
Abstract
Aims/hypothesis
ATPase copper transporting α (ATP7A), also known as Menkes disease protein, is a P-type ATPase that transports copper across cell membranes. The critical role of ATP7A-mediated copper homeostasis has been well recognised in various organs, such as the intestine, macrophages and the nervous system. However, the importance of adipocyte ATP7A-mediated copper homeostasis on fat metabolism is not well understood. Here, we sought to reveal the contribution of adipose ATP7A to whole-body fat metabolism in mice.
Methods
We generated adipocyte-specific Atp7a-knockout (ASKO) mice using the Cre/loxP system, with Cre expression driven by the adiponectin promoter. ASKO mice and littermate control mice were aged on a chow diet or fed with a high-fat diet (HFD); body weight, fat mass, and glucose and insulin metabolism were analysed. Histological analysis, transmission electron microscopy and RNA-sequencing (RNA-Seq) analysis of white adipose tissue (WAT) were used to understand the physiological and molecular changes associated with loss of copper homeostasis in adipocytes.
Results
Significantly increased copper concentrations were observed in adipose tissues of ASKO mice compared with control mice. Aged or HFD-fed ASKO mice manifested a lipoatrophic phenotype characterised by a progressive generalised loss of WAT. Dysfunction of adipose tissues in these ASKO mice was confirmed by decreased levels of both serum leptin and adiponectin and increased levels of triacylglycerol and insulin. Systemic metabolism was also impaired in these mice, as evidenced by a pronounced glucose intolerance, insulin resistance and hepatic steatosis. Moreover, we demonstrate a significant induction of lipolysis and DNA-damage signalling pathways in gonadal WAT from aged and HFD-fed ASKO mice. In vitro studies suggest that copper overload is responsible for increased lipolysis and DNA damage.
Conclusions/interpretation
Our results show a previously unappreciated role of adipocyte Atp7a in the regulation of ageing-related metabolic disease and identify new metallophysiologies in whole-body fat metabolism.
Data availability
The datasets generated during the current study are available in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number CRA001769 (http://bigd.big.ac.cn/gsa).
Similar content being viewed by others



Introduction
Copper is an essential metal nutrient, the homeostasis of which must be precisely regulated by copper transporters throughout the lifespan of all eukaryotes. Copper-dependent enzymes are essential for various cellular processes, such as redox balance, angiogenesis, energy production, iron homeostasis, the biosynthesis of neuromodulators, and many other processes [1]. Mutations in copper transporters lead to serious diseases of copper metabolism, such as Menkes disease and Wilson’s disease [2]. The former is a fatal neurodegenerative disorder caused by the mutation of the gene encoding ATPase copper-transporting α (ATP7A), while the latter is a hepatoneurological disorder caused by mutations in ATPase copper-transporting β (ATP7B).
A tight link between copper homeostasis and lipid metabolism has been well recognised [3]. Insufficient copper is involved in the aetiology of non-alcoholic fatty liver disease (NAFLD) [4, 5] and atherogenic dyslipidaemia [6] in humans. Studies in rats have shown that a copper-deficient diet is linked to hepatic steatosis, insulin resistance [4], hypertension, elevated triacylglycerol and cholesterol levels and abnormal lipoprotein profiles [7, 8]. Investigations into the underlying mechanisms suggest that a copper-deficient diet promotes lipid biosynthesis [3], and interactions between a copper-deficient diet and dietary fructose may contribute to these pathophysiological phenotypes [9]. Furthermore, individuals with, and rodent models of, Wilson’s disease exhibit lipid dysregulation, suggesting that the association between copper dyshomeostasis and the dysregulation of lipids occurs in humans [10,11,12]. Studies in fish have also revealed a decrease in the whole-body lipid content following the consumption of increased dietary copper concentrations [13, 14]. Higher lipolysis rates of subcutaneous white adipose tissue (sWAT) have been observed in copper-supplemented steer and finishing pigs [15]. Taken together, these findings reveal a close, although poorly understood, interaction between copper homeostasis and whole-body lipid metabolism.
Adipose tissues are among the largest and most dynamic organs in the human body [16]. The dysfunction of adipose tissue contributes to lipodystrophy [17, 18], obesity and obesity-associated chronic metabolic diseases, including diabetes [19] and cardiovascular diseases [20]. The biological role of copper homeostasis in adipocytes has become the focus of increasing attention recently. The Rodríguez group reported that an increase in the extracellular copper concentration promotes the differentiation of mesenchymal stem cells (MSCs) into adipocytes [21]. Copper was recently shown to regulate cAMP-dependent lipolysis by altering the activity of the cAMP-degrading phosphodiesterase 3B, cGMP-inhibited (PDE3B) in 3T3-L1 cells, a well-recognised pre-adipocyte cell line [22]. Moreover, the inactivation of the copper-dependent enzyme, semicarbazide-sensitive amine oxidase (SSAO), was found to increase lipid storage resulting in adipocyte hypertrophy and triacylglycerol accumulation in adipocytes [23]. These data suggest a close interconnected relationship between adipocyte function and copper homeostasis; however, the underlying mechanisms remain poorly understood.
The ubiquitously expressed copper-transporting ATPase, ATP7A, plays a critical role in copper export, a process that protects most cells from the overaccumulation of potentially toxic copper. The physiological consequences of a loss of ATP7A activity are well documented in Menkes disease, a systemic copper deficiency disorder. Because null mutations in Atp7a are lethal in embryonic mice, tissue-specific mouse models of Atp7a deletion have been used to define the role of ATP7A and copper homeostasis in the physiological processes of specific organs. Targeted deletion of Atp7a in mouse enterocytes causes entrapment of copper in the intestines and a systemic copper deficiency, thus revealing the role of ATP7A in dietary copper transport [24]. Ablation of Atp7a in macrophages increases the susceptibility of mice to bacterial infection, revealing novel roles for copper in innate immunity [25]. In motor neurons, specific deletion of Atp7a results neuromuscular phenotypes that model the human disease, X-linked spinal muscular atrophy [26]. Other studies have hinted at roles for ATP7A in lipid metabolism. ATP7A-mediated copper transport has been implicated in the macrophage-mediated oxidation of LDL-cholesterol, potentially contributing to the pathogenesis of atherosclerosis [27]. Cold-stimulated brown adipose tissue (BAT) showed the elevated copper levels and significantly increased ATP7A levels [28]. In view of these studies, we hypothesised that deletion of ATP7A in adipocytes would shed light on the role of copper on adipocyte lipid metabolism and related physiological processes at the whole-body level.
