
Abstract
Owing to their overall low energy scales, flexible molecular architectures, and ease of chemical substitution, molecule-based multiferroics are extraordinarily responsive to external stimuli and exhibit remarkably rich phase diagrams. Even so, the stability and microscopic properties of various magnetic states in close proximity to quantum critical points are highly under-explored in these materials. Inspired by these opportunities, we combined pulsed-field magnetization, first-principles calculations, and numerical simulations to reveal the magnetic field–temperature (B–T) phase diagram of multiferroic (NH4)2FeCl5⋅H2O. In this system, a network of intermolecular hydrogen and halogen bonds creates a competing set of exchange interactions that generates additional structure in the phase diagram—both in the vicinity of the spin flop and near the 30 T transition to the fully saturated state. Consequently, the phase diagrams of (NH4)2FeCl5⋅H2O and its deuterated analog are much more complex than those of other molecule-based multiferroics. The entire series of coupled electric and magnetic transitions can be accessed with a powered magnet, opening the door to exploration and control of properties in this and related materials.
Similar content being viewed by others

Introduction
With their strong coupling between charge, structure, and magnetism, multiferroics are ideal platforms for exploring quantum phase transitions.1,2 These transitions are controlled by external stimuli such as voltage, magnetic field, pressure, and light—quite different from traditional phase transitions that rely upon thermal fluctuations.3,4,5,6,7,8,9 Molecule-based multiferroics offer several advantages over their oxide counterparts for the study of quantum phase transitions. These include overall low-energy scales, flexible molecular architectures, and ease of chemical substitution.10,11,12,13 Thus, in addition to topological similarities with oxide multiferroics, the molecular analogs tend to have lower critical fields that are more easily reached in the laboratory.14,15,16,17 A significant bottleneck to the investigation of molecule-based multiferroics and their application for ultra low-power multi-state memory, switching devices, and novel computing architectures18,19,20,21 is the inadequate understanding of the different equilibrium states and their proximity to quantum critical points. Uncovering the full magnetic field–temperature (B–T) phase diagram is a significant step toward addressing these issues.10,22 At the same time, revealing the nature of the various magnetic quantum phase transitions and the character of the competing phases provides important opportunities for property control and tests our theoretical understanding of quantum magnets.10,15,16,17,23
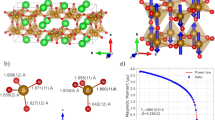
These ideas can be explored in the erythrosiderites, the class of compounds with chemical formula A2FeX5⋅H2O, where A is ammonium or an alkali metal and X is a halide. We focus on multiferroic (NH4)2FeCl5⋅H2O and its deuterated analog due to their spontaneous electric polarization and unusual magnetic behavior.24,25 The orthorhombic unit cell consists of four octahedral [FeCl5⋅H2O]2− groups and eight \({\mathrm{NH}}_4^ +\) ions that are all symmetrically equivalent.24 Fig. 1c summarizes the temperature-driven transitions. These include (i) an order–disorder transition involving \({\mathrm{NH}}_4^ +\) [TO/D ≈ 79 K], (ii) the Néel transition below which incommensurate-collinear sinusoidal magnetic order appears [TN ≈ 7.25 K], and (iii) a transition to an incommensurate-cycloidal spin state that supports a spontaneous electric polarization primarily along the a-axis [TFE ≈ 6.9 K].26,27,28,29 A fascinating series of spin and polarization flops occur with increasing magnetic field.24,25 These magnetically-driven transitions will be discussed in greater detail below. There is an abundance of hydrogen and halogen bonding between neighboring [FeCl5⋅H2O]2− octahedra. As shown in Fig. 1a, b, these intermolecular interactions form a set of superexchange pathways linking the Fe3+\(\left( {S = \frac{5}{2}} \right)\) centers.28,29,30,31 The full set of magnetic interaction pathways includes Fe-O-H⋯Cl-Fe, Fe-Cl⋯Cl-Fe, and Fe-Cl⋯O-Fe linkages. The magnetic interaction through O-H⋯Cl is the strongest and forms a zig-zag chain along the b-axis.24 As (NH4)2FeCl5⋅H2O is a type-II multiferroic, polarization derives from the spin configuration. Therefore, one cannot begin to understand polarization, polarization flops, and magnetoelectric coupling without first examining the underlying spin system and determining the B–T phase diagram.

