
Abstract
This review is an effort to summarize recent developments in synthesis of O-glycosides and N-, C-glycosyl molecules with promising antidiabetic potential. Articles published after 2000 are included. First, the O-glycosides used in the treatment of diabetes are presented, followed by the N-glycosides and finally the C-glycosides constituting the largest group of antidiabetic drugs are described. Within each group of glycosides, we presented how the structure of compounds representing potential drugs changes and when discussing chemical compounds of a similar structure, achievements are presented in the chronological order. C-Glycosyl compounds mimicking O-glycosides structure, exhibit the best features in terms of pharmacodynamics and pharmacokinetics. Therefore, the largest part of the article is concerned with the description of the synthesis and biological studies of various C-glycosides. Also N-glycosides such as N-(β-d-glucopyranosyl)-amides, N-(β-d-glucopyranosyl)-ureas, and 1,2,3-triazolyl derivatives belong to the most potent classes of antidiabetic agents. In order to indicate which of the compounds presented in the given sections have the best inhibitory properties, a list of the best inhibitors is presented at the end of each section. In summary, the best inhibitors were selected from each of the summarizing figures and the results of the ranking were placed. In this way, the reader can learn about the structure of the compounds having the best antidiabetic activity. The compounds, whose synthesis was described in the article but did not appear on the figures presenting the structures of the most active inhibitors, did not show proper activity as inhibitors. Thus, the article also presents studies that have not yielded the desired results and show directions of research that should not be followed. In order to show the directions of the latest research, articles from 2018 to 2019 are described in a separate Sect. 5. In Sect. 6, biological mechanisms of action of the glycosides and patents of marketed drugs are described.
Similar content being viewed by others

1 Introduction
Diabetes mellitus is a disease closely associated with the metabolic syndrome and in developed countries it is a major public health problem [1,2,3]. There are three main types of diabetes mellitus: type 1 (insulin-dependent), type 2 (insulin resistance), and gestational diabetes. Type 2 diabetes mellitus (T2DM) accounts for 90–95% of the diabetic cases. In T2DM, insulin resistance is the major problem. Chronic hyperglycemia is associated with long-term damage, dysfunction and failure of various organs such as eyes, kidneys, nerves, heart and blood vessels. While type 1 diabetics can be treated by the administration of exogenous insulin, for type 2 patients generally diet, exercise, and oral hypoglycemic agents are prescribed. A large number of oral antidiabetic drugs aimed to eliminate three major metabolic disorders leading to hyperglycemia-dysfunction of β-cells, peripheral insulin resistance, excessive hepatic glucose production [4, 5]. Current pharmacological treatments are symptomatic and aim at maintaining the blood glucose levels close to the fasting normoglycemic range of 3.5–6 mM/l. This can be achieved by an array of small molecule drugs (e.g., biguanides, sulfonylureas, thiazolidinediones, glycosidase inhibitors) and ultimately by administration of insulin.
Metformin is a biguanide, which is now the most widely prescribed antidiabetic drug (Fig. 1). Metformin is the first-line medication for the treatment of type 2 diabetes particularly in people who are overweight and is believed to be the most widely used medication for diabetes, which is taken by mouth. However, for 30–40% of T2DM patients, combination therapy is frequently applied as pharmacological treatments.

Metformin—the most widely prescribed antidiabetic drug
Glycogen is a polymer of α-1,4- and α-1,6-linked glucose units that provides a readily available source of energy in living organisms. Glycogen synthase (GS) and glycogen phosphorylase (GP) are the two enzymes that control the synthesis and degradation of this polysaccharide. A key role in glycogen metabolism plays GP [6, 7]. With the rapid increase of type 2 diabetic patients recently, it is becoming an interesting field to discover GP inhibitors for potential antidiabetic drugs. As GP is a typical allosteric protein with several key inhibitor-binding sites including the inhibitor, the catalytic, the allosteric, and the new allosteric sites, the research works were mainly focused on compounds that can bind these sites and show selective inhibitory effect [6,7,8,9,10]. So, GP transfers a glucose unit from the non-reducing end of the storage polysaccharide glycogen to an inorganic phosphate. Three isoforms of GP exist in the brain, muscle, and liver tissue. The liver is capable of storing glucose as glycogen and producing and releasing glucose to the bloodstream [6, 7]. GP is an allosteric enzyme, which exists in two interconvertible forms GPa (phosphorylated, active, high substrate affinity) and GPb (unphosphorylated, inactive, low substrate affinity). Design of GP inhibitors is a target for a better control of hyperglycemia. The inhibitors targeting the seven binding sites of GP show a large molecular diversity. Among them, various glucose derivatives bind mostly to the catalytic site of the enzyme. N-Acyl-β-d-glucopyranosylamines, N-acyl-N′-β-d-glucopyranosyl ureas, glucopyranosylidene-spiro-heterocycles, as well as N- and C-glucosylated heterocycles belong to the most potent classes of this inhibitor family [6, 7]. In 2001, So and Karplus designed a number of potential GP inhibitors with a variety of computational approaches [11]. 2D and 3D similarity-based QSAR models were used to identify novel molecules that may bind to the glucose-binding site. The designed ligands were evaluated by a multiple screening method [12]. In this way, a total of 301 candidate ligands for GP have been designed using an array of computational approaches.
Several kinds of mimics of O-glycosides, first of all S-, N-, and C-glycosyl derivatives, may display similar biological activities; however, due to their significantly distinct chemical properties, such molecules can be valuable tools in deciphering the biological roles of natural sugars, and may also serve as leads for new drugs. Among glycomimetics, C-glycosides have attracted much attention due to the existence of a number of naturally occurring representatives. Comparing to O-glycosides, the C-glycosides are structurally more stable against acidic and enzymatic cleavage due to the existence of their C–C glycosidic bond. Bristol-Myers Squibb [13] and Kotobuki [14] disclosed C-aryl glucosides in 2001, which appear to have potent inhibition and good stability in vivo.
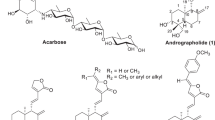
Many efforts devoted to develop carbohydrate-based therapeutics aim at finding inhibitors of glycoprocessing enzymes and discovering their structure–activity relationships (SAR). In therapies of diabetes, sugar derived or glycomimetic structures, such as acarbose, miglitol, or voglibose, have been applied (Fig. 2) [15].