Methods
Animals
All mouse husbandry and euthanasia procedures were performed in accordance with the guidelines of the Animal Care and Use Committee of the Institute of Zoology. Mice with adiponectin-Cre-driven deletion of the floxed Atp7a allele were generated and maintained as described previously [29]. A detailed description of the methods is included in the electronic supplementary material (ESM Methods).
Cell culture
3T3-L1 pre-adipocytes were originally purchased from ATCC, Manassas, VA, USA, and then maintained in the laboratory. No mycoplasma contamination was found. Cells were cultured in DMEM containing 10% (vol./vol.) newborn calf serum and were grown to confluence. Cells were then differentiated by the addition of insulin, dexamethasone and 3-isobutyl-1-methylxanthine (IBMX), as described previously [22]. For copper-supplementation experiments, differentiated 3T3-L1 cells were incubated with DMEM supplemented with 2% BSA (vol./vol.) for 10 h, followed by a 2 h treatment with 50 μmol/l CuCl2 in DMEM.
Blood analysis
Blood was obtained via the orbital vein before the mice were killed. The triacylglycerol content was measured using enzymatic assay kits from Applygen (Beijing, China). Plasma insulin, leptin and adiponectin levels were measured using ELISAs (Millipore, Bedford, MA, USA). The free glycerol content and NEFA were measured with colorimetric assays (Sigma-Aldrich, St Louis, MO, USA, and Wako Chemical, Osaka, Japan, respectively).
Measurements of the tissue metal contents
Tissues harvested for trace metal analysis were collected in 1.5 ml tubes washed in nitric acid. Tissues were dissolved in 70% (vol./vol.) HNO3; metal contents were inductively measured using inductively coupled plasma MS (ICP-MS) in the core facility of Peking University Medical School. Values were acquired in triplicate for each sample and the results were normalised to the wet tissue weight.
Metabolic studies
GTTs (i.p. injection of 2 g glucose/kg body weight) were performed in mice after a 16 h overnight fast (17:00–09:00 hours), and ITTs (i.p. injection of 0.75 U/kg body weight, Humulin, Eli Lilly, Indianapolis, IN, USA) were performed after a 4 h fast (09:00–13:00 hours). Glucose concentrations were measured in blood collected from the tail vein at the indicated time points after the injection using a glucose monitor and strips (OneTouch, Milpitas, CA, USA). Oxygen consumption was measured using the TSE LABmaster system (TSE Systems, Bad Homburg, Germany), as described previously [30].
Lipolysis analysis in vitro
For the in vitro lipolysis analysis, epididymal fat was removed, cut into 10 mg fat pads and stimulated with or without 1 μmol/l isoprenaline (Sigma-Aldrich), as described previously [31]. The medium was collected for determination of glycerol levels.
Histological studies
The WAT, BAT and liver were dissected and fixed with 4% (wt/vol.) paraformaldehyde overnight. Fixed specimens were embedded in paraffin blocks, sectioned at 5 μm thickness and stained with H&E. The size of H&E-stained WAT adipocytes was calculated using ImageJ software (https://imagej.nih.gov/ij/, version 1.46r). The adipocyte size was determined in five different sections of the fat pads from each mouse (four mice per group). For transmission electron microscopy (TEM), adipose tissues were fixed with 2% (vol./vol.) glutaraldehyde for 12 h, post fixed with osmium tetroxide, dehydrated, embedded, cut into ultra-thin sections and stained using previously described methods [32].
Quantitative PCR analysis
Real-time quantitative (q)PCR was used to determine relative gene expression levels. Tissues, including fat and liver, were dissected and total RNAs were extracted. For further details, see ESM Methods. 18S was used as a housekeeping gene and sequence of primers are shown in ESM Table 1.
Immunoblot analysis
Tissues or cells were lysed in lysis buffer and subjected to western blotting as described previously, with minor modifications [24]. The proteins were detected with antibodies against ATP7A, hormone-sensitive lipase (HSL), phospho-HSL Ser563 (p563HSL), phospho-HSL Ser565 (p565HSL), adipose triglyceride lipase (ATGL), adrenoceptor β3 (ADRB3), cyclin-dependent kinase inhibitor 1A (p21), phosphorylated histone H2AX (γ-H2AX), tumour protein p53 (p53), peroxisome proliferator activated receptor γ (PPARγ), CCAAT enhancer binding protein α (CEBPα), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and tubulin. For details, see ESM Methods.
RNA sequencing and identification of differentially expressed genes
These procedures were performed as described previously [32].
Statistical analysis
No randomisation or blinding was carried out as part of the study, and none of the data were excluded. Statistical comparisons between two groups were made using the two-tailed Student’s t test; comparisons between three or more groups were made by two-way ANOVA, followed by a Tukey’s post hoc test, using GraphPad Prism v6 (San Diego, CA, USA). Energy expenditure was analysed by ANCOVA, with body weight as a covariate. The data are presented as means ± SEM. Levels of statistical significance were set at *p < 0.05, **p < 0.01 and ***p < 0.001.
Results
ASKO mice exhibit persistently elevated copper in adipose tissues and progressive fat loss with ageing
Hemizygous male Atp7afl/Y-Adipo-Cre+ (hereafter called ASKO mice [note that Adipo is also known as Adipoq]) progeny showed significant lower levels of ATP7A in the sWAT, gonadal (g)WAT and BAT compared with littermate control male mice, Atp7afl/Y-Adipo-Cre− (hereafter referred to as wild-type [WT]) mice (Fig. 1a). No differences in ATP7A expression were present in other tissues such as the kidney and muscle (ESM Fig. 1). Copper and iron concentrations in sWAT and BAT from WT and ASKO mice were measured using ICP-MS. Consistent with a critical role for ATP7A in cellular copper efflux, copper levels were significantly increased in the adipose tissues from ASKO mice at 6, 12 and 16 months of age compared with those from WT mice (Fig. 1b,c). In contrast, iron levels were not significantly altered (ESM Fig. 2a,b).

