTO is susceptible to the reduction of tantalum (Ta5+ to Ta4+). The usage of m-LaTaO4 as a precursor for in situ ammonolysis results in LaTaON2, neither involving intermediate phases nor reduction of Ta(V) to Ta(IV) (no black powder indicating a reduction). Instead, the initial formation of oxygen vacancies (yellow LaTaO3.87(7)□0.12(3)) is followed by a one-step mass change of −5.5% (onset at 820 °C) indicating the conversion to LaTaON2 (Fig. 4).

In situ TGA ammonolysis of m-LaTaO4. In situ ammonolysis (10 vol% Ar in NH3) of m-LaTaO4. The insets show PXRD patterns at selected termination points and the anionic composition of the product received by termination at 820 °C determined by HGE. The coloured powders of the termination experiments are shown beneath the respective measuring point. The strong topotactic relation between m-LaTaO4 and LaTaON2 is represented by the respectively orientated sections of the crystal structures.
To be more precise, the in situ ammonolysis of m-LaTaO4 starts with a mass change of –0.5% between 25 °C and 820 °C (Fig. 4). According to the PXRD results of termination products, the crystal structure of m-LaTaO4 remained unchanged in this temperature range. However, a colour change from white (25 °C) to yellow (820 °C) indicated a change of the chemical composition including oxygen vacancy formation, which was confirmed via hot gas extraction (Supplementary Table 5). Further heating to 950 °C led to a change of the powder colour from yellow to purple accompanied by a massive mass change of −5.5%. The detected mass change is in accordance with the expected mass change of Δmcalc. = −5.2% calculated for the formation of LaTaON2 from LaTaO4. Termination experiments at 820 °C and at 950 °C together with hot gas extraction measurements revealed the conversion of microcrystalline LaTaO4 to LaTaON2 above 820 °C by nitrogen incorporation. An isothermal step with a near-constant mass was followed by a second ammonolysis cycle at 1000 °C for 14 h. The PXRD pattern of the final product clearly showed the presence of a perovskite-type phase (insets, Fig. 4). The mass changes during these heating and cooling steps might be attributed to the reversible adsorption and desorption of ammonia and water molecules as previously mentioned for n-LTO. We assume that a strong topotactical relation between crystalline ABO4 and crystalline ABON2 often described in literature18 also applies to the transformation of m-LaTaO4 to LaTaON2. Based on this assumption, the octahedron chains of defective m-LaTa(O,□)4 shown in Fig. 4 are supposed to create a pattern similar to LaTaON2 by rotating around the c-axis. During oxygen vacancy formation and subsequent nitrogen incorporation they connect to the neighbouring chains in a “zipper-type” manner18. However, the microstructure of n-LTO is different and exhibits therewith a “soft” topotactic relation to the resulting oxynitride along the interspace of the octahedron chains (Fig. 1) inducing a faster reduction of Ta(V) to Ta(IV). Afterwards, a “soft” topotactic reaction with a simultaneous “zipper-type” octahedral connection18 enables the afore mentioned filling of oxygen vacancies by nitrogen.
The lower nitrogen content of LaTa(O,N)3 in comparison to LaTaON2 when using n-LTO for in situ ammonolysis can be explained by the specific surface area and the primary particle size and, thus, the microstructure of the oxide precursors. The specific surface area of n-LTO is SBET = 7 m²/g (primary particle size: nm-range) and that of m-LaTaO4 SBET = 2 m²/g (primary particle size: µm-range). Further details about the oxide precursors can be found in Supplementary Note 1. The higher surface area and the smaller primary particle size of the nanocrystalline precursor compared to m-LaTaO4 makes it more susceptible to Ta reduction, since the diffusion of reducing agents is simplified (at higher temperatures NH3 decomposes into nitrogen- and hydrogen-containing species and molecular hydrogen9. A detailed explanation of the interaction of the reducing species with n-LTO can be found in the Supplementary Note 7: Reduction of Ta. This is confirmed by the finding that in situ ammonolysis of n-LTO that has been previously converted to m-LaTaO4 also leads to LaTaON2 (Fig. 5 and Supplementary Note 2).