Carbohydrate derivatives and glycomimetic compounds in therapies of T2DM
In 2017, Bokor et al. presented a review [16] where they described the syntheses and diverse bioactivities of C-glycopyranosyl arenes and heteroarenes. They provided a classification of the preparative routes to these synthetic targets according to methodologies and compound categories. Several of these compounds display antidiabetic properties due to enzyme inhibition and are used in the pharmacological treatment of type 2 diabetes. Figure 3 shows the glycoside structures that are discussed in this article. O-Glycosides, N-glycosides, and C-glycosides as antidiabetic drugs have been described in the following sections.

O-Glycosides and N-, C-glycosyl antidiabetic molecules that are discussed in this review
2 O-Glycosides as Antidiabetic Agents
It is known that an O-glycoside—natural product phlorizin (Fig. 4) can lower plasma glucose levels and improve insulin resistance by increasing renal glucose excretion [17]. However, its sensitivity toward hydrolysis by glucosidases, unselective inhibition of both SGLTs (sodium glucose transporters), and unfavorable effects of its aglycon phloretin on other glucose transporters prevented this compound from use as an antidiabetic drug.

Structure of the natural product phlorizin
More recently, guava leaves have gained attention in the control of T2DM [18, 19]. In 2013, Eidenberger and coworkers investigated in vitro the effect of extracts from Psidium guajava L. leaves containing the flavonol-glycoside components [20]. An ethanolic extract was prepared from dried, powdered leaves of guava and was found to contain seven main flavonol-glycosides, which were isolated by semi-preparative HPLC and tested individually. All isolated flavonol-glycosides were tested for their antidiabetic potential. Peltatoside 1, hyperoside 2, isoquercitrin 3, and guaijaverin 4 (Fig. 5) show an inhibitory effect 5–10 times higher than that obtained for the three other partially characterized flavonol-glycosides. It seems therefore that most of the inhibitory action of the guava extract is due to the four identified flavonol constituents 1–4 [20].

Structures of the flavonol-glycosides: peltatoside 1, hyperoside 2, isoquercitrin 3, guaijaverin 4 [20]
In 2015, Diaz-Lobo et al. [21] reported on the synthesis and biological evaluation of O-glycoside—a selective inhibitor that consists of an azobenzene moiety glycosidically linked to the anomeric carbon of a glucose molecule. The molecule incorporates an azobenzene photoswitch whose conformation can be significantly altered by irradiation with UV light. Synthesis of compound 9 (Scheme 1) started with the quantitative peracetylation of d-glucose 5 with acetic anhydride in pyridine. Next, the anomeric acetyl group of 1,2,3,4,6-penta-O-acetyl-d-glucopyranoside 6 was selectively cleaved using benzylamine in THF to furnish 7 that was employed for the glycosylation of 4-hydroxyazobenzene by the Mitsunobu reaction. The resulting 4-(phenylazo)phenyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside 8 was deacetylated with MeONa/MeOH to give 4-(phenylazo)phenyl-d-glucopyranoside 9. The azoglucoside 9 was obtained as a mixture of the α and β anomers. UV light induced E→Z photoisomerization of the azobenzene glucoside 9 was observed. In the ground state, the more stable (E)-isomer of the azobenzene glucoside 9 had a slight inhibitory effect on rat muscle GP (RMGP, IC50 = 4.9 mM) and Escherichia coli GS (EcGS, IC50 = 1.6 mM). After irradiation and subsequent conversion to the (Z)-form, the inhibitory potency of the azobenzene O-glucoside did not significantly change for RMGP (IC50 = 2.4 mM), while its effect on EcGS increased 50-fold (IC50 = 32 μM). Although compound 9 was synthesized as a 1:4 mixture of the α- and β-anomers, analysis suggested that the more abundant β-anomer is the one responsible for the observed inhibition. So, Diaz-Lobo et al. showed that the ability to selectively photocontrol the catalytic activity of key enzymes of glycogen metabolism might represent a new approach for the treatment of glycogen metabolism disorders [21].