ASKO mice displayed a persistently high copper content and progressive reduction in fat mass during ageing. (a) Immunoblot showing ATP7A levels in the sWAT, gWAT and BAT from WT and ASKO mice. One representative immunoblot from three independent experiments is shown. (b, c) Copper content in the sWAT (b) and BAT (c) from WT and ASKO mice at 6, 12 and 16 months of age (n=4 mice/group). (d) MRI analysis of the body compositions of 16-month-old WT and ASKO mice (n=8 mice/group). (e) Representative image of WT and ASKO mice showing reduced adiposity in the 16-month-old ASKO mice. (f) Representative images of the sWAT, gWAT, BAT and livers from 16-month-old WT and ASKO mice. (g) Relative organ weights of 16-month-old WT and ASKO mice (n=8 mice/group). (h, i) Weight of sWAT (h) and gWAT (i) from WT and ASKO mice at 6, 12 and 16 months of age (n=8 mice/group). Grey bars/black lines, WT mice; red bars/lines, ASKO mice. All quantitative data are presented as the means ± SEM. *p<0.05, **p<0.01 and ***p<0.001 for differences between WT and ASKO mice. †p<0.05 for differences in the effect of ageing in ASKO mice (vs 12 months)

Plasma biochemistry and insulin sensitivity indices in aged ASKO mice. (a–d) Concentrations of plasma adiponectin (a), leptin (b), triacylglycerol (c) and insulin (d) in chow-fed 16-month-old WT and ASKO mice (n=6 mice/group). (e) Food intake of chow-fed 16-month-old WT and ASKO mice (n=6 mice/group). (f, g) Results of the GTT (f) and ITT (g) performed on 12-month-old WT and ASKO mice (n=6 mice/group). (h) H&E staining of gWAT, BAT and liver sections from WT and ASKO mice fed chow diet at different ages. Scale bars, 50 μm. Grey bars/black lines, WT mice; red bars/lines, ASKO mice. All quantitative data are presented as the means ± SEM. *p<0.05, **p<0.01 and ***p<0.001 for differences between WT and ASKO mice. TG, triacylglycerol
ASKO and WT mice were kept up to 16 months on normal chow diet to examine the effect of chronic copper accumulation on whole-body fat metabolism. No differences in body weight, body composition or relative organ weights were observed at 6 months of age (ESM Fig. 3a–c). At 1 year of age, the mean body weight of ASKO mice was slightly decreased without statistical significance (ESM Fig. 3a). This was accompanied by a significant reduction in WAT depots, increased BAT deposits and increased liver weights in 1-year-old ASKO mice (ESM Fig. 3d). These effects of adipocyte-specific loss of ATP7A were more pronounced in 16-month-old mice. According to MRI analyses, 16-month-old ASKO mice weighed approximately 18% less than age-matched WT mice, with the bulk of this difference due to a reduced fat mass (Fig. 1d). The analysis of harvested tissues further confirmed a dramatically reduced fat mass in 16-month-old ASKO mice (Fig. 1e–g), which was accompanied by BAT and liver hypertrophy (Fig. 1f,g). Furthermore, the weights of sWAT and gWAT from WT mice were significantly increased with age, while those from ASKO mice exhibited an age-dependent fat loss (Fig. 1h,i). Taken together, these data demonstrate that loss of adipocyte ATP7A promotes age-dependent decreases in fat mass.

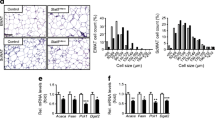
Decreased body weight and fat content in ASKO mice fed an HFD. (a) Body weight of the WT and ASKO mice fed the HFD (n≥8 mice/group). (b) Representative photograph of mice exhibiting HFD-induced obesity. (c) Body composition of WT and ASKO mice (n=8 mice/group) from MRI analysis. (d) Photographs of the sWAT, gWAT, pWAT and BAT from the WT and ASKO mice fed the HFD. (e) Relative organ weights of HFD-fed mice (n=10 mice/group). (f) H&E staining and TEM images of the sWAT from ASKO and WT mice fed the HFD. (g) The distribution of adipocyte size in the sWAT from WT and ASKO mice fed the HFD. The area of 1000 adipocytes was measured. (h) H&E staining and TEM images of BAT from ASKO and WT mice fed the HFD. (i, j) Serum adiponectin (i) and leptin (j) levels in WT and ASKO mice fed the HFD (n=6 mice/group). (k) Expression of the adiponectin and leptin mRNA in WT and ASKO mice fed the HFD (n=6 mice/group). Scale bars, 50 and 20 μm for H&E and TEM, respectively. Grey bars/black lines, WT mice; red bars/lines, ASKO mice. All quantitative data are presented as the means ± SEM *p<0.05 and ***p<0.001 for differences between WT and ASKO mice
Impaired glucose tolerance and insulin resistance in aged ASKO mice
To assess the effects of ATP7A deletion on systemic metabolism, we compared circulating levels of adiponectin, leptin, triacylglycerol and insulin in aged WT and ASKO mice. Consistent with the dramatically reduced fat depots in 16-month-old ASKO mice, serum levels of adiponectin and leptin were also significantly reduced (Fig. 2a,b). In addition, significantly higher serum triacylglycerol and insulin levels were observed in 16-month-old ASKO mice (Fig. 2c,d). As these changes are closely associated with energy balance and glucose homeostasis, food intake and glucose tolerance were investigated in aged mice. Our data showed that the ASKO mice were hyperphagic, consuming approximately 130% as much food as the controls (Fig. 2e). At 6 months of age, there were no differences in blood glucose between WT and ASKO mice in response to exogenous glucose challenge or insulin administration (ESM Fig. 4a,b). However, the ASKO mice at 12 months of age exhibited significantly delayed glucose clearance and impaired insulin sensitivity (Fig. 2f,g). This was associated with smaller but morphologically normal white adipocytes in aged ASKO mice compared with WT mice (Fig. 2h). Brown adipocytes from aged mice showed signs of hypertrophy (Fig. 2h) and their livers showed significant steatosis (Fig. 2h). Taken together, these data suggest that ASKO mice exhibit age-related metabolic dysfunction and insulin resistance.