Conversion of n-LTO to m-LaTaO4 and subsequent in situ TGA ammonolysis to LaTaON2. The first mass change of –1.8% is caused by desorption of water. The second negative mass change between 803 °C and 923 °C is due to pyrolysis of organic residues from the preceded soft chemistry synthesis. Thermogravimetric measurements coupled with mass spectrometry (TGA-MS) revealed the release of CO2, NO2 and organic fragments. The measurement results are displayed in Supplementary Fig. 4 and discussed in Supplementary Note 3. The measurement was first carried out in synthetic (syn.) air prior to the ammonolysis step.
Moreover, scanning electron microscopy (SEM) images of the termination products of the annealing process of n-LTO in syn. air prior to in situ ammonolysis (Fig. 6a) show a continuous microstructural change with increasing temperature leading to a morphology very similar to m-LaTaO4 (Fig. 6c).

SEM images of the termination products of all in situ ammonolysis studies. a Precursor conversion from n-LTO to m-LaTaO4 in synthetic (syn.) air prior to ammonolysis. b SEM images of the termination products of in situ ammonolysis of n-LTO and c SEM images of the in situ ammonolysis of m-LaTaO4. The temperatures mark the termination temperature at which the products were observed. Scalebar of the SEM images: 1 µm.
In parallel, the onset temperature of the ammonolysis reaction steadily increases from 588 °C to 820 °C with increasing primary particle size and crystallinity (n-LTO to m-LaTaO4, Figs 2, 5 and 6). Hence, a well-designed microstructure allows to enable or supress significant Ta reduction.
Ex situ ammonolysis of lanthanum tantalum oxides
Eventually, in situ ammonolysis of n-LTO did not deliver LaTaO2N which might be attributed to the less reductive atmosphere caused by the 10 vol% Ar in NH3 needed to protect the device. The application of nearly 100 vol% instead of 90 vol% NH3 in a classical thermal gas flow ammonolysis setup turned the in situ into an ex situ ammonolysis. The ex situ ammonolysis only slightly differs from the in situ setup with respect to the sample environment. Using the same temperature profile as in the in situ ammonolysis and an NH3 flow rate of 300 mL/min we finally converted n-LTO into bright red LaTaO1.98(7)N0.98(5) (SBET = 25 m2/g) after 10 h at 950 °C via ex situ ammonolysis (anionic composition obtained by HGE, Supplementary Table 6). However, when using the microcrystalline instead of the nanocrystalline precursor for ex situ ammonolysis with the same temperature profile and ammonia flow as in in situ ammonolysis, purple-coloured LaTaO1.05(3)N2.02(3) is obtained (very similar to the in situ ammonolysis product). This indicates a very similar parameter selection in ex situ and in situ ammonolysis. Considering the results of all in situ and ex situ ammonolysis experiments, we have identified the reduction sensitivity due to microstructural differences and the ammonia concentration as main parameters to adjust the O:N ratio in LaTa(O,N)3.
The aforementioned red LaTaO1.98(7)N0.98(5) and the purple LaTaO1.05(3)N2.02(3) phases prepared by ex situ ammonolysis are subjected to a second ex situ ammonolysis cycle (same temperature profile as in in situ ammonolysis) with KCl flux addition in order to heal possible defects25. After the second cycle the compositions slightly change to a bright red LaTaO1.97(3)N1.02(5) (SBET = 8 m2/g) and to purple LaTaO0.99(1)N2.00(9) (SBET = 3 m2/g), respectively. LaTaO1.97(3)N1.02(5) (LaTaO2N) reveals less vacancies compared to LaTaO1.98(7)N0.98(5) (obtained from first cycle) indicating a defect healing effect via KCl flux. Additionally, a reoxidation study (Supplementary Note 8, Supplementary Fig. 7 and Supplementary Table 7) validated the formation of LaTaO2N.