Synthesis of 4-(phenylazo)phenyl-d-glucopyranoside 9 [21]
Functional foods can be used alone or in combination with existing therapies in preventing and treating type 2 diabetes. Trans-2,3,5,4′-tetrahydroxystilbene 2-O-β-glucopyranoside (trans-THSG) 10 (Fig. 6), a dominant bioactive compound from Polygonum multiflorum (PM), has attracted increasing research interests due to its strong antioxidant activity. The content of naturally occurring cis-THSG (cis-2,3,5,4′-tetrahydroxystilbene 2-O-β-glucopyranoside) 11 (Fig. 6) is very low in PM root, therefore W. Tang et al. prepared in 2017 cis-THSG by mimicking the traditional process of PM [22]. The anti-diabetic effects of trans- and cis-THSG were evaluated in type 2 diabetes. Cis-THSG 11 was found to be more effective than trans-THSG 10 in hypoglycemic effect [22].

Structures of trans-THSG 10 and cis-THSG 11 [22]
Figure 7 presents information on the antidiabetic activity of O-glycosides discussed in Sect. 2. Below the structural formula of each inhibitor, the number of the compound and the scheme number on which it is located and the corresponding reference are provided. The most important information on the action of a given compound as a specific inhibitor is also included. Analyzing the structure of the compounds shown in Fig. 7, it can be seen that the phenyl groups are a structural element that is repeated in each compound. In the case of three compounds, they are phenolic derivatives. An interesting approach to the issue of active inhibitor structure is the idea presented by Diaz-Lobo and coworkers [21], in which they turned their attention to the ability to selectively photocontrol the catalytic activity of key enzymes of glycogen metabolism.

Antidiabetic activity of O-glycosides described in Sect. 2
3 N-Glycosides as Antidiabetic Agents
3.1 N-(β-d-Glucopyranosyl) Amides and N-(β-d-Glucopyranosyl)-Urea Derivatives
Since O-glycosides are usually hydrolytically unstable, many carbohydrate analogues such as N- or C-glycosides have been synthesized as therapeutic agents. Inhibition of GP is one of several intensively investigated approaches to find novel treatments for type 2 diabetes mellitus. Some N-glycosides such as N-(β-d-glucopyranosyl) amides 12–14 (Fig. 8) were examined as inhibitors of GP [23]. N-(β-d-glucopyranosyl)-N’-acyl urea derivatives 15 and 16 are also inhibitors of GP. Compound 17 represents the most efficient glucose analogue inhibitor [24].

N-(β-d-glucopyranosyl) amides 12–14 and N-(β-d-glucopyranosyl)-N’-acyl urea derivatives 15–17 as inhibitors of GP [23]
In 2004, Gyorgydeak et al. transformed 2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl- and 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl azides 18 into the corresponding per-O-acetylated N-(β-d-glycopyranosyl) amides 19 by Staudinger protocol (Scheme 2) [25]. Removal of the protecting groups carried out by Zemplén deacetylation furnished compounds 20. Compounds 19 and 20 were tested against rabbit muscle glycogen phosphorylase. The best inhibitor of this series was N-(β-d-glucopyranosyl) 3-(2-naphthyl)-propenoic amide (Ki = 3.5 μM). It was shown that the acyl urea moiety is essential for the strong inhibition. A properly positioned and large enough hydrophobic group attached to the amide moiety makes the inhibition one order of magnitude stronger than that of the best amide inhibitor known earlier [N-(β-d-glucopyranosyl) acetamide 12, Fig. 8]. However, N-(β-d-glucopyranosyl) 3-(2-naphthyl)-propenoic amide is still much less efficient than the best-known inhibitor urea derivative 17 (Ki = 0.4 μM) [25].

Synthesis of N-(β-d-glucopyranosyl)- and N-(2-acetamido-2-deoxy-β-d-glucopyranosyl) amides 20 [25]
In 2006, Czifrak et al. used extension of the modified Staudinger methodology to the synthesis of N-(β-d-glucopyranosyl) monoamides of various dicarboxylic acids [26]. Such compounds offer the possibility to place a strongly polar group (COOH) at different distances from the sugar moiety while the ability to form the important H-bond from the amide can be maintained. O-Peracetylated N-(β-d-glucopyranosyl)imino trimethylphosphorane obtained in situ from 2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl azide 21 and PMe3 (Scheme 3) was reacted with saturated and unsaturated aliphatic and aromatic dicarboxylic acids, or their anhydrides, or monoesters to give the corresponding N-(β-d-glucopyranosyl) monoamides of dicarboxylic acids or derivatives (e.g., derivative 23, Scheme 3). The acetyl protecting groups were removed according to the Zemplén protocol to give a series of compounds, which were moderate inhibitors against rabbit muscle glycogen phosphorylase b. The best inhibitor was 3-(N-β-d-glucopyranosyl-carbamoyl) propanoic acid 23 (n = 2) with Ki = 20 μM [26].

Synthesis of N-(β-d-glucopyranosyl) monoamides 23 of various dicarboxylic acids [26]
In 2008, Somsak et al. showed that the synthesis of the highly efficient glycogen phosphorylase inhibitors N-(β-d-glucopyranosyl)-N′-substituted ureas has been significantly improved by using of glucopyranosylammonium carbamate [27]. This compound allowed the preparation of N-(β-D-glucopyranosyl)-N′-substituted ureas, -thioureas and selenourea 27 in two steps from d-glucose 24 (Scheme 4).