ATP7A deficiency in adipose tissue exacerbates diet-induced hepatic steatosis and insulin resistance. (a) Representative photograph of liver tissues from WT and ASKO mice fed the HFD. (b) Liver histology of WT and ASKO mice; scale bar, 50 μm. (c) Hepatic triacylglycerol content in WT and ASKO mice (n=7 mice/group). (d) Plasma triacylglycerol level in mice fed the HFD (n=6 mice/group). (e) qPCR analysis of gene expression in liver tissues from HFD-fed WT and ASKO mice (n=6 mice/group), expressed as relative units using the \( {2}^{-\varDelta \varDelta {\mathrm{C}}_{\mathrm{t}}} \) method; (Srebp1c, Srebp2, Pparγ, Lxrα, Lxrβ, Tnfα and Mcp1 are also known as Srebf1, Srebf2, Pparg, Nr1h3, Nr1h2, Tnf and Ccl2, respectively). (f, g) Results of the GTT (f) and ITT (g) performed in HFD-fed WT and ASKO mice (n=6 mice/group). (h) Plasma insulin levels in mice fed the HFD (n=6 mice/group). Grey bars/black lines, WT mice; red bars/lines, ASKO mice. All quantitative data are presented as the means ± SEM *p<0.05 and **p<0.01 for differences between WT and ASKO mice. TG, triacylglycerol
ASKO mice are resistant to diet-induced obesity
To assess the effect of a high-fat diet (HFD) on mutant mice, both WT and ASKO mice were fed an HFD for 20 weeks on reaching 20 g in weight. Compared with WT mice, HFD-induced weight gain was significantly reduced in ASKO mice (Fig. 3a,b). MRI analyses revealed an unchanged lean mass and a markedly reduced fat mass gain in HFD-fed ASKO mice (Fig. 3c). Moreover, HFD-fed ASKO mice showed significantly reduced masses of sWAT, gWAT and perinephric (p)WAT (Fig. 3d,e). Further histological analysis revealed that adipocyte-specific loss of ATP7A markedly attenuated HFD-induced sWAT hypertrophy, which was confirmed using TEM (Fig. 3f). Area measurements of adipocytes from the sWAT of HFD-fed mice further confirmed the reduction in adipocyte hypertrophy in HFD-fed mutant mice (Fig. 3g). In addition, HFD-fed ASKO mice exhibited a significant increase in the BAT mass (Fig. 3d,e). Histological and TEM analyses revealed increased hypertrophy of BAT adipocytes from HFD-fed ASKO mice, as evidenced by the larger size of BAT adipocytes, the increased abundance of lipid droplets and the infiltration of oligolocular and unilocular adipocytes (Fig. 3h). Furthermore, the density of nuclei (number of nuclei per unit area) was also significantly decreased in the ASKO mice (Fig. 3h). Fasting levels of adiponectin and leptin in serum were measured and the HFD-fed ASKO mice displayed significantly lower serum adiponectin and leptin levels (Fig. 3i,j). These were associated with decreased levels of mRNA (Fig. 3k). Based on these data, we conclude that the specific deletion of Atp7a in adipose tissues protects against HFD-induced obesity.
ASKO mice are susceptible to HFD-diet-induced liver steatosis and insulin resistance
Although mutant mice were resistant to HFD-induced obesity, the sizes and weights of livers from HFD-fed ASKO mice were significantly greater than those of HFD-fed WT mice (Figs 3e and 4a). Histological staining revealed that the livers from HFD-fed ASKO mice contained markedly enlarged lipid droplets compared with control mice, suggesting a more advanced steatosis (Fig. 4b). Consistent with this finding, hepatic triacylglycerol levels were significantly increased in the ASKO mice (Fig. 4c). Significantly higher serum triacylglycerol levels were also observed in HFD-fed ASKO mice compared with WT mice (Fig. 4d).
Next, we examined the expression of genes involved in lipogenesis, oxidative phosphorylation, transcriptional regulation and inflammation in the livers of HFD-fed mice using qPCR. The expression of genes related to hepatic de novo lipogenesis (Acc1 [also known as Acaca] and Elovl6) and their major transcriptional regulator, sterol regulatory element binding transcription factor 1 (SREBP1c; encoded by Srebp1c), was slightly increased in HFD-fed ASKO mice (Fig. 4e). In contrast, the expression of genes involved in fatty acid oxidation and oxidative phosphorylation (Cpt1a, Cpt2, Cox4 [also known as Cox4i1], Cycs, Acadl and Acadm) was significantly downregulated in the liver of HFD-fed ASKO mice (Fig. 4e).
GTTs and ITTs were also performed for HFD-fed WT and ASKO mice. Compared with the WT littermates, ASKO mice exhibited higher blood glucose levels after an i.p. injection of glucose (Fig. 4f), suggesting that these animals were glucose intolerant. In addition, significantly slower insulin-stimulated glucose clearance was observed in ASKO mice relative to WT mice (Fig. 4g). Furthermore, a significant increased serum insulin level was observed in fasted ASKO mice that had been fed the HFD (Fig. 4h). These data suggest that adipocyte-specific deletion of Atp7a compromised systemic insulin sensitivity and reduced glucose tolerance in mice fed the HFD.
Increased DNA damage and lipolysis in gWAT from HFD-fed ASKO mice
A genome-wide RNA-sequencing (RNA-Seq) analysis was performed to determine the molecular signature of the response of gWAT on chronic high copper exposure in HFD-fed ASKO mice. Differentially expressed genes were screened based on the criteria of p < 0.01 and fold change >1.5 and the volcano plots showed a broad overview of the changes in gene expression in both mice fed the HFD (Fig. 5a).