XPS measurements of LaTaO1.97(3)N1.02(5) and LaTaO0.99(1)N2.00(9) obtained by ex situ ammonolysis (Fig. 3c, survey spectra cf. Supplementary Note 9, Supplementary Fig. 8) show significant differences of the Ta oxidation states between both compounds. The data reveal a Ta(IV)–(O,N) binding character with a certain amount of Ta(III)–(O,N) in LaTaO1.97(3)N1.02(5) and a Ta(V)–(O,N) and a Ta(III)–(O,N) binding character in LaTaO0.99(1)N2.00(9) (Fig. 3b and Supplementary Table 8). Since the binding energy at EB(Ta 4f7/2) = 24.4 eV in LaTaO1.97(3)N1.02(5) corresponds to that in LaTaO3.44(4)N0.41(2)□0.14(6), the same binding character due to a similar chemical environment ([Ta(O,N)6]z- octahedron) can be assumed. However, the fitted binding energy EB(Ta 4f7/2) = 25.0 eV in LaTaO0.99(1)N2.00(9) reveals a Ta(V)–(O,N) binding character which can be explained by a simple chemical shift of 0.3 eV to lower binding energies due to nitrogen substitution (m-LaTaO4, EB(Ta 4f7/2) = 25.3 eV (P21/c)). The Ta(III)–(O,N) binding character is the result of the reducing conditions during ammonolysis favouring the reduction of Ta on the surface compared to the bulk. XPS is highly surface-sensitive owing to the small mean free path of photo-emitted electrons26. Therefore, the concentrations of Ta(IV)–(O,N) and Ta(V)–(O,N) in the bulk are assumed to be higher than on the surface. The Ta(III)–N binding character in LaTaO0.99(1)N2.00(9) at EB(Ta 4f7/2) = 22.9 eV is attributed to the secondary phase TaN since the reference binding energy of this nitride is EB(Ta 4f7/2) = 23.0 eV27. Additionally, XPS measurements of LaTaO2N (LaTaO1.97(3)N1.02(5)) also confirmed the absence of Ta5+ (observed, however, in LaTaON2 (LaTaO0.99(1)N2.00(9)) eliminating the possibility of a 1:1 mixture of Ta3+ and Ta5+ in LaTaO2N.
Investigations of the magnetic properties of LaTaO2N and LaTaON2 by superconducting quantum interference device (SQUID) measurements were carried out in order to confirm the presence of Ta4+ although challenging due to the presence of magnetic impurities such as Ta3+ as observed from XPS analysis (Fig. 3) and the in general limited knowledge about the magnetism of 5d transition metal compounds (compared to their 3d counterparts)28,29. The respective zero field cooled (ZFC) curves at 500 Oe down to 2 K are shown in Supplementary Note 10 and Supplementary Fig. 9. The low temperature regions (below ~70 K) can be described by a paramagnetic Curie–Weiss-like behaviour with very small effective moments (1.3 × 10−4 µB/Ta at 2 K for the LaTaO2N sample and 9.8 × 10−5 µB/Ta for the LaTaON2 sample, respectively) pointing to an activation of magnetic impurity states rather than an intrinsic materials property. The effective magnetic moment is further drastically reduced at increasing temperature. Overall the observed magnetisation is much lower than 1 µB/Ta as expected from the simplest paramagnetic model. The small magnetic moment and the fact that Ta is a 5d element suggests that the largely extended 5d orbitals in LaTaO2N are strongly hybridised and overlapping with the O/N 2p orbitals30 leading to weakly localised electrons hampering the up-built of a significant magnetisation. It is well-known for 3d transition metal containing perovskites ABO3 and ABF3 that with decreasing electronegativity of the anion the hybridisation (or in other words the covalency of the B–X bond) is enhanced resulting in the aforementioned stronger delocalisation of the electrons and hence the spin30. We believe the same is valid for 5d transition metal containing perovskites. Besides, two further factors could contribute diminishing the effective magnetic moment: (i) the spin-orbit coupling being expected to be much stronger in 5d than in 3d materials, (ii) the experimentally observed presence of stretched octahedra (Supplementary Note 11 and Supplementary Table 9e) results in a splitting of the initially degenerated t2g levels. This splitting leads to the formation of four-fold degenerated 5dxz and 5dyz states lower in energy than the initial state only occupied by one electron, making it difficult to develop magnetic ordering31.