Crystal structure, exchange interactions, and energy scales. a Crystal structure of (NH4)2FeCl5⋅H2O at 300 K with five different exchange interactions are represented with colored dash line. J1 pass between two water ligands (Fe-O-H⋯Cl-Fe). J2 and J3 pass between two Cl− ions (Fe-Cl⋯Cl-Fe). J4 and J5 pass between two \({\mathrm{NH}}_4^ +\) ions (Fe-Cl⋯O-Fe). The magnetic coupling is three dimensional: strong quasi-two-dimensional interactions with antiferromagnetically coupled planes. b Calculated J values against distance between Fe3+ ions. c Schematic depicting the sequence of temperature and magnetic field transitions that occur in (NH4)2FeCl5⋅H2O. d Ratio of J interactions with respect to J1 depicting how the collinear and non-collinear states are stabilized with respect to the nature of the counterion. The blue star in the non-collinear state corresponds to (NH4)2FeCl5⋅H2O. The red star in the collinear state corresponds to K2FeCl5⋅H2O whose exchange constants are taken from ref. 31
We combined pulsed-field magnetization with complementary first-principles calculations and numerical simulations to reveal the magnetic properties and complete B–T phase diagram of multiferroic (NH4)2FeCl5⋅H2O. Significantly, all of the magnetically driven transitions—including that to the fully saturated state—are within the range of experimentally available powered magnets. The overall complexity of the phase diagram arises from the competing exchange pathways (J1–J5) produced by an elaborate network of intermolecular hydrogen and halogen bonds. The magnetic quantum phase transition and its satellites are the most striking of the sequence, and the discovery of these structures is one of the major factors that make this system a quantum material. That they occur in a compound where the exchange interactions consist entirely of intermolecular hydrogen and halogen bonds but still sport similarities with other well-known multiferroics such as TbMnO3 and MnWO4 is remarkable.32,33,34,35,36,37 Structure–property relations involving counterion substitution and spin-state noncollinearity are also discussed.
Results and discussion
Magnetic properties and field-induced transitions in (NH4)2FeCl5⋅H2O
Figure 2 summarizes the pulsed-field magnetization of (NH4)2FeCl5⋅H2O and its deuterated analog. There are numerous field-induced transitions depending on the isotropic decoration and the direction of the applied field. They naturally separate into two groups: (i) a series of low field spin reorientations below 6 T and (ii) the saturation field and its satellites near 30 T. These satellites are evident in Fig. 2e, f and are associated with competing exchange pathways. They are discussed in detail below. The low field features are small but clearly revealed for B || c, whereas they are more complicated for B || a. The transition to the fully polarized state (BSat) is very distinct and occurs near 30 T in each case. This is the energy scale above which all frustration is relieved. Depending on the field direction, we find one or two small satellites in the vicinity of BSat—evidence for a series of quasi-isoenergetic phases near the quantum phase transition. These values are summarized in Table 1.

Pulsed-field magnetization of (NH4)2FeCl5⋅H2O and locating the critical fields. a, b Low temperature magnetization of (NH4)2FeCl5⋅H2O and its deuterated analog for B || c. c, d Pulsed-field magnetization of (NH4)2FeCl5⋅H2O and its deuterated analog for B || a. The insets show a close-up view of ∂M/∂B at low fields—in the vicinity of the series of metamagnetic transitions. Derivative of the magnetization in the vicinity of the spin reorientation (e) and saturation fields (f) for (NH4)2FeCl5⋅H2O with fields applied || c
The overall shape of the magnetization is consistent with expectations for a three-dimensional (reasonably isotropic with \(S \;>\; \frac{1}{2}\)) material, with a linear rather than concave rise on approach to BSat.17 This is in line with our numerical modeling (discussed below) from which we extract the various exchange interactions. We can also estimate the value of the primary exchange interaction from the size of the ultimate critical field. Taking BSat = 30.3 T for the field along the c direction in the hydrogenated sample and using a simple Hamiltonian38 with one exchange constant \({\cal{H}} = - (J/2) \sum\nolimits_{i \ne j} {{\mathbf{S}}_i \cdot {\mathbf{S}}_j} - g\mu _{\mathrm{B}}B \sum\nolimits_i {S_i^z}\), we extract J = −1.32 K. This value is in reasonable agreement with J1 from our modeling calculations. Here, the primary exchange interaction corresponds to the Fe-O-H⋯Cl-Fe pathway as shown in Fig. 1b. It also compares well with that of the isomorphic K2FeCl5⋅H2O analog (J1 = −1.65 K).31
The saturation fields in Table 1 reveal several interesting structure–property relations. In each case, BSat ≈ 30 T, with modest variations that depend upon isotopic substitution and field direction. This suggests that the anisotropies and Dyaloshinski–Moriya interactions are not large in (NH4)2FeCl5⋅H2O. This is consistent with our predictions for small anisotropies, summarized below. Additional evidence for overall three-dimensional antiferromagnetism comes from the linear magnetization between 5 and 30 T. We also find that BSat,D < BSat,H. This is a very common trend—similar isotope effects were reported in [Ni(HF2)(pyz-d4)2]SbF6 and CuF2(pyz)(H2O)2.39,40 Deuteration typically reduces the exchange interactions by a few percent, because a heavier atom has a smaller excursion from its equilibrium position in the anharmonic potential.39,40 Deuterium substitution also tends to smear out the multiple lower field transitions. Moreover, we find that BSat,a > BSat,c. This deviation from isotopic behavior is probably related to g-factor anisotropy.17 There is also a more complex set of magnetically driven transitions when B || a.