Synthesis of GP inhibitors N-(β-d-glucopyranosyl)-N’-substituted ureas 27 with using of glucopyranosylammonium carbamate 26 [27]
In 2012, Nagy et al. synthesized N-(4-substituted-benzoyl)-N′-(β-d-glucopyranosyl) urea derivatives 32 by addition of O-peracetylated β-d-glucopyranosylamine 28 to acyl-isocyanates 29 and subsequent deprotection (Scheme 5) [28]. Some compounds 32 were obtained by reactions of β-d-glucopyranosylammonium carbamate 31 with acyl-isocyanates 29. Most of the new compounds 32 were low micromolar inhibitors of rabbit muscle glycogen phosphorylase b. There was no significant improvement of the inhibitory efficiency for N-(4-substituted-benzoyl)-urea 32 in comparison to N-benzoyl-urea. These results indicated the lack of a specific and crucial interaction from four position in phenyl ring within the catalytic site. The best inhibitors were compounds 32 with substituents R=4-CH3–C6H4 (Ki = 2.3 μM) and R=4-NO2–C6H4 (Ki = 3.3 μM) [28].

Synthesis of N-(4-substituted-benzoyl)-N′-(β-d-glucopyranosyl) urea derivatives 32 [28]
In 2012, Konya et al. synthesized new glucose derivatives for the inhibition of GP [29]. They have reported on the synthesis and enzymatic evaluation of a series O-peracetylated N-(β-d-glucopyranosyl)-carboxamides with isoxazole or 1,2,3-triazole rings. In a DCC-mediated coupling 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosylamine 34 and propiolic acid gave N-propynoyl-2,3,4,6-tetra-O-acetyl-β-d-glucopyranosylamine 35, which was transformed by 1,3-dipolar cycloadditions with aromatic azides and nitrile-oxides to the corresponding O-peracetylated N-(β-d-glucopyranosyl)-1-substituted-1,2,3-triazole-4-carboxamides 36 and N-(β-d-glucopyranosyl)-3-substitutedisoxazole-5-carboxamides 38, respectively (Scheme 6). These compounds were O-deacetylated by Zemplén protocol to compounds 37 and 39, which were tested as inhibitors of rabbit muscle glycogen phosphorylase b. Deacylated compounds 37 and 39 inhibited rabbit muscle glycogen phosphorylase b in the low micromolar range. The best inhibitors of the two series were N-(β-d-glucopyranosyl)-1-(3,5-dimethyl-phenyl)-1,2,3-triazole-4-carboxamide 37 (Ar=3,5-di-Me-C6H3, Ki = 34 μM) and N-(β-d-glucopyranosyl)-3-(indol-2-yl)-isoxazole-5-carboxamide 39 (Ar=indol-2-yl, Ki = 164 μM).

Synthesis of N-(β-d-glucopyranosyl)-1-substituted-1,2,3-triazole-4-carboxamides 37 and N-(β-d-glucopyranosyl)-3-substituted-isoxazole-5-carboxamides 39 [29]
In 2014, Parmenopoulou and coworkers reported the in silico screening in the Zinc database of 1888 N-acyl-β-d-glucopyranosylamines as potential GP inhibitors [30]. Six selected candidates from the screening were then synthesized and their inhibitory potency was assessed both in vitro and ex vivo. The direct acylation of 2,3,4,6-tetra-O-acetyl-β-d-glucopyranosylamine 41, easily prepared from the per-O-acetylated β-d-glucopyranosyl azide 40 upon catalytic hydrogenation, with a diverse set of commercially available acyl chlorides RCOCl, furnished the protected N-acyl-β-d-glucopyranosylamines 42. Removal of the acetyl groups of the derivatives 42, performed either by saturated methanolic ammonia or by the Zemplén method yielded analogues 43 (Scheme 7). Their inhibition constants’ values Ki in vitro ranged from 5 to 377 μM while two of them were effective at causing inactivation of GP in rat hepatocytes at low μM concentrations [30].

Synthesis of N-acyl-β-d-glucopyranosylamines 43 [30]
Figure 9 presents the best GPb inhibitors from the N-(β-copper-catalyzed azide–alkyne cycloaddition-glucopyranosyl) amides and N-(β- d -glucopyranosyl)-urea derivatives described in Sect. 3.1. The structural formula of each inhibitor, the number of the compound, the scheme number and the corresponding reference are provided. In the figure, the inhibitors are arranged in order from the strongest characterized by the lowest inhibitory constant Ki value to the weaker one with the highest Ki value. It can be seen that all inhibitors accumulated in Fig. 8 are derivatives of glucose and compound 17 represents the most efficient glucose analogue inhibitor.

Values of inhibitory constants Ki of the best GPb inhibitors from the N-glycosides described in Sect. 3.1
3.2 1,2,3-Triazolyl N-Glycosides
The medicinal importance of triazoles is due to their bioisosterism with peptide bonds as they can actively participate in hydrogen bonding, and due to their strong dipole moments, the triazoles are extremely stable to hydrolysis and oxidative/reductive conditions. In 2011, Anand and coworkers described an efficient synthesis of 1,2,3-1H-triazolyl glycohybrids with two sugar units via copper-catalyzed azide-alkyne cycloaddition (CuAAC) [31]. Potential inhibitors were prepared by a 1,3-dipolar cycloaddition of glycosyl azides 47 and 49 to 2,3-unsaturated alkynyl glycosides 46 (Scheme 8). The synthesized glycohybrids were screened for their α-glucosidase, glycogen phosphorylase, and glucose-6-phosphatase inhibitory activities. A few of the glycohybrids showed promising inhibitory activities against these enzymes [31].