Phase purity of LaTaO2N is proven by high resolution (HR)-PXRD. LaTaON2 has already been reported in the space groups Imma and C2/m17,32,33 and both are considered for LaTaO2N as well. Since we have observed no clear evidence for a monoclinic distortion and the Imma space group gives a slightly lower χ2 residual than C2/m, we propose Imma as space group for LaTaO2N. The same applies to LaTaON2 (Supplementary Fig. 10 and Supplementary Table 9 for complete crystal structure analysis). The unit cell volumes of LaTaO2N (Vcell = 264.78(3) Å3) and LaTaON2 (Vcell = 264.68(2) Å3) are very similar. The slightly larger unit cell of LaTaO2N can be explained by the increase of the effective ionic radius of Ta from 0.64 Å (Ta5+)34 to 0.68 Å (Ta4+)34 after ammonolysis. This expansion is mostly compensated by the different O:N ratio. The partial replacement of the larger nitrogen (1.46 Å)34 by the smaller oxygen (1.40 Å)34 leads to a contraction of the unit cell. Furthermore, LaTaO2N is phase pure, while in LaTaON2 a small amount of Ta3N5 (Cmcm) was detected through HR-PXRD (Supplementary Fig. 10b and Supplementary Table 9d). Since XPS reveals TaN on the surface and HR-PXRD Ta3N5 in the bulk, a nitrogen gradient combined with a decreased susceptibility for the reduction of Ta (from Ta5+ to Ta3+) can be assumed. However, TaN can also be amorphous or the amount below the HR-PXRD detection limit.
Scanning electron microscopy gives insight into the morphology of the oxynitrides synthesised by ex situ ammonolysis. LaTaO2N shows primary particles in the nm-range, while LaTaON2 exhibits porous and sintered particles in the µm-range (Fig. 7a).

SEM images and Kubelka–Munk curves of ex situ-prepared LaTaO2N and LaTaON2. a SEM images of LaTaO2N and LaTaON2 show the different morphologies of the materials in the µm-range. Scalebar of the SEM images: 1 µm. b Kubelka–Munk curves of LaTaO2N and LaTaON2 together with the respective powder images. The bright red colour indicates a much lower optically active defect concentration compared to LaTaON2.
In addition to the determination of the specific surface area (Supplementary Table 10), an increased pore formation in LaTaON2 during ammonolysis can also be confirmed by comparing the SEM images of the oxide with its respective oxynitride. Furthermore, the particle size increases during ammonolysis of n-LTO, whereas using m-LaTaO4 the particle size remains constant (Fig. 6c). The respective SEM images of the oxides can be found in the SI (Supplementary Fig. 2c).
As the colours of the obtained oxynitrides differ from each other, diffuse reflectance spectroscopy (DRS) measurements were performed. The data – converted to Kubelka–Munk35 curves (Fig. 7b) – show a difference of ∆Eg = 0.1 eV between the optical bandgaps of the LaTaO2N (1.9 eV) and LaTaON2 (1.8 eV). Detailed investigation shows that the Kubelka–Munk curve of LaTaO2N converges to zero in contrast to that of LaTaON2, indicating a much lower optically active defect concentration. In addition, after the optical bandgap of Eg = 1.8 eV an intensity increase as observed for LaTiO2N36, which exhibited undesired defects was not detected for LaTaO2N. LaTaO2N possess an unusual colour (bright red), which has been expected to be darker (bluish37) due to the reduction of Ta5+ to Ta4+. Additionally, the larger optical bandgap of LaTa(IV)O2N compared to LaTa(V)ON2 is noticeable. Based on both observations, we assume a larger crystal field splitting for LaTaO2N caused by the d1 state of Ta4+, since Ta is coordinated in a distorted octahedral environment (Jahn–Teller effect31) indicated by bond length analysis via Rietveld refinements. Specific visible light-driven applications require materials with appropriate defect concentrations38,39,40 and suitable bandgaps41,42,43. Therefore, LaTaO2N could be an interesting candidate for further investigations in this application field.
Discussion
In this in situ and ex situ ammonolysis study the key parameters to tailor the oxidation state and to synthesise a perovskite family member LaTa(IV)O2N containing unusual Ta4+ were identified. The oxynitride has an optical bandgap of 1.9 eV, a bright red colour, and a low optically active defect concentration providing promising physical properties for light-driven applications. The formation of LaTa(IV)O2N is boosted by the oxide precursor’s microstructure (primary particle size, specific surface area and crystallinity) and the ammonia concentration (nearly 100%). In previous studies, the ammonolysis mechanism was already investigated and mostly clarified for the topotactic case. The findings shown here make a substantial contribution to the elucidation of the ammonolysis mechanism in general and to that of amorphous/nanocrystalline oxide precursors in particular. This opens up new perspectives and possibilities for the synthesis of further perovskite-type oxynitrides.