DNA damage and lipolysis are increased in WAT of HFD-fed ASKO mice. (a) Volcano plot of genes displaying a significant difference in expression (fold change) in the gWAT from ASKO and WT mice (n=4 mice/group). Coloured dots label genes on the indicated pathways. The vertical dashed lines represent 1.5-fold change; the horizontal red dashed line represents p=0.01. (b) Ingenuity pathway analysis (IPA) of differentially expressed genes in ASKO adipose tissue compared with WT adipose tissue. (c, d) Quantification of the mRNA levels of the DNA damage genes (c) and lipolysis genes (d) in gWAT from mice fed the HFD for 15 weeks (n=6 mice/group), expressed as relative units using the \( {2}^{-\varDelta \varDelta {\mathrm{C}}_{\mathrm{t}}} \) method. (e) Western blot showing the levels of DNA-damage and lipolysis markers in gWAT of HFD-fed mice. One representative blot from three independent experiments is shown. (f) Basal and isoprenaline-stimulated glycerol release in gWAT explants from WT (n=5 mice) and ASKO (n=4 mice) mice fed the HFD. (g) Western blots for DNA-damage and lipolysis markers in lysates of copper-treated 3T3-L1 cells. (h) Schematic diagram of the differentially expressed genes/proteins in the lipolysis signalling pathway. Red arrows indicate increased expression and blue arrows indicate decreased expression. (i) The whole-body oxygen consumption rate (\( \dot{V}{\mathrm{O}}_2 \)) was measured throughout the day and night and is presented as the mean \( \dot{V}{\mathrm{O}}_2 \) over 24 h (WT, n=6 mice; ASKO, n=5 mice). (j) Statistical analysis by ANCOVA of the energy expenditure of HFD-fed WT and ASKO mice (p=0.105) (WT, n=6 mice; ASKO, n=5 mice). Grey bars/black lines, WT mice; red bars/lines, ASKO mice. All quantitative data are presented as the means ± SEM; *p<0.05 for differences between WT and ASKO mice. Glut4 is also known as Slc2a4. AC, adenylate cyclase; AR, adrenergic receptor; ATM, ATM serine/threonine kinase; FA, fatty acid; GADD45 growth arrest and DNA-damage-inducible 45; GPCR, G-protein-coupled receptor; LXR, liver X receptor; PDE, phosphodiesterase; PKA, protein kinase A; PKIA, protein kinase inhibitor, α; RXR retinoid X receptor; VDR, vitamin D receptor
A gene ontology analysis was performed with differentially expressed genes and, notably, the gene ontology terms of DNA damage and cAMP-mediated signalling were significantly enriched (Fig. 5a,b). The heatmaps of the expression of these genes were profiled based on RNA-Seq data (Fig. 5a [blue and purple dots] and ESM Fig. 5a,b). Further, DNA-damage- and lipolysis-related genes, including p21 (also known as Cdkn1a), Ccnb1, Cks1b, Cks2, Cdca5, Adrb3, Atgl (also known as Pnpla2) and Hsl, were confirmed by qPCR analysis, while the expression of lipogenesis genes was unchanged (Fig. 5c,d). Levels of proteins associated with DNA damage (γ-H2AX, p53 and p21) and lipolysis (ADRB3, p563HSL, p565HSL and ATGL) were also elevated in the gWAT of HFD-fed ASKO mice compared with WT control mice (Fig. 5e). Consistent with higher expression of lipolysis-related genes, isoprenaline stimulation significantly increased the release of glycerol from gWAT explants isolated from HFD-fed ASKO mice (Fig. 5f).
We speculated that lipolysis and DNA damage in the ASKO mice may be related to the hyperaccumulation of copper in adipocytes. To explore this relationship further, we examined the effect of excess copper on lipolysis and DNA-damage markers using a cultured 3T3-L1 adipocyte model. The 3T3-L1 cells were differentiated into adipocytes in vitro, confirmed by the formation of lipid droplets and the induction of PPARγ and CEBPα (ESM Fig. 6). Treatment of mature 3T3-L1 adipocytes with copper was found to stimulate the expression of proteins associated with DNA damage and lipolysis (Fig. 5g). A cellular model for copper-overload-induced lipolysis is summarised in Fig. 5h. It is of interest to compare energy expenditure between both groups of mice on HFD. Metabolic cage data revealed higher relative oxygen consumption rates in ASKO mice (Fig. 5i); however, no significant difference in oxygen consumption of both mice was found when body weight was a covariate (p = 0.105 [Fig. 5j]).

Deletion of Atp7a is associated with differential expression of genes involved in DNA damage and lipid metabolism in the sWAT of aged mice fed with chow diet. Expression levels of genes related to DNA damage (a–c), and adipokine, lipogenesis and lipolysis (d–f) genes, in ASKO and WT mice fed a chow diet for 6 months (a, d; n=6 mice/group), 12 months (b, e; WT, n=7 mice; ASKO, n=5 mice) and 16 months (c, f; n=6 mice/group). Grey bars, WT mice; red bars, ASKO mice. Data are expressed as relative units using the \( {2}^{-\varDelta \varDelta {\mathrm{C}}_{\mathrm{t}}} \)method and presented as the means ± SEM; *p<0.05, **p<0.01 and ***p<0.001 for differences between WT and ASKO mice
Accelerated DNA damage in WAT of aged ASKO mice
As described above, excessive energy intake led to a significant increase in the expression of genes involved in repairing DNA damage in the adipose tissues of ASKO mice. This prompted us to investigate the age dependency of this response. The expression of DNA damage genes (e.g. Cks2, Cdca8, Mad2l1) was significantly elevated in ASKO mice by 6 months of age (Fig. 6a), and was further increased in ASKO mice at 12 months (Fig. 6b) and 16 months of age (Fig. 6c). The expression of genes encoding proteins involved in lipogenesis and lipolysis and adipokines was also examined in the sWAT of WT and ASKO mice at 6, 12 and 16 months of age. No significant differences in gene expression were observed between WT and ASKO mice at 6 months of age (Fig. 6d). However, by 12 and 16 months of age there were significant decreases in the expression of genes for adiponectin and leptin, and an induction of resistin and lipolysis-related genes in the sWAT of ASKO mice (Fig. 6e,f). We also measured the serum glycerol and NEFA in both WT and ASKO at different ages. Neither glycerol nor NEFA was changed in 6- and 12-month-old ASKO mice, but levels were slightly decreased in 16-month-old ASKO mice (ESM Fig. 7a,b).