Uncovering the spin structure and magnetic exchange interactions
To further explore these ideas, we carried out several different numerical simulations, first to understand the zero-field ground state and then to interpret the field-induced transitions. In zero field, the Hamiltonian can be written as
where Jij is the isotropic exchange interaction between Si and Sj, Kb is the single-ion anisotropy, and Si are classical spin vectors. The actual value of the parameters used in this simulation were obtained from density functional theory (DFT). Details are available in the Methods section and Supplementary Information.
Our numerical simulations produce spin structures that are remarkably consistent with the experimental results (Fig. 3a). Moreover, the experimentally observed wave vector of the spin spiral is Q ≈ 0.228 ± 0.02 reciprocal lattice units26, which compares well with the numerically calculated value of Q ≈ 0.205 ± 0.001 (the small difference in Q may be attributed to the effects of single-ion anisotropy along the a- and c-axis). These results demonstrate the validity of the exchange interaction parameters obtained from DFT and our model Hamiltonian. To determine the origin of non-collinear spin structure, we investigated how exchange interactions change the magnetic ground state. Interestingly, even in the absence of J1, J3, and J5, we can reproduce the non-collinear spin structure (Supplementary Fig. S3). However, in the absence of J2 or J4, which form a triangular plaquette (Fig. 1a), a collinear spin structure is stable. Therefore, geometrical frustration with competing antiferromagnetic J2 and J4 in a triangular lattice stabilizes the non-collinear spin state propagating along the c direction.

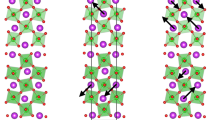
Numerical simulation results for the spin ground state and spin density across the magnetic quantum phase transition. a The red arrows represent the spin structures experimentally observed from the neutron diffraction.26,27 The blue arrows are the spin structure obtained from the numerical simulations with exchange interaction parameters from DFT calculations. b, c Spin density in the non-collinear state as viewed along a and c. d Calculated spin density in the ferromagnetic state as viewed along the c direction. The sign is given by the color bar
The collinear spin structures observed in certain other A2FeX5⋅D2O erythrosiderites can also be explained within this framework, as J4 is mediated by the ammonium ions. The latter have very different characteristics compared with alkaline ions. The absence of ammonium ions relieves magnetic frustration by making either J2 and/or J4 dominant. Moreover, the interactions between triangular lattice planes are mediated by the strongest antiferromagnetic exchange J1. This makes the spin spirals on the different triangular lattice planes almost antiferromagnetic, a prediction that has been experimentally confirmed.26 In triangular lattice antiferromagnets, exotic magnetic ground states and successive magnetic phase transitions have been observed.41 Thus, the series of magnetic phase transitions observed in field also arises from the subtly competing exchange interactions in this system. These calculations also suggest that pressure may provide a pathway between the different A2FeX5⋅H2O materials (Fig. 1d). As the phase diagram indicates, the spin-state transition occurs with increasing J3/J1. We therefore speculate that uniaxial pressure applied along J3 could induce a transition from a non-collinear to collinear spin structure. On the other hand, hydrostatic pressure is unlikely to induce a magnetoelastic phase transition, as the J-ratios would be more or less the same.
We also carried out first-principles calculations to obtain the spin densities in the non-collinear antiferromagnetic and ferromagnetic states of (NH4)2FeCl5⋅H2O. These results are summarized in Fig. 3. In the zero-field (non-collinear antiferromagnetic) state, the spin density resides primarily on the Fe3+ centers and to a lesser extent on the chlorine and water ligands. The phase of the spin density alternates characteristically42 and the empty space between ligands is the node between the up- and down-spin states. In the full-field (ferromagnetic) state, the spin-density pattern of Fe3+ is similar to that of the ligands but with an overall in-phase (rather than out-of-phase) arrangement. Another difference in the spin density can be found on the water ligand extending toward the nearest chlorine through the H-O⋯Cl hydrogen bonding pathway. As discussed previously, the H-O⋯Cl pathway produces the primary exchange interaction J1. The additional spin density across this intermolecular hydrogen bond suggests that J1 increases in the fully saturated state. Hence, the field-induced change in spin density is linked to modifications of the magnetic exchange interactions. This spin density redistribution also contributes to the lack of inversion symmetry by exacerbating the anti-alignment of polarization and magnetization.
Revealing the phase diagram of (NH4)2FeCl5⋅H2O
We can use the pulsed-field magnetization data in Fig. 2 to develop the B–T phase diagram. In order to precisely determine the location of the various phase boundaries and track their dependence on the external stimuli, we calculated (∂M/∂B)T and plotted these curves vs. field (Fig. 2e, f). The position of the spin flop and transition to the fully polarized state is clear. The use of derivative techniques is particularly advantageous when following trends in small features. For instance, careful analysis of the derivative structure in the vicinity of the spin-flop transitions yields three phase boundaries (Fig. 2e). Analysis in the vicinity of the saturation field (indicated with an arrow) reveals a series of transitions as well (Fig. 2f). (∂M/∂B)T also indicates the relative importance of magnetism at each phase boundary. These derivative techniques uncover two important regions: (i) the low-field spin reorientations below 6 T and (ii) the high-field region approaching (and including) the fully polarized state at BSat ≈ 30 T. With increasing temperature, the position and relative importance of the different peaks in the derivative response evolves.