Methods
Synthesis of LaTaO2N and LaTaON2
The oxynitrides LaTaO2N and LaTaON2 were prepared from the respective nanocrystalline and microcrystalline oxide precursors (cf. Supplementary Methods) via thermal ammonolysis (synthesis of the respective oxides is described in the SI). The precursor oxides (200 mg) were transferred to an alumina boat and ammonolysed at 950 °C for 10 h with a NH3 gas flow rate of 300 mL/min (Westfalen AG, > 99.98%). A second ammonolysis step together with KCl flux (Roth, ≥ 99%, Ph. Eur.) was carried out in a 1:1 weight ratio of sample and flux at 1000 °C for 14 h.
Sample characterisation
In order to clarify the phase purity and crystal structure of the produced oxides and oxynitrides, powder X-ray diffraction (PXRD) measurements were carried out on a Rigaku Smartlab powder X-ray diffractometer using Cu-K α1,2 radiation. To avoid a contribution of Cu-Kβ radiation a thin nickel foil as filter was used (efficiency is ~90%). The continuous scan covered an angular range of 10° ≤ 2θ ≤ 90° with an angular step interval of 0.025°. For selected oxide and oxynitride samples additional high-resolution synchrotron radiation PXRD measurements were performed at the beam line ID22 of the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. The powders were filled into 0.7 mm diameter Kapton® capillaries (wavelength, see specific refinements). The collected diffraction data were evaluated via Rietveld refinements44,45 using FullProf. 2k46. A pseudo-Voigt function was selected to describe the reflection profile and the background was linearly interpolated between a set of background points with refinable heights. The anionic composition of the oxynitrides were fixed according to the respective compositions determined by HGE because the virtually equal form factors make it impossible to discriminate between O2− and N3− by means of X-rays. O and N were statistically assigned to the two independent crystallographic sites.
The chemical composition of the produced samples was investigated via inductively coupled plasma optical emission spectroscopy (ICP-OES) using a Spectro Ciros CCD ICP-OES instrument for cations and hot gas extraction technique (HGE) using an Eltra ONH-2000 analyser for the anions.
The investigation of oxynitride formation from nanocrystalline and microcrystalline oxides and the reverse reaction of LaTaO2N to the corresponding oxide was performed by thermogravimetric analysis (TGA) using a Netzsch STA 449 F3 Jupiter. In situ ammonolysis experiments were carried out under flowing NH3 (80 mL/min NH3 + 8 mL/min Ar) on alumina plates with a heating rate of 10 °C/min up to 1000 °C. To protect the TGA device from corrosion the measurements were performed in 10 vol% Ar in NH3. A fast cooling rate of 40 °C/min was used to successfully quench the intermediates since the mass changes observed by TGA and HGE were in good agreement. Reoxidation was carried out on an alumina plate under flowing synthetic air (20.5 vol% O2 in N2, Westfalen AG, 50 mL/min) to study the anionic composition of LaTaO2N. The sample was first heated up to 200 °C and maintained at this temperature for half an hour to remove surface adsorbed water. Thereafter, heating was continued up to 1400 °C at a heating rate of 10 °C/min. The temperature was maintained for 2 h in order to achieve full conversion of oxynitride to respective single-phase oxide. TGA-MS to determine possible organic residues in n-LTO was carried out with a Netzsch STA 449 C Jupiter coupled with a GAM 200 (InProcess Instruments) mass spectrometer. The oxide was heated to 1200 °C at a rate of 10 °C/min in a crucible under syn. air (20.5 vol% O2 in N2, Westfalen AG, 50 mL/min) and then cooled down to 25 °C.