Decreased expression of genes/proteins involved in the thermogenic programme and lipolysis in BAT of aged mice on chow diet. (a, b) mRNA expression levels of BAT-activation-related genes (a) and lipolysis genes (b) in BAT of mice at 6, 12 and 16 months of age, expressed as relative units using the \( {2}^{-\varDelta \varDelta {\mathrm{C}}_{\mathrm{t}}} \) method. (c) Western blots for uncoupling protein 1 (mitochondrial, proton carrier) (UCP1) and lipolysis markers in the BAT of 16-month-old ASKO and WT mice. One representative blot from three independent experiments is shown. White bars, 6-month-old ASKO mice; pink bars, 12-month-old ASKO mice; blue bars, 16-month-old ASKO mice. All quantitative data are presented as the means ± SEM; *p<0.05 and **p<0.01 for differences between WT and ASKO mice
BAT activation genes are suppressed in aged ASKO mice
Our results reveal that adipocyte-specific deletion of Atp7a stimulated an age-dependent increase in BAT mass which was associated with tissue hypertrophy. We tested whether this might be associated with age-dependent changes in the expression of BAT-activation-related genes. Our data indicate that the expression of Ucp1, Elovl3, Dio2 and Pgc1α (also known as Ppargc1a) was progressively downregulated in the BAT of aged ASKO mice (Fig. 7a). In addition, there was a significant alteration in the expression of lipolysis-related genes at both the RNA and protein levels in the BAT of 12- and 16-month-old ASKO mice (ESM Fig. 8 and Fig. 7b,c).

Model depicting the effect of adipose Atp7a deficiency on the regulation of ageing-related metabolic disease. Inactivation of adipocyte ATP7A results in massive copper accumulation, activation of DNA-damage signalling, fat metabolism dysregulation and abnormal secretion of adipokines, including HSL and ATGL, in aged and HFD-fed mice
Discussion
The goal of this study was to investigate the requirement for ATP7A-mediated copper transport in the whole-body fat metabolism of mice. Our results show that mature adipocytes lacking ATP7A exhibited a significant loss of adipose tissue in an age-dependent manner and in response to HFD intake, leading to increased susceptibility to hepatic steatosis and insulin resistance. The increase in copper level in adipocytes due to the loss of ATP7A was associated with changes in the expression of genes involved in lipolysis and DNA damage checkpoint regulation, resulting in adipose tissue dysfunction and changes in metabolic variables in ASKO mice (Fig. 8).
Although our ASKO mice exhibited progressive adipose tissue loss, the changes in adiposity did not significantly manifest until 12 months of age. This phenotype is notably different from the reported phenotypes of mouse models of lipodystrophy, such as Agpat2-knockout mice [33], adipocyte-specific-Bscl2-knockout mice [17, 34] and mice null for Fsp27 (also known as Cidec) on an ob/ob background [35]. Unlike our ASKO mice, these models exhibit significantly reduced fat deposition at early time points, culminating in more severe metabolic consequences. In addition, the severe histological phenotypes associated in these lipodystrophic models (abnormal adipocyte morphology with enlarged cytoplasm and irregular lipid droplets) was generally absent in the adipocytes of ASKO mice. Furthermore, as it is well-known that copper overload induces cellular DNA damage accumulation [36], the upregulation of DNA-damage-related genes in the WAT of either aged or HFD-fed ASKO mice leading to weight loss and ageing-related metabolic phenotypes suggests that these changes are associated with copper hyperaccumulation. Taken together, these observations suggest that the ASKO mice are a model of accelerated lipoatrophy.
Adipose tissue dysfunction is often accompanied by a fatty liver. Although increased fat lipolysis and energy expenditure can partially compensate for the defects in lipid storage in ASKO mice, the modest increase in energy expenditure was apparently not sufficient to dispose of all the excess lipid/energy delivered to the WAT under conditions of high-fat intake, as there was a significant accumulation of lipids in the liver of ASKO mice. In addition, it is likely that the decreased leptin levels in ASKO mice were responsible for the hyperphagia and elevated ectopic fat deposition in the livers of these mutant mice.
A notable finding of our study was the development of insulin resistance in aged and HFD-fed ASKO mice. This may have been caused, in part, by the decrease in adiponectin and leptin secretion, the higher serum triacylglycerol levels and the fatty liver in the ASKO mice. In addition, previous studies have suggested that ageing of adipose tissue, in particular that associated with elevated DNA damage (higher levels of p53 and p21), plays key roles in the development of insulin resistance [37, 38]. Moreover, the induction of DNA damage in adipocytes has been shown to increase adipocyte insulin resistance and lipolytic activity in vitro [39] and in vivo [37]. The DNA-damage markers p53 and p21 were significantly elevated in the WAT of aged and HFD-fed ASKO mice; we speculate that the insulin resistance in these mice may have arisen from DNA damage. It will be important in future studies to investigate the mechanisms by which copper accumulation contributes to these phenotypes.
It has been reported that endogenous copper promotes lipolysis by altering the activity of the cAMP-degrading phosphodiesterase PDE3B in vitro [22]. Consistent with this concept, we observed an increased lipolysis in the copper-loaded adipose tissues from aged and HFD-fed ASKO mice. Conversely, a copper-deficient diet is thought to promote lipid biosynthesis, which has raised the possibility that copper-related cell signalling might be a potential therapeutic target in the development of treatments for obesity and type 2 diabetes [40]. However, in the context of age or high-fat intake, our results suggest that higher cellular copper concentrations in adipocytes can accelerate the ‘lipoatrophic’ insulin-resistant state, rather than a ‘lean and healthy’ state in animals. Therefore, well-designed in vitro studies and ‘lean’ animal models will be important for developing copper-centred treatment strategies for human metabolic diseases, such as obesity and diabetes.