Figure 4 displays the B–T phase diagram of (NH4)2FeCl5⋅H2O for B || c. We label the different regimes in accord with prior magnetization, polarization, magnetostriction, and neutron scattering.24,25,26,27,29 Here, AFM = antiferromagnetic, PM = paramagnetic, ICC = incommensurate cycloidal, CS = cycloidal spiral, C1 = distorted cycloid, C2 = quasi-collinear, CLS = collinear sinusodial, FE = ferroelectric, and NE = non-electric. Of course, the transition to the fully polarized state (BSat ≈ 30 T) along with two additional reorientation transitions immediately preceding it are entirely new—providing the first glimpse of the full complexity of the B–T diagram in (NH4)2FeCl5⋅H2O. Similar phase diagrams for B || a and for the deuterated crystals are available in the Supplementary Information.

Developing the phase diagram. a B–T phase diagram of (NH4)2FeCl5⋅H2O for B || c obtained from an analysis of the magnetization at various temperatures. The zero-field transitions are taken from ref. 24. b Close-up view of the low-field region of the diagram showing the numerous phases labeled according to polarization, magnetostriction, and neutron scattering.24,25,26,27 Here, AFM = antiferromagnetic, PM = paramagnetic, ICC = incommensurate cycloidal, CS = cycloidal spiral, C1 = distorted cycloid, C2 = quasi-collinear, CLS = collinear sinusodial, FE = ferroelectric, and NE = non-electric. Data points from prior studies up to 15 T are indicated by open white circles.24
With increasing magnetic field, (NH4)2FeCl5H2O undergoes a sequence of transitions where the quantum fluctuations present in the antiferromagnetic ground state are suppressed as the system is driven into the fully polarized state. The primary high-field transition at 30 T is a quantum phase transition driven by external stimuli rather than thermal fluctuations.8,9,10,22 The magnitude of BSat is linked to the largest exchange interaction (J1), with a pathway of Fe-O-H ⋯ Cl-Fe. Moreover, the spin density redistributes to favor this pathway in the ferromagnetic state (Fig. 3d). The other exchange interactions are similar in magnitude but smaller. Thus, although we cannot explicitly link J2–5 to specific phase boundaries, it stands to reason that the more subtle transitions at 26.9 and 28.6 T can be attributed to the lesser, competing J’s. For instance, J2 and J4 are the next largest superexchange interactions and may be correlated with the weak 28.6 T phase boundary, whereas J3 and J5 may be linked with the 26.9 T boundary. Obviously, all of the expected five spin re-orientations should be resolved at the lowest temperatures.
In the lower field regime, (NH4)2FeCl5⋅H2O undergoes a series of transitions in the vicinity of the 3.7 T spin-flop transition (Fig. 4b). As before, this cascade of transitions is due to the many competing exchange pathways in this compound. This grouping can be linked to the five exchange pathways, although resolution is lost due to similarities in magnitude and thermal broadening. Triangular frustration created by J2 and J4 (Fig. 1a) is alleviated as magnetic field increases. With increasing temperature, the low-field phase boundaries converge toward a pair of triple points between AFM 1, AFM 2, and AFM 3, as well as AFM 1, AFM 3, and CLS/NE phases. Overall, the low-field portion of the magnetic phase diagram is in excellent agreement with that of Ackermann et al.24 The main exception is the splitting of the 3.7 T spin flop at base temperature (Fig. 2e).
Although the properties and complex multiferroic phases of (NH4)2FeCl5⋅H2O have been extensively studied in the low-field regime,24,25,26,27,29 it is interesting to anticipate the high-field response. In general, for type-II multiferroics, the spins need to break spatial inversion symmetry in order to create an electric polarization. Common spin structures that break spatial inversion symmetry are various kinds of spirals.43,44,45 If the spins are fully aligned with the magnetic field, then there is no spatial inversion symmetry-breaking spin structure available to create electric polarization. In other words, above the 30 T transition to the fully saturated spin state, the polarization is likely to be quenched. The high-field state is therefore unlikely to be magnetoelectric. Things are different at 5 T. Here, the spin configuration is quasi-collinear AFM 3 and the system is in the FEIII phase with polarization along c.24,27 Whether the spin structure, polarization, and magnetoelectric coupling at 5 T24,25,26,27,29 are similar to those further away from equilibrium—for instance, at 27 T—awaits further study. That ferroelectricity depends so intimately upon the spin configuration is the overarching motivation for revealing the magnetic phases of (NH4)2FeCl5⋅H2O and unveiling the overall structure of the phase diagram.