X-ray photoelectron spectroscopy (XPS) on LaTaO2N, LaTaON2, n-LTO, m-LaTaO4 and termination products was carried out to investigate the oxidation states using a Thermo VG Theta Probe 300 XPS system from Thermo Fisher Scientific. The incident beam provided monochromatic and micro-focused Al Kα radiation and a spot size of 400 µm. The powders were fixed on a carbon tape and a flood gun was used to avoid charging effects. For background subtraction a Shirley-type inelastic background was utilised and the zero-shift correction was done by normalizing the measured C 1 s peak to 284.5 eV. The peak fitting was implemented by carefully considering quantum mechanical requirements for the intensity and energy relations of Ta 4f7/2 and Ta 4f5/2 orbitals with ∆EB = 1.91 eV22 and identical full width at half maximum (FWHM) for both orbital contributions.
The morphology of the produced oxynitrides and oxides was analysed via scanning electron microscopy (SEM) (ZEISS GeminiSEM 500, 5 kV). The in-lens detector was used for imaging.
UV–visible diffuse reflectance spectra (DRS) were obtained using a Carry 5000 UV–VIS NIR spectrophotometer. The baseline was measured with BaSO4. The spectra were recorded in the range of 200–800 nm. The Kubelka–Munk35 conversion was applied to the obtained reflectance spectra and the optical bandgap was estimated by extrapolating the onset of absorption to the abscissa.
The specific surface area was obtained via nitrogen sorption, first annealing the samples at 120 °C to remove adsorbed water. Adsorption and desorption isotherms were collected at liquid nitrogen temperature using an Autosorb-1-MP (Detection limit: SBET > 1 m2/g) from Quantachrome Instruments. The specific surface area was determined via the Brunauer–Emmet–Teller47 (BET) method.
SQUID measurements to investigate the magnetic behaviour of Ta in LaTaO2N and LaTaON2 were carried out with a commercial VSM MPMS3 Superconducting Quantum Interference Device (SQUID) from Quantum Design. For zero field cooling (ZFC) measurements the magnetic field was set to 500 Oe.
Data availability
The authors declare that all other data supporting the findings of this study are available within the paper and its Supplementary Information.
Change history
06 March 2020
A Correction to this paper has been published: https://doi.org/10.1038/s42004-020-0280-7
References
Yajima, T. et al. A labile hydride strategy for the synthesis of heavily nitridized BaTiO3. Nat. Chem. 7, 1017–1023 (2015).
Ebbinghaus, S. G. et al. Perovskite-related oxynitrides - Recent developments in synthesis, characterisation and investigations of physical properties. Prog. Solid State Chem. 37, 173–205 (2009).
Yang, M. et al. Anion order in perovskite oxynitrides. Nat. Chem. 3, 47–52 (2011).
Jansen, M. & Letschert, H. P. Inorganic yellow-red pigments without toxic metals. Nature 404, 980–982 (2000).
Hojamberdiev, M. et al. Elucidating the impact of A-site cation change on photocatalytic H2 and O2 evolution activities of perovskite-type LnTaON2 (Ln = La and Pr). Phys. Chem. Chem. Phys. 19, 22210–22220 (2017).
Tessier, F., Maillard, P., Cheviré, F., Domen, K. & Kikkawa, S. Optical properties of oxynitride powders. Ceram. Soc. Jpn 117, 1–5 (2009).
Hisatomi, T., Kubota, J. & Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 43, 7520–7535 (2014).
Seo, J. et al. Efficient solar-driven water oxidation over perovskite-type BaNbO2N photoanodes absorbing visible light up to 740 nm. Adv. Energy Mater. 8, 1800094 (2018).
Kageyama, H. et al. Expanding frontiers in materials chemistry and physics with multiple anions. Nat. Commun. 9, 772 (2018).
Fuertes, A. Synthetic approaches in oxynitride chemistry. Prog. Solid State Chem. 51, 63–70 (2018).
Suemoto, Y. et al. Intergrowth between the oxynitride perovskite SrTaO2N and the Ruddlesden-Popper phase Sr2TaO3N. Inorg. Chem. 57, 9086–9095 (2018).
Kikkawa, S., Hosono, A. & Masubuchi, Y. Remarkable effects of local structure in tantalum and niobium oxynitrides. Prog. Solid State Chem. 51, 71–80 (2018).
Zhang, L. et al. Photoelectrochemical water oxidation of LaTaON2 under visible-light irradiation. Int. J. Hydrog. Energy 39, 7697–7704 (2014).