Synthesis of 1,2,3-1H-triazolyl glycohybrids 48 and 50 by 1,3-dipolar cycloaddition of glycosyl azides 47 and 49 to 2,3-unsaturated alkynyl glycosides 46 [31]
In 2014, Goyard et al. examined CuAAC between acetylated β-d-glucosyl azide 51 and alkyl or phenyl acetylenes 52, which led to the corresponding 4-substituted 1-glucosyl-1,2,3-triazoles 54 (Scheme 9) [32]. 5-Halogeno analogues 56 were prepared in similar conditions but with 2 equiv CuI or CuBr. In reactions with two equiv CuCl and either propargyl acetate or phenyl acetylene, the major products 59 displayed two 5,5′-linked triazole rings resulting from homocoupling of the 1-glucosyl-4-substituted 1,2,3-triazoles (Scheme 10). The cycloaddition of 51 and 52 afforded four different products 53, 55, 57, and 59 with only minor amounts of the expected chlorinated derivatives 55 and with the dimeric products 59 being isolated as the major component. The two 4-phenyl substituted structures of compound 59 were unambiguously identified as atropisomers with aR stereochemistry. All O-unprotected derivatives (Schemes 9, 10) were tested as inhibitors of GP. The modest inhibition activities measured showed that 4,5-disubstituted 1-glucosyl-1,2,3-triazoles bind weakly to the enzyme. This suggests that such ligands do not fit the catalytic site or any other binding site of the GP [32].

Synthesis of 4-substituted-1-glucosyl-1,2,3-triazoles 54 and 56 by copper-catalyzed azide–alkyne cycloaddition [32]

Cycloaddition of azide 51 and acetylenes 52 in the presence of 2 equiv CuCl [32]
In order to gain additional data for structure–activity studies of the inhibition glycogen phosphorylase, in 2015 Goyard et al. prepared a series of eight GP inhibitor candidates from peracetylglucopyranosyl azide 61 by 1,3-dipolar cycloaddition (Scheme 11) [33]. The need for a N-Boc-protected propargylamine was identified in the CuAAC with azide 61 under Meldal’s conditions, while Sharpless’ conditions were better adapted to the CuAAC of azide 61 with propargyl bromide. Cycloaddition of Boc-propargylamine with azide 61 afforded the N-Boc precursor of a 4-aminomethyl-1-glucosyl-1,2,3-triazole 62, which gave access to a series of amide and sulfonamide derivatives (Scheme 11). The Boc-protected amine 62 was converted to the free amine 64, which were functionalized with acyl chlorides R2COCl affording the amides 67. The amine 64 was also converted to the sulfonamide derivative 69 using p-toluenesulfonyl chloride TsCl. The sulfonamide 70 was synthesized in order to take advantage of hydrophobic contact in the β-channel of GP and also to have potential additional contacts with the sulfonamide group and the side chain amino acids of the enzyme. Arbuzov reaction of the brominated derivative 71 with triethylphosphite under microwave activation allowed for formation of the acetylated phosphonate 72, which was converted to phosphonate 73 (Scheme 11). Enzymatic studies revealed poor to moderate inhibitions of deacetylated derivatives toward glycogen GP. The N-Boc-protected amine 63 was the best inhibitor (IC50 = 620 μM) unexpectedly slightly better than the 2-naphthylamido 68 substituted analogue (IC50 = 650 μM) [33].

Synthesis of 4-amidomethyl-1-glucosyl-1,2,3-triazoles [33]
SGLT2 (sodium-dependent glucose co-transporter 2) is a glucose transporter that is responsible for 90% of the renal glucose reabsorption. For the treatment of type 2 diabetes, suppression of glucose reabsorption through the inhibition of SGLT2 is a promising therapeutic approach. Therefore in 2015, Bai et al. developed a convenient approach to the synthesis of novel triazole-N-glycoside derivatives 78 via CuSCN-catalyzed click reaction and Ullmann-type coupling reaction (Scheme 12) and examined the SGLT2 inhibitory activities of prepared N-glycosides [34]. For carbohydrate azides 74: glucosyl azide, galactosyl azide, ribosyl azide and aminoglucosyl azide, reactions were performed and the triazole-N-glycosides 76 were generated with high selectivity, while mannosylazide and lactosyl azide showed moderate selectivity (side product 77). After the successful construction of [6] ring-fused triazole-N-glycosides, the preparation of [6, 7] or [6, 8] ring-fused triazole-N-glycosides via this protocol was also tried. Deprotection was carried out in the presence of BCl3 or 1,2-diaminoethane and finally the acetyl-containing N-glycosides were treated with sodium methoxide to obtain the corresponding target compounds 78 and side product 79 (Scheme 12). The SGLT2 inhibitory activities of N-glycosides 78 were evaluated and some compounds showed moderate SGLT2 inhibition activities at 100 nM [34].