In summary, our study reveals a previously unappreciated role of adipocyte Atp7a expression in regulating whole-body fat metabolism. The adipocyte-specific ablation of ATP7A significantly reduces WAT deposition under conditions of energy overload, thereby leading to an increased susceptibility to hepatic steatosis and insulin resistance. It will be important to understand the possible mechanisms by which copper-induced DNA damage in fat depots leads to accelerated age-related lipoatrophy. This study not only improves our understanding of the physiological role of adipocyte copper levels in fat metabolism in vivo, but also provides a novel and tractable animal model of accelerated lipoatrophy for better understanding of premature ageing and senescence of adipose tissue.
Data availability
The datasets generated during the current study are available in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number CRA001769 (http://bigd.big.ac.cn/gsa).
Abbreviations
- ADRB3:
-
Adrenoceptor β3
- ASKO:
-
Adipocyte-specific Atp7a-knockout
- ATGL:
-
Adipose triglyceride lipase
- ATP7A:
-
ATPase copper-transporting α
- BAT:
-
Brown adipose tissue
- CEBPα:
-
CCAAT enhancer binding protein α
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- gWAT:
-
Gonadal white adipose tissue
- γ-H2AX:
-
Phosphorylated histone H2AX
- HFD:
-
High-fat diet
- HSL:
-
Hormone-sensitive lipase
- ICP-MS:
-
Inductively coupled plasma MS
- pWAT:
-
Perinephric white adipose tissue
- p21:
-
Cyclin-dependent kinase inhibitor 1A
- p53:
-
Tumour protein p53
- p563HSL:
-
Phospho-HSL Ser563
- p565HSL:
-
Phospho-HSL Ser565
- PPARγ:
-
Peroxisome proliferator activated receptor γ
- qPCR:
-
Quantitative PCR
- RNA-Seq:
-
RNA sequencing
- sWAT:
-
Subcutaneous white adipose tissue
- TEM:
-
Transmission electron microscopy
- WAT:
-
White adipose tissue
- WT:
-
Wild-type
References
Kardos J, Héja L, Simon Á, Jablonkai I, Kovács R, Jemnitz K (2018) Copper signalling: causes and consequences. Cell Commun Signal 16(1):71. https://doi.org/10.1186/s12964-018-0277-3
Kodama H, Fujisawa C (2009) Copper metabolism and inherited copper transport disorders: molecular mechanisms, screening, and treatment. Metallomics 1(1):42–52. https://doi.org/10.1039/B816011M
Morrell A, Tallino S, Yu L, Burkhead JL (2017) The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life 69(4):263–270. https://doi.org/10.1002/iub.1613
Aigner E, Strasser M, Haufe H et al (2010) A role for low hepatic copper concentrations in nonalcoholic fatty liver disease. Am J Gastroenterol 105(9):1978–1985. https://doi.org/10.1038/ajg.2010.170
Feldman A, Aigner E, Weghuber D, Paulmichl K (2015) The potential role of iron and copper in pediatric obesity and nonalcoholic fatty liver disease. Biomed Res Int. www.hindawi.com/journals/bmri/2015/287401/. Accessed 7 Jan 2019 2015:1–7. https://doi.org/10.1155/2015/287401
Klevay LM (2011) Is the Western diet adequate in copper? J Trace Elem Med Biol 25(4):204–212. https://doi.org/10.1016/j.jtemb.2011.08.146
Al-Othman AA, Rosenstein F, Lei KY (1992) Copper deficiency alters plasma pool size, percent composition and concentration of lipoprotein components in rats. J Nutr 122(6):1199–1204. https://doi.org/10.1093/jn/122.6.1199
Al-Othman AA, Rosenstein F, Lei KY (1993) Copper deficiency increases in vivo hepatic synthesis of fatty acids, triacylglycerols, and phospholipids in rats. Proc Soc Exp Biol Med 204(1):97–103. https://doi.org/10.3181/00379727-204-43640
Song M, Vos MB, McClain CJ (2018) Copper-fructose interactions: a novel mechanism in the pathogenesis of NAFLD. Nutrients 10(11). https://doi.org/10.3390/nu10111815
Levy E, Brunet S, Alvarez F et al (2007) Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of Wilson’s disease. Life Sci 80(16):1472–1483. https://doi.org/10.1016/j.lfs.2007.01.017
Seessle J, Gohdes A, Gotthardt DN et al (2011) Alterations of lipid metabolism in Wilson disease. Lipids Health Dis 10(1):83. https://doi.org/10.1186/1476-511X-10-83
Hamilton JP, Koganti L, Muchenditsi A et al (2016) Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B−/− (Wilson disease) mice. Hepatology 63(6):1828–1841. https://doi.org/10.1002/hep.28406
Tan X-Y, Luo Z, Liu X, Xie C-X (2011) Dietary copper requirement of juvenile yellow catfish Pelteobagrus fulvidraco. Aquac Nutr 17(2):170–176. https://doi.org/10.1111/j.1365-2095.2009.00720.x
Chen Q-L, Luo Z, Wu K et al (2015) Differential effects of dietary copper deficiency and excess on lipid metabolism in yellow catfish Pelteobagrus fulvidraco. Comp Biochem Physiol B: Biochem Mol Biol 184:19–28. https://doi.org/10.1016/j.cbpb.2015.02.004
Engle TE (2011) Copper and lipid metabolism in beef cattle: A review. J Anim Sci 89(2):591–596. https://doi.org/10.2527/jas.2010-3395
Scherer PE (2006) Adipose tissue from lipid storage compartment to endocrine organ. Diabetes 55(6):1537–1545. https://doi.org/10.2337/db06-0263
Liu L, Jiang Q, Wang X et al (2014) Adipose-specific knockout of Seipin/Bscl2 results in progressive lipodystrophy. Diabetes 63(7):2320–2331. https://doi.org/10.2337/db13-0729
Softic S, Boucher J, Solheim MH et al (2016) Lipodystrophy due to adipose tissue-specific insulin receptor knockout results in progressive NAFLD. Diabetes 65(8):2187–2200. https://doi.org/10.2337/db16-0213
Guilherme A, Virbasius JV, Puri V, Czech MP (2008) Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9(5):367–377. https://doi.org/10.1038/nrm2391