Si, W. et al. Investigating the behavior of various cocatalysts on LaTaON2 photoanode for visible light water splitting. Phys. Chem. Chem. Phys. 19, 656–662 (2017).
Weidenkaff, A. Preparation and application of nanostructured perovskite phases. Adv. Eng. Mater. 6, 709–714 (2004).
Ueda, K., Kato, H., Kobayashi, M., Hara, M. & Kakihana, M. Control of valence band potential and photocatalytic properties of NaxLa1-xTaO1+2xN2-2x oxynitride solid solutions. J. Mater. Chem. A 1, 3667–3674 (2013).
Clark, L., Oró-Solé, J., Knight, K. S., Fuertes, A. & Attfield, J. P. Thermally robust anion-chain order in oxynitride perovskites. Chem. Mater. 25, 5004–5011 (2013).
Ebbinghaus, S. G., Aguiar, R., Weidenkaff, A., Gsell, S. & Reller, A. Topotactical growth of thick perovskite oxynitride layers by nitridation of single crystalline oxides. Solid State Sci. 10, 709–716 (2008).
Vullum, F., Nitsche, F., Selbach, S. M. & Grande, T. Solid solubility and phase transitions in the system LaNb1−xTaxO4. J. Solid State Chem. 181, 2580–2585 (2008).
Nordling, C. ESCA: Elektronen-Spektroskopie fur chemische Analyse. Angew. Chem. 4, 144–153 (1972).
Briggs, D. & Grant, J. T. Surface Analysis by Auger and X-ray Photoelectron Spectroscopy. (IM Publications, 2003).
Moulder, J. F., Stickle, W. F., Sobol, P. E. & Bomben, K. D. Handbook of X-ray Photoelectron Spectroscopy. (Perkin-Elmer Corporation Physical Electronics Division, 1992).
Logvinovich, D. et al. Synthesis and physical chemical properties of Ca-substituted LaTiO2N. Prog. Solid State Chem. 35, 281–290 (2007).
Park, N. Y. & Kim, Y. Il Morphology and band gap variations of oxynitride LaTaON2 depending on the ammonolysis temperature and precursor. J. Mater. Sci. 47, 5333–5340 (2012).
Kim, Y. Il Effects of KCl flux on the morphology, anion composition, and chromaticity of perovskite oxynitrides, CaTaO2N, SrTaO2N, and LaTaON2. Ceram. Int. 40, 5275–5281 (2014).
Henderson, G. S., de Groot, F. M. F. & Moulton, B. J. A. X-ray Absorption Near-Edge Structure (XANES). Spectrosc. Rev. Mineral. Geochem. 78, 75–138 (2014).
Zaman, A. & Meletis, E. Microstructure and mechanical properties of TaN thin films prepared by reactive magnetron sputtering. Coatings 7, 209, 1–16 (2017).
Erickson, A. S. et al. Ferromagnetism in the Mott insulator Ba2NaOsO6. Phys. Rev. Lett. 99, 016404 (2007).
Wang, X. Y., Avendaño, C. & Dunbar, K. R. Molecular magnetic materials based on 4d and 5d transition metals. Chem. Soc. Rev. 40, 3213–3238 (2011).
Varignon, J., Bibes, M. & Zunger, A. Origins Vs. fingerprints of the Jahn-Teller effect in d-electron ABX 3. 1–26. Available at: http://arxiv.org/abs/1906.07587.
Kugel, K. & Khomskiĭ, D. The Jahn-Teller effect and magnetism: transition metal compounds. Sov. Phys. Uspekhi 25, 231–256 (1982).
Günther, E., Hagenmayer, R. & Jansen, M. Strukturuntersuchungen an den Oxidnitriden SrTaO2N, CaTaO2N und LaTaON2 mittels Neutronen- und Röntgenbeugung. Z. Anorg. Allg. Chem. 626, 1519–1525 (2000).
Cordes, N. & Schnick, W. Ammonothermal synthesis of crystalline oxonitride perovskites LnTaON2 (Ln=La, Ce, Pr, Nd, Sm, Gd). Chem. Eur. J. 2, 11410–11415 (2017).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A32, 751–767 (1976).
Kortüm, G., Braun, W. & Herzog, G. Principles and techniques of diffuse-reflectance spectroscopy. Angew. Chem. Int. Ed. 2, 333–341 (1963).