Synthesis of triazole-N-glycoside derivatives 78 via CuSCN-catalyzed click reaction and Ullmann-type coupling reaction [34]
3.3 Other N-Glycosides
In 2016, Chu et al. investigated the effect of C6-substitution on inhibition of SGLT2 by N-indolylglucosides 83 (Scheme 13) [35]. As they investigated in a previous article [36], results suggested that the C6 position of the sugar moiety may play a critical role in the suppression of SGLT2. Therefore, Chu et al. led optimization of N-glycosides proceeded via modification at the C6 position only, the aglycone unit 4-chloro-3-(4-cyclopropylbenzyl)-1H-indole being fixed. N-Indolylglucosides 83 were prepared according to a synthetic method depicted in Scheme 13, using N-indolylglucoside 80 as the starting material. Treatment of 80 with methanesulfonyl chloride (MsCl) in pyridine gave the corresponding 6-OMs N-glycoside, which was sequentially reacted with sodium azide (NaN3) to afford the 6-azido compound 81. Next, the synthesis of amides 83 was carried out via the amine 82, generated by the reduction of azido group in 81 with Zn/AcOH in THF. Amine 82 underwent direct amide bond formation with a variety of acyl chlorides to furnish 6-amide derivatives 83 (Scheme 13). After SAR study 6-amide derivatives 83 (R=acetyl and 3-methoxy-3-oxopropanoyl) were identified as potent SGLT2 inhibitors. The data obtained indicated that 83 (R=acetyl and 3-methoxy-3-oxopropanoyl) are mildly to moderately selective for SGLT2 over SGLT1. Both compounds were also evaluated in a urinary glucose excretion test and pharmacokinetic study. Compound 83 (R=acetyl) was found capable of inducing urinary glucose excretion in rats [35].

Synthesis of N-indolylglucosides 83 [35]
A key point for the design of efficient drugs is the characterization of the interactions governing its binding to the enzyme. In 2017, Mamais et al. designed and prepared a glucose-based acridone derivative (GLAC), which is a potent inhibitor of GP and it allows probing subtle interactions in catalytic site [37]. The design of a catalytic site inhibitor was based on C-4 modification of β-d-glucopyranosyluracil and introducing of flat aromatic substituent, which can increase the binding affinity in the β-channel. Authors utilized an acridone moiety as a possible chromophore as well as fluorophore. β-d-Glucopyranosyluracil 84 was converted to 4-triazolyl derivative 85 and next substitution of 85 by 2-aminoacridone provided the protected adducts 86 (Scheme 14). Final deprotection furnished desired product GLAC 87. Authors reveal that the part of the catalytic site of GP behaves as a highly basic environment in which GLAC 87 exists as a bis-anion. Authors reassumed that solvent structure of GLAC and the water-bridged hydrogen bonding interactions formed with the catalytic site residues in the β-channel are responsible for the observed inhibition potency [37].

Synthesis of GLAC 87 (N1-(β-d-glucopyranosyl)-N4-[2-acridin-9(10H)-onyl]-cytosine) [37]
Figure 10 shows inhibitory properties of the best GPb and SGLT inhibitors from the N-glycosides: 1,2,3-triazolyl N-glycosides, N-indolylglycosides, and N-uracil glycoside presented in Sects. 3.2 and 3.3. The GLAC compound 87 is one of the best GPb inhibitors described so far (Ki 31 nM). All compounds are derivatives of glucose. The presence of the cyclopropane ring in the structure of two of the active inhibitors is also noteworthy.

4 C-Glycosides as Antidiabetic Agents
4.1 Aromatic C-Glycosyl Derivatives
C-Glycosides are more metabolically stable than O-glycosides and tend to have higher oral bioavailability and plasma exposure without needing to be converted to a prodrug [4]. Among C-glycosyl derivatives, C-glycosylarenes have attracted much attention. In 2007, Praly et al. underwent kinetic and X-ray crystallographic study of two enzyme complexes of rabbit muscle unphosphorylated glycogen phosphorylase b (GPb) with ligands of the C-glucosylbenzo(hydro)quinone type [38]. The synthesis of quinones was accomplished from C-β-d-glycopyranosyl-1,4-dimethoxybenzenes 89, which were prepared by reaction of penta-O-acetyl-β-d-glycopyranoses 88 and 1,4-dimethoxybenzene (Scheme 15). Next, compounds 89 were converted to the corresponding C-glycosylhydro and C-glycosylbenzoquinones, with either an acetylated or deprotected sugar moiety. C-β-d-Glucosylbenzoquinone 91 (R1=H, R2=OH) and C-β-d-glucosylhydroquinone 95 (R1=H, R2=OH) (Scheme 15) were found to be competitive inhibitors of rabbit muscle GPb with Ki values of 1.3 and 0.9 mM, respectively. In order to elucidate the structural basis of inhibition, the authors determined the crystal structures of 91 and 95 in complex with GPb. The complex structures reveal that the inhibitors can be accommodated at the catalytic site at approximately the same position as α-d-glucose and stabilize the transition state conformation of the 280 s loop by making several favorable contacts to Asp283 and Asn284 of this loop [38].