Hajer GR, van Haeften TW, Visseren FLJ (2008) Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J 29(24):2959–2971. https://doi.org/10.1093/eurheartj/ehn387
Rodríguez JP, Ríos S, González M (2002) Modulation of the proliferation and differentiation of human mesenchymal stem cells by copper. J Cell Biochem 85(1):92–100. https://doi.org/10.1002/jcb.10111
Krishnamoorthy L, Cotruvo JA Jr et al (2016) Copper regulates cyclic AMP-dependent lipolysis. Nat Chem Biol 12(8):586–592. https://doi.org/10.1038/nchembio.2098
Yang H, Ralle M, Wolfgang MJ et al (2018) Copper-dependent amino oxidase 3 governs selection of metabolic fuels in adipocytes. PLoS Biol 16(9):e2006519. https://doi.org/10.1371/journal.pbio.2006519
Wang Y, Zhu S, Hodgkinson V et al (2012) Maternofetal and neonatal copper requirements revealed by enterocyte-specific deletion of the Menkes disease protein. Am J Physiol Gastrointest Liver Physiol 303(11):G1236–G1244. https://doi.org/10.1152/ajpgi.00339.2012
Ladomersky E, Khan A, Shanbhag V et al (2017) Host and pathogen copper-transporting P-type ATPases function antagonistically during salmonella infection. Infect Immun 85(9):e00351–e00317. https://doi.org/10.1128/IAI.00351-17
Hodgkinson VL, Dale JM, Garcia ML et al (2015) X-linked spinal muscular atrophy in mice caused by autonomous loss of ATP7A in the motor neuron. J Pathol 236(2):241–250. https://doi.org/10.1002/path.4511
Qin Z, Konaniah ES, Neltner B, Nemenoff RA, Hui DY, Weintraub NL (2010) Participation of ATP7A in macrophage mediated oxidation of LDL. J Lipid Res 51(6):1471–1477. https://doi.org/10.1194/jlr.M003426
Wang C, Liang X, Tao C et al (2017) Induction of copper and iron in acute cold-stimulated brown adipose tissues. Biochem Biophys Res Commun 488(3):496–500. https://doi.org/10.1016/j.bbrc.2017.05.073
Wang Y, Zhu S, Weisman GA, Gitlin JD, Petris MJ (2012) Conditional knockout of the Menkes disease copper transporter demonstrates its critical role in embryogenesis. PLoS One 7(8):e43039. https://doi.org/10.1371/journal.pone.0043039
Yuan X, Wei G, You Y et al (2016) Rutin ameliorates obesity through brown fat activation. FASEB J 31(1):333–345. https://doi.org/10.1096/fj.201600459RR
Yang X, Lu X, Lombès M et al (2010) The G0/G1 switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab 11(3):194–205. https://doi.org/10.1016/j.cmet.2010.02.003
Lin J, Cao C, Tao C et al (2017) Cold adaptation in pigs depends on UCP3 in beige adipocytes. J Mol Cell Biol 9(5):364–375. https://doi.org/10.1093/jmcb/mjx018
Cortés VA, Curtis DE, Sukumaran S et al (2009) Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab 9(2):165–176. https://doi.org/10.1016/j.cmet.2009.01.002
Mcilroy GD, Suchacki K, Roelofs AJ et al (2018) Adipose specific disruption of seipin causes early-onset generalised lipodystrophy and altered fuel utilisation without severe metabolic disease. Mol Metab 10:55–65. https://doi.org/10.1016/j.molmet.2018.01.019
Zhou L, Park S-Y, Xu L et al (2015) Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27-deficient mice. Nat Commun 6(1):5949. https://doi.org/10.1038/ncomms6949
Linder MC (2012) The relationship of copper to DNA damage and damage prevention in humans. Mutat Res Fundam Mol Mech Mutagen 733(1):83–91. https://doi.org/10.1016/j.mrfmmm.2012.03.010
Minamino T, Orimo M, Shimizu I et al (2009) A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 15(9):1082–1087. https://doi.org/10.1038/nm.2014
Stout MB, Justice JN, Nicklas BJ, Kirkland JL (2016) Physiological aging: links among adipose tissue dysfunction, diabetes, and frailty. Physiology 32(1):9–19. https://doi.org/10.1152/physiol.00012.2016
Vergoni B, Cornejo P-J, Gilleron J et al (2016) DNA damage and the activation of the p53 pathway mediate alterations in metabolic and secretory functions of adipocytes. Diabetes 65(10):3062–3074. https://doi.org/10.2337/db16-0014
Burkhead JL, Lutsenko S (2013) The role of copper as a modifier of lipid metabolism. Available from www.intechopen.com/books/lipid-metabolism/the-role-of-copper-as-a-modifier-of-lipid-metabolism. https://doi.org/10.5772/51819
Acknowledgements
We are grateful to W. Jin from the Institute of Zoology for his generous gift of adiponectin-Cre mice and to J. Lin from the Institute of Zoology for his great help with the metabolic cage analysis.
Funding
This research was funded by the Major National Scientific Research Projects (2015CB943101), the National Natural Science Foundation of China (31672387 and 31601929), the Elite Youth Programme of the Chinese Academy of Agricultural Sciences (ASTIP-IAS05) and the Fundamental Research Funds for Central Non-profit Scientific Institution (2016ywf-yb-1).
Author information
Authors and Affiliations
Contributions
YanW and KL initiated and designed the study. CT, YajW, YZ, JP, YF and XL acquired the data. CT, JP and CC analysed the data. JZ, MJP and KL were involved in analysis and interpretation of the data. CT and YanW interpreted the data and drafted the manuscript. JZ, MJP and KL revised the article. All authors revised and approved the final version of the manuscript. YanW is responsible for the integrity of this work.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM
(PDF 526 kb)
Rights and permissions
About this article

Cite this article
Tao, C., Wang, Y., Zhao, Y. et al. Adipocyte-specific disruption of ATPase copper transporting α in mice accelerates lipoatrophy. Diabetologia 62, 2340–2353 (2019). https://doi.org/10.1007/s00125-019-4966-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-4966-2