What differentiates the B–T phase diagram of (NH4)2FeCl5⋅H2O from that of other molecule-based multiferroics is the exceptional level of detail. For instance, in metal-organic framework compounds such as (CH3)2NH2]Mn(HCOO)3, the phase diagram exhibits a spin flop near 0.3 T, a broad canted phase, and a transition to the fully saturated state at 15.3 T.22 Likewise, in CaCo2As2, the critical fields are 3.7 and 7.5 T.46 The overall simplicity of these B–T phase diagrams arises from the superexchange pathways through formate ligands or Co⋯As interactions, respectively. Magnetic exchange, of course, can also take place through hydrogen and halide bonds. This is the situation in (NH4)2FeCl5⋅H2O and a number of other molecule-based materials.47,48,49,50,51 What makes (NH4)2FeCl5⋅H2O so unusual is that there are five isoenergetic intermolecular hydrogen and halide bonds that function as superexchange pathways. In addition to the extraordinary softness, this gives rise to an unusual degree of frustration that is manifested as a series field-driven transitions and an extremely complex B–T phase diagram. Traditionally, these systems have been under-explored, but our work on (NH4)2FeCl5⋅H2O is a major step toward changing this situation. Other members of the erythrosiderite family such as K2FeCl5⋅H2O do not seem to be as soft as the ammonium compound. The K+-containing system is also a collinear antiferromagnet rather than a multiferroic. This difference has been a puzzle for some time and appears to be related to the character of the superexchange network as discussed in the Supplementary Information. Unexpectedly, the spin structure of TbMnO3 is similar to that in (NH4)2FeCl5⋅H2O,24,29,32,33,34 providing yet another reason to more deeply examine the properties in this unusual type-II molecule-based multiferroic.
We combined pulsed-field magnetization measurements with first-principles calculations and numerical simulations of exchange interactions to reveal the magnetic properties of the type-II multiferroic (NH4)2FeCl5⋅H2O. The B–T phase diagram is surprisingly complex with a spin-flop transition between 3 and 5 T, and a transition to the fully saturated state near 30 T—each of which is preceded by a series of weak reorientation transitions that reflect the many quasi-isoenergetic exchange interactions in this material. The latter arise from an elaborate intermolecular hydrogen and halogen bonding network, and in addition to a full description of the non-collinear ground state, we evaluate field-induced changes to the spin density pattern in terms of these intermolecular exchange interactions across the entire field regime. Structure–property relations in (NH4)2FeCl5⋅H2O are discussed with an emphasis on how isotopic decoration impacts the intermolecular hydrogen and halogen bond network and saturation fields. In addition to distinguishing (NH4)2FeCl5⋅H2O from other members of the erythrosiderite family, we compare the phase diagram and character of the exchange interactions with other well-known multiferroics such as [(CH3)2NH2]Mn(HCOO)3 where the metal centers are linked by formate ligands, TbMnO3, which has a similar non-collinear spin structure, and MnWO4 where the different field-induced spin configurations flop the polarization. The development of the phase diagram and our uniquely detailed understanding of the magnetically-driven transitions in (NH4)2FeCl5⋅H2O open the door to the exploration of frustrated multiferroics and other quantum materials in which hydrogen and halogen bonds support magnetic exchange.
Methods
Crystal growth
Hydrogentated and deuterated (NH4)2FeCl5⋅H2O single crystals were grown by solution method using HCl/DCl, FeCl3, and NH4Cl/ND4Cl, respectively.24,29 A sealed saturated solution was kept in a sample environment chamber at 38 °C and allowed to slowly evaporate. Large crystals were obtained. The crystals were characterized by magnetic susceptibility and specific heat measurements, and no significant deuteration-induced effects were recorded between the hydrogenated and deuterated samples. Samples were oriented using morphological faces as references.24
Magnetization measurements
High-field magnetization measurements were performed using a 65 T short-pulse magnet at the National High Magnetic Field Laboratory in Los Alamos using a 1.5 mm bore, 1.5 mm long, 1500-turn compensated-coil susceptometer, constructed from 50-gauge high-purity copper wire as described in ref. 22. In addition to full field pulses, we carried out 10 and 35 T shots to resolve the spin flop and saturation. The critical fields (BSF, BSat, and all satellite transitions) were determined using first derivative techniques. The spin transitions were brought together to create the B–T phase diagram.
First-principles density functional theory
First-principles calculations were performed using DFT within the local density approximation LDA + U method as implemented in the Vienna ab initio simulation package (VASP 5.4.1).52,53,54 We use the Dudarev55 implementation with on-site Coulomb interaction U = 2.0 eV to treat the localized 3d electron states in Fe. Our value for U is chosen to bring Q closest to the experimental value. The projector augmented wave (PAW) potentials56,57 explicitly include eight valence electrons for Fe (3d7 4s1), five for N (2s3 2p3), one for H(1s1), seven for Cl (3s2 3p5), and six for O (2s2 2p4). Before calculating the exchange interactions and spin density, the crystal structure was taken from ref. 58 and the Crystallography open database (COD ID: 9012597). The atomic structure was optimized for both lattice parameters and atomic positions with collinear-antiferromagnetic spin ordering. A plane-wave basis set with a cutoff energy of 500 eV was used. The k-point sampling used the Monkhorst–Pack scheme59 and employed 2 × 3 × 4 and 2 × 3 × 2 meshes for the unit cell and the supercell of (NH4)2FeCl5⋅H2O, respectively. The atomic positions were optimized until the interatomic forces were smaller than 1 meV/Å.