Maegli, A. E. et al. Perovskite-type LaTiO2N oxynitrides for solar water splitting: Influence of the synthesis conditions. Energy Procedia 22, 61–66 (2011).
Siegrist, T., Cava, R. J. & Krajewski, J. J. Reduced alkaline earth tantalates. Mater. Res. Bull. 32, 881–887 (1997).
Zhang, G., Liu, G., Wang, L. & Irvine, J. T. S. Inorganic perovskite photocatalysts for solar energy utilization. Chem. Soc. Rev. 45, 5951–5984 (2016).
Xiao, M., Wang, S., Thaweesak, S., Luo, B. & Wang, L. Tantalum (Oxy)nitride: narrow bandgap photocatalysts for solar hydrogen generation. Engineering 3, 365–378 (2017).
Osterloh, F. E. & Parkinson, B. A. Recent developments in solar water-splitting photocatalysis. MRS Bull. 36, 17–22 (2011).
Castelli, I. E. et al. New cubic perovskites for one- and two-photon water splitting using the computational materials repository. Energy Environ. Sci. 5, 9034–9043 (2012).
Rajeshwar, K., McConnell, R. & Licht, S. Solar Hydrogen Generation. (Springer Berlin Heidelberg, 2008).
Lohaus, C., Klein, A. & Jaegermann, W. Limitation of Fermi level shifts by polaron defect states in hematite photoelectrodes. Nat. Commun. 9, 4309 (2018).
Rietveld, H. M. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Crystallogr. 22, 151–152 (1967).
Rietveld, H. M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 2, 65–71 (1969).
Rodriguez-Carvajal, J. FullProf. 2k, version 5.30, ILL. 2012 version 5., (2012).
Brunauer, S., Emmett, P. H. & Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 60, 309–319 (1938).
Acknowledgements
We thank Mrs. Annette Fuchs, Prof. Dr. Joachim Maier and Prof. Dr. Bettina Lotsch (Max Planck Institute for Solid State Research, Stuttgart), Mr. Peter Schützendübe (Max Planck Institute for Intelligent Systems, Stuttgart), and Mr. Samir Hammoud (Max Planck Institute for Intelligent Systems, Stuttgart) for nitrogen sorption, DRS, XPS, and chemical analysis, respectively, and Alan Fernandes-Dias for producing the microcrystalline oxide precursor. We also thank Dr. Benjamin Balke (Fraunhofer Research Institution Materials Recycling and Resource Strategies, Hanau), Dr. Andrea Knöller, Dr. Angelika Veziridis (Institute for Materials Science, University of Stuttgart), MSc. Sven Fecher (Max Planck Institute for Solid State Research, Stuttgart), and Jun.-Prof. Hongbin Zhang (Department of Materials and Earth Sciences, Technische Universität Darmstadt) for fruitful discussions. We acknowledge the financial support of the European Synchrotron Radiation Facility, Grenoble, France and the synchrotron radiation beam time. This work was supported by the Deutsche Forschungsgemeinschaft within the priority program SPP 1613 “Solar H2” (WE 2803/7–1).
Author information
Authors and Affiliations
Contributions
C.B. developed the parameters for the synthesis of the oxide precursors and the perovskite-type oxynitrides and the in situ and ex situ ammonolysis study. C.B. performed and pre-evaluated the PXRD, SEM, DRS and TGA measurements. C.B., M.W., and A.W. collectively elaborated the analysis of the synthesis, in situ and ex situ ammonolysis data. G.R. and C.B. analysed the XPS data. M.W. and M.C. collected the HR-PXRD data at the ESRF. C.B., M.W., S.Y., and M.C. analysed the HR-PXRD data, which had been refined by C.B. and M.W. C.B. and E.G. measured and analysed together the SQUID data. C.B. wrote the paper with input from M.W., G.R., M.C., and S.Y. and A.W. provided final contributions to the conclusions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bubeck, C., Widenmeyer, M., Richter, G. et al. Tailoring of an unusual oxidation state in a lanthanum tantalum(IV) oxynitride via precursor microstructure design. Commun Chem 2, 134 (2019). https://doi.org/10.1038/s42004-019-0237-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0237-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