Synthesis of C-β-d-glucosylbenzoquinones 91 and C-β-d-glucosylhydroquinones 95 [38]
Protein tyrosine phosphatase 1B (PTP1B) has recently been identified as a new drug target for type 2 diabetes [39]. In 2008. Lin et al. synthesized β-C-glycosiduronic acid quinones and β-C-glycosyl compounds as sugar-based PTP1B inhibitors [40]. To prepare 2-carbamoylbenzoic acid derivatives 100 (Scheme 16) and 106 (Scheme 17), β-C-aryl glucosides 96 and 102 were first tritylated at 6-position, followed by protection of secondary hydroxyl function as benzoyl ester. To avoid intramolecular transesterification reaction, detritylation has been realized under acidic condition with TFA to afford 98 and 104. The 6-hydroxy group was transformed into azide via mesylate. Staudinger protocol was then employed to convert azido sugars to carbamoylbenzoic acid derivatives. Reaction of 99 with phthalic anhydride led to a mixture of the desired compound 100 and N-phthalimide derivative 101 (Scheme 16). Treatment of 105 with phthalic anhydride in THF afforded 106 (Scheme 17). Benzoyl protected quinone derivatives as well as aryl β-C-glycosyl compounds showed IC50 values of 0.77–5.27 μM against PTP1B, with compounds 100 and 106 bearing an acidic function being the most potent [40].

Synthesis 2-carbamoylbenzoic acid derivative of β-C-glycosyl compound 100 as sugar-based PTP1B inhibitor [40]

Synthesis 2-carbamoylbenzoic acid derivative of β-C-glycosyl compound 106 as sugar-based PTP1B inhibitor [40]
In 2008, Meng et al. discovered dapagliflozin, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes [41]. Synthesis of dapagliflozin started from Friedel–Crafts acylation of phenetole with 5-bromo-2-chlorobenzoyl chloride, which was formed from 5-bromo-2-chlorobenzoic acid 107 in reaction with oxalyl chloride (Scheme 18). Reduction of p-benzophenone 108 by triethylsilane and BF3·OEt2 provided aglycon 109. Lithium halogen exchange, followed by the addition of the nascent lithiated aromatic to 110, gave a mixture of lactols, which were converted in situ to the desilylated O-methyl lactols 111 by treatment with methanesulfonic acid in methanol. Reduction of the anomeric methoxy group of 111 using triethylsilane and BF3·OEt2, followed by peracetylation, yielded tetraacetate 112. Hydrolysis of 112 with lithium hydroxide generated 113 (Scheme 18). Authors resumed that dapagliflozin 113 is a potent, selective SGLT2 inhibitor that is not subject to O-glucosidase degradation. Compound 113 is a much more potent stimulator of glucosuria in normal rats than other SGLT2 inhibitors. The promising significant reduction of blood glucose levels in diabetic rats prompted further evaluation of 113 in the clinic for the treatment of type 2 diabetes [41].

Synthesis of dapagliflozin 113 SGLT2 inhibitor [41]
In 2010, Kato and Kawabata synthesized an isoflavone C-glucoside puerarin and several derivatives, which were candidate for treatment of diabetes mellitus [42]. Treatment of TMSOTf to the mixture of glucosyl imidate 114 and an acetophenone 115 afforded a C-glucoside 116 (Scheme 19). Group 6-OH of compound 116 was selectively protected by a benzyl group. Aldol condensation of 117 with an aldehyde 118 gave chalcones 119. Compounds 119 were treated with Tl(NO3)3 and heated in an acidic medium to form isoflavone structure 120. The benzyl group protecting OH was selectively removed and after trifluoromethanesulfonylation and subsequent reaction with Pd(OAc)2 derivative 121a was prepared. Finally, benzyl groups were removed by treatment with BBr3 to give puerarin 122 in 13% overall yield and regioisomer of 122, genistein 6-C-glucoside 123 (Scheme 19). The compound 122 was applied for the structure–activity relationship study. The results of research indicated that the C-glucoside part of the compound 122 was not much involved in the activity. The structure responsible for the glucose uptake enhancing activity was the isoflavone moiety. However, the C-glucose may involve in physical properties of 122 and raises solubility in water [42].

Synthesis of puerarin 122 and its regioisomer 123 [42]
In 2010, Nomura et al. discovered that C-glucosides bearing heterocyclic ring formed metabolically more stable inhibitors for sodium-dependent glucose cotransporter 2 (SGLT2) than the O-glucoside [43]. To synthesize appropriate compounds, aglycones 124 were dissolved in tetrahydrofuran and toluene, and treated with n-butyllithium at − 78 °C to generate aryllithium, followed by addition of 2,3,4,6-tetra-O-trimethylsilyl-β-d-gluconolactone (Scheme 20). The resulting anomeric mixture of lactols was converted into desilylated methyl ethers 125 by addition of methanesulfonic acid in methanol. C-Glucoside derivatives 126 were obtained by stereoselective reduction of 125 using a combination of triethylsilane and boron trifluoride etherate in methylene chloride. Thiophene derivative 126 canagliflozin (R1=Me in the para position relative to glucose moiety, Het=tiophene with R2=C6H4-4-F in position 2) was a highly potent and selective SGLT2 inhibitor and showed pronounced anti-hyperglycemic effects in high-fat diet-fed mice (IC50 = 2.2 nM). Canagliflozin is the first SGLT2 inhibitor to be approved in the USA and is under regulatory review in the EU [44, 45].