To calculate exchange interactions, we used an energy-mapping analysis for localized spins without spin–orbit coupling.60,61 A 1 × 1 × 2 supercell was chosen to evaluate the exchange interaction J4, which cannot be included in a smaller unit cell. A detailed explanation for the computation of Jij is provided in section 2 of the Supplementary Information. In order to simulate the spin state with Q ≈ 0.23 along the c direction,26,27 a very large supercell would be required. To make this problem more tractable, we assumed that Q = 0.25 and used a 1 × 1 × 4 supercell to reproduce the non-collinear spin ordering. On the other hand, the ferromagnetic spin density was obtained from a collinear spin-polarized scf calculation. The k-point sampling uses the Monkhorst–Pack scheme59 and employs a 2 × 3 × 1 and a 2 × 3 × 4 mesh for non-collinear and collinear spin density calculations, respectively.
Numerical simulations
To find the spin ground state at zero magnetic field from Eq. (1), the exchange interactions were taken from the DFT calculations. In zero magnetic field and with no anisotropy in the cycloidal plane, the spin cycloid can be approximated by a single sinusoidal function.62,63 As single-ion anisotropy along the b-axis preserves the harmonic spin cycloid in the ac plane, no \(S_i^b\left( {\mathbf{R}} \right)\) component was considered. We can safely assume that the spins are classical vectors, as the Fe3+ ions have a large spin value 5/2. Thus, the trial functions for Si(R) are
with \(S_i^y({\mathbf{R}}) = 0\). The index i labels the four distinct spins in the magnetic unit cell (see ref. 26 for spin number assignment in the magnetic unit cell) and ϕi are the phases. For our model, ϕ1 = ϕ2 and ϕ3 = ϕ4.
To evaluate Q and ϕi, we minimized the energy \(E = \left\langle {\cal{H}} \right\rangle\) using periodic boundary conditions along all directions while fixing ϕ1 = 0. For the incommensurate spin state, we minimized the energy within a magnetic unit cell with 2000 sites along c. After minimization, we checked that the classical forces on each spin vanish and we tried multiple Q and ϕi values as initial values in the optimization to avoid metastable states. The detailed procedure to determine the field-dependent magnetization is described in section 3 of the Supplementary Information.
Data Availability
The data that support the findings of this study are available from corresponding authors upon reasonable request.
References
Narayan, A., Cano, A., Balatsky, A. V. & Spaldin, N. A. Multiferroic quantum criticality. Nat. Mater. 18, 223–228 (2019).
Cheong, S.-W. & Mostovoy, M. Multiferroics: a magnetic twist for ferroelectricity. Nat. Mater. 6, 13–20 (2007).
Basov, D. N., Averitt, R. D. & Hsieh, D. Towards properties on demand in quantum materials. Nat. Mater. 16, 1077–1088 (2017).
Coleman, P. & Schofield, A. J. Quantum criticality. Nature 433, 226–229 (2005).
Sachdev, S. & Keimer, B. Quantum criticality. Phys. Today 64, 29–35 (2011).
Wang, Y. et al. Strongly-coupled quantum critical point in an all-in-all-out antiferromagnet. Nat. Commun. 9, 2953 (2018).
Gegenwart, P., Si, Q. & Steglich, F. Quantum criticality in heavy-fermion metals. Nat. Phys. 4, 186–197 (2008).
Goko, T. et al. Restoration of quantum critical behavior by disorder in pressure-tuned (Mn, Fe)Si. npj Quantum Mater. 2, 44 (2017).
Wehinger, B. et al. Giant pressure dependence and dimensionality switching in a metal-organic quantum antiferromagnet. Phys. Rev. Lett. 121, 117201 (2018).
Hughey, K. D. et al. Structure-property relations in multiferroic [(CH3)2NH2]M(HCOO)3 (M = Mn, Co, Ni). Inorg. Chem. 57, 11569–11577 (2018).
Tian, Y. et al. Cross coupling between electric and magnetic orders in a multiferroic metal-organic framework. Sci. Rep. 4, 6062 (2014).
Polyakov, A. O. et al. Coexisting ferromagnetic and ferroelectric order in a CuCl4-based organic-inorganic hybrid. Chem. Mater. 24, 133–139 (2012).
Jain, P. et al. Switchable electric polarization and ferroelectric domains in a metal-organic-framework. npj Quantum Mater. 1, 16012 (2016).
Gómez-Aguirre, L. C. et al. Magnetic ordering-induced multiferroic behavior in [CH3NH3][Co(HCOO)3] metal-organic framework. J. Am. Chem. Soc. 138, 1122–1125 (2016).
Lancaster, T. et al. Controlling magnetic order and quantum disorder in molecule-based magnets. Phys. Rev. Lett. 112, 207201 (2014).
Goddard, P. A. et al. Dimensionality selection in a molecule-based magnet. Phys. Rev. Lett. 108, 077208 (2012).
Goddard, P. A. et al. Experimentally determining the exchange parameters of quasi-two-dimensional Heisenberg magnets. New J. Phys. 10, 083025 (2008).
Mittal, S. A survey of techniques for architecting processor components using domain-wall memory. ACM J. Emerg. Technol. Comput. Syst. 13, 1–25 (2017).
Wang, Y. et al. Unveiling hidden ferrimagnetism and giant magnetoelectricity in polar magnet Fe2Mo3O8. Sci. Rep. 5, 12268 (2015).
Heron, J. T. et al. Deterministic switching of ferromagnetism at room temperature using an electric field. Nature 516, 370–373 (2014).
Clarkson, J. D. et al. Hidden magnetic states emergent under electric field, in a room temperature composite magnetoelectric multiferroic. Sci. Rep. 7, 15460 (2017).
Clune, A. J. et al. Magnetic field-temperature phase diagram of multiferroic [(CH3)2NH2]Mn(HCOO)3. Phys. Rev. B 96, 104424 (2017).
Stroppa, A. et al. Electric control of magnetization and interplay between orbital ordering and ferroelectricity in a multiferroic metal-organic framework. Angew. Chem. Int. Ed. 50, 5847–5850 (2011).
Ackermann, M., Brüning, D., Lorenz, T., Becker, P. & Bohatý, L. Thermodynamic properties of the new multiferroic material (NH4)2FeCl5⋅H2O. New J. Phys. 15, 123001 (2013).
Tian, W. et al. Electronic phase separation and magnetic-field-induced phenomena in molecular multiferroic (ND4)2FeCl5·D2O. Phys. Rev. B 98, 054407 (2018).
Rodríguez-Velamazán, J. A. et al. Magnetically-induced ferroelectricity in the (ND4)2[FeCl5(D2O)] molecular compound. Sci. Rep. 5, 14475 (2015).
Rodríguez-Velamazán, J. A. et al. Magnetic-field-induced change of magnetoelectric coupling in the hybrid multiferroic (ND4)2[FeCl5·D2O]. Phys. Rev. B 95, 174439 (2017).
Carlin, R. L. & Palacio, F. The A2[FeX5(H2O)] series of antiferromagnets. Coord. Chem. Rev. 65, 141 (1985).
Tian, W. et al. Spin-lattice coupling mediated multiferroicity in (ND4)2FeCl5D2O. Phys. Rev. B 94, 214405 (2016).
Luzón, J., Campo, J., Palacio, F., McIntyre, G. J. & Millán, A. Understanding magnetic interactions in the series A2FeX5 · H2O (A = K, Rb; X = Cl, Br). I. Spin densities by polarized neutron diffraction and DFT calculations. Phys. Rev. B 78, 054414 (2008).
Campo, J. et al. Understanding magnetic interactions in the series A2FeX5·H2O (A = K, Rb; X = Cl, Br). II. Inelastic neutron scattering and DFT studies. Phys. Rev. B 78, 054415 (2008).
Argyriou, D. N. et al. Melting of incommensurate-ferroelectric phase with magnetic field in multiferroic TbMnO3. Phys. Rev. B 75, 020101(R) (2007).
Kimura, T. et al. Magnetic control of ferroelectric polarization. Nature 426, 55–58 (2003).
Solovyev, I. V. Spin-spiral inhomogeneity as the origin of ferroelectric activity in orthorhombic manganites. Phys. Rev. B 83, 054404 (2011).
Taniguchi, K., Abe, N., Takenobu, T., Iwasa, Y. & Arima, T. Ferroelectric polarization flop in a frustrated magnet MnWO4 induced by a magnetic field. Phys. Rev. Lett. 97, 097203 (2006).
Lorenz, B. Hexagonal manganites - (RMnO3): class (I) multiferroics with strong coupling of magnetism and ferroelectricity. ISRN Condensed Matter Phys. 2013, 497073 (2013).
Wang, J. et al. Comprehensive inelastic neutron scattering studdy of the multiferroic Mn1−xCoxWO4. Phys. Rev. B 98, 214425 (2018).
Brinzari, T. V. et al. Electron-phonon and magnetoelastic interactions in ferromagnetic Co[N(CN)2]2. Phys. Rev. Lett. 111, 047202 (2013).
Brambleby, J. et al. Combining microscopic and macroscopic probes to untangle the single-ion anisotropy and exchange energies in an S = 1 quantum antiferromagnet. Phys. Rev. B 95, 134435 (2017).
Goddard, P. A. et al. Isotope effect in quasi-two-dimensional metal-organic antiferromagnets. Phys. Rev. B 78, 052408 (2008).
Lee, M. et al. Magnetic phase diagram and multiferroicity of Ba3MnNb2O9: A spin-5/2 triangular lattice antiferromagnet with weak easy-axis anisotropy. Phys. Rev. B 90, 224402 (2014).
Musfeldt, J. L. et al. Magnetoelastic coupling through the antiferromagnet-to-ferromagnet transition of quasi-two-dimensional [Cu(HF2)(pyz)2]BF4 using infrared spectroscopy. Phys. Rev. Lett. 103, 157401 (2009).
Tokura, Y., Seki, S. & Nagaosa, N. Multiferroics of spin origin. Rep. Prog. Phys. 77, 076501 (2014).
Khomskii, D. Classifying multiferroics: mechanisms and effects. Physics 2, 20 (2009).
Kimura, T. Spiral magnets as magnetoelectrics. Annu. Rev. Mater. Res. 37, 387–413 (2007).
Zhang, W. et al. Spin-flop transition and magnetic phase diagram in CaCo2As2 revealed by torque measurements. Phys. Rev. B 92, 144416 (2015).
O’Neal, K. R. et al. Spin-lattice coupling in [Ni(HF2)(pyrazine)2]SbF6 involving the \({\mathrm{HF}}_2^ -\) superexchange pathway. Inorg. Chem. 55, 12172 (2016).
Nemec, I., Herchel, R., Šilha, T. & Trácenícek, Z. Towards a better understanding of magnetic exchange mediated by hydrogen bonds in Mn(III)/Fe(III) salen-type supramolecular dimers. Dalton Trans. 43, 15602 (2014).
Ghannadzadeh, S. et al. Evolution of magnetic interactions in a pressure-induced Jahn-Teller driven magnetic dimensionality switch. Phys. Rev. B 87, 241102(R) (2013).
Manson, J. L. et al. Magnetic structure and exchange interactions in quasi-one-dimensional MnCl2(urea)2. Inorg. Chem. 54, 11897–11905 (2015).
Manson, J. L. et al. [Cu(HF2)2(pyrazine)]n: a rectangular antiferromagnetic lattice with a spin exchange path made up of two different FHF− bridges. Angew. Chem. Int. Ed. 50, 1573–1576 (2011).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558(R) (1993).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048 (1981).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA + U study. Phys. Rev. B 57, 1505 (1998).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Figgis, B. N., Raston, C. L., Sharma, R. P. & White, A. H. Crystal structure of diammonium aquapentachloroferrate(III). Aust. J. Chem. 31, 2717–2720 (1978).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Xiang, H. J., Kan, E. J., Wei, S.-H., Whangbo, M.-H. & Gong, X. G. Predicting the spin-lattice order of frustrated systems from first principles. Phys. Rev. B 84, 224429 (2011).
Xiang, H. J., Lee, C. H., Koo, H. J., Gong, X. G. & Whangbo, M.-H. Magnetic properties and energy-mapping analysis. Dalton Trans. 42, 823 (2013).
Zhitomirsky, M. E. & Zaliznyak, I. A. Static properties of a quasi-one-dimensional antiferromagnet in a magnetic field. Phys. Rev. B 53, 3428 (1996).
Fishman, R. S. & Okamoto, S. Noncollinear magnetic phases of a triangular-lattice antiferromagnet and of doped CuFeO2. Phys. Rev. B 81, 020402(R) (2010).
Acknowledgements
Research at Tennessee is supported by the National Science Foundation (DMR-1707846) and the Materials Research Fund at the University of Tennessee. A portion of this work was performed at the National High Magnetic Field Laboratory, which is supported by the National Science Foundation through DMR-1644779 and the States of Florida and New Mexico. J.S. appreciates funding from Basic Energy Sciences, U.S. Department of Energy FWP “Science in 100T program”. Research at Oak Ridge National Laboratory is sponsored by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering, and Scientific User Facilities Division. J.H.L. at UNIST is supported by Basic Research Laboratory (NRF2017R1A4A1015323), Creative Materials Discovery Program (2017M3D1A1040828) of NRF, the MOTIE (Ministry of Trade, Industry Energy) (No. 10080657), and KRSC (Korea Semiconductor Research Consortium) support program. We appreciate useful conversations with Brian J. Donahoe.
Author information
Authors and Affiliations
Contributions
The project was conceived by A.J.C. and J.L.M. The sample was synthesized by W.T. High-field measurements were performed and analyzed by A.J.C., K.D.H., J.F.-B., J.S. and J.L.M. First-principles calculations and numerical simulations were carried out by J.N., M.L., J.H.L. and R.S.F. All authors discussed the data. The manuscript was written by A.J.C., M.L., J.N., J.S., J.H.L. and J.L.M., and all authors commented on the document.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Clune, A.J., Nam, J., Lee, M. et al. Magnetic field-temperature phase diagram of multiferroic (NH4)2FeCl5·H2O. npj Quantum Mater. 4, 44 (2019). https://doi.org/10.1038/s41535-019-0180-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41535-019-0180-1
This article is cited by
-
Tunable spin textures in polar antiferromagnetic hybrid organic–inorganic perovskites by electric and magnetic fields
npj Computational Materials (2020)