Synthesis of C-glucoside derivatives 126 selective SGLT2 inhibitors [43]
In 2011, Xu and coworkers synthesized a series of C-aryl glucosides with various substituents at the 4′-position of the distal aryl ring and evaluated for inhibition of human hSGLT1 and hSGLT2 [46]. Scheme 21 depicts the construction of aglycone 132, which is a part of bexagliflozin 134 selective SGLT2 inhibitor that reached phase III clinical trials [47]. Friedel–Crafts acylation of benzene with the benzoyl chloride derived from benzoic acid 127 by treatment with oxalyl chloride provided the corresponding benzophenone 128, which were reduced by triethylsilane in the presence of TFA and catalytic trifluoromethanesulfonic acid to generate bromodiarylmethane 129. Finally, aglycone 132 was constructed by vinyl ether formation of the alcohol 130 with vinyl acetate in the presence of sodium carbonate and a catalytic amount of [IrCl(COD)]2 (Scheme 21). Lithium-bromide exchange of bromodiarylmethane 132 and addition of the resulting aryllithium to 2,3,4,6-tetra-O-trimethylsilyl-d-gluconolactone 133 followed by etherification with methanol in the presence of methylsulfonic acid provided desilylated O-methyl lactols, which were reduced with triethylsilane and BF3.OEt2 to give desired C-aryl glucoside 134. The IC50 values for bexagliflozin against human SGLT1 and SGLT2 are 5.6 μM and 2 nM, respectively [47].

Synthesis of bexagliflozin 134 selective SGLT2 inhibitor [47]
In 2012, Imamura and coworkers discovered a novel benzothiophene derivatives 139, among them ipragliflozin (R1=H, R2=F), which are a highly potent and selective SGLT2 inhibitors (Scheme 22) [48]. Lithiation of 135 followed by the addition of aromatic benzaldehydes, yielded alcohols those were reduced with Et3SiH and BF3.OEt2 to give aglycones 136. Lithium halogen exchange followed by the addition of a lithiated aromatic to compound 137 yielded lactols those were reduced by treatment with Et3SiH and BF3 etherate to give compounds 138. Successive removal of the benzyl groups generated compounds 139. Ipragliflozin (R1=H, R2=F) was a highly potent and selective human SGLT2 inhibitor (IC50 = 7.4 nM).

Synthesis of benzothiophene derivatives 139 selective SGLT2 inhibitors [48]
Figure 11 contains the best inhibitors GPb, PTP1B, and SGLT2 having the aromatic C-glycoside structure that are described in Sect. 4.1. The analysis of the structure of the C-glycosyl derivatives presented in Fig. 11 shows that the presence of a chlorine or fluorine atom as well as the presence of sulfur-containing heteroaromatic ring guarantees an increased antidiabetic activity of the compound. All inhibitors shown in Fig. 11 are derivatives of glucose. Thus, also taking into account the structures of previously presented the best inhibitors, it can be concluded that this glucose molecule guarantees high inhibitor activity. Referring to the structure of the best inhibitors shown in Fig. 10, it can also be concluded that the cyclopropane ring is a structural element that guarantees high inhibitor activity. The repeating structural element is also the 1,4-dihydroxy or dimethoxyphenyl system.

Inhibitory properties of the best inhibitors from the aromatic C-glycosyl derivatives described in Sect. 4.1
4.2 Heteroaromatic C-Glycosyl Derivatives
In 2001, Somsák et al. described highly chemo-, regio-, and stereoselective procedure that allows for the preparation of d-gluco- and d-xylopyranosylidene-spiro-hydantoins and thiohydantoins in six steps from the corresponding free sugar [49]. In the key step of the syntheses C-(1-bromo-1-deoxy-β-d-glycopyranosyl)formamides 142 and 143 were reacted with cyanate ion to give spiro-hydantoins 144 and 145 with a retained configuration at the anomeric center as the major products (Scheme 23). Thiocyanate ions gave spiro-thiohydantoins 144 with an inverted anomeric carbon as the only products. The acetylated compounds were deprotected by the Zemplen procedure. Enzyme assays with a and b forms of muscle and liver glycogen phosphorylases showed spiro-hydantoin 144 (R1=CH2OH, R2=H, X=O) and spirothiohydantoin 144 (R1=CH2OH, R2=H, X=S) to be the best and equipotent inhibitors with Ki values in the low micromolar range. The study of epimeric pairs of d-gluco and d-xylo spiro-hydantoins and N-(d-glucopyranosyl)amides indicated the role of specific hydrogen bridges in binding the inhibitors to the enzyme [49].

Synthesis of glycopyranosylidenespiro-hydantoins and thiohydantoins 144 and 145 [49]
In 2004, Somsak et al. have decided to prepare C-(β-d-glucopyranosyl) heterocycles exhibiting acidic, basic, and neutral properties in the heterocyclic moieties [50]. They transformed per-O-acetylated and -benzoylated β-d-glucopyranosyl cyanides 146 into the corresponding 5-(β-d-glucopyranosyl)tetrazoles 147, 2-(β-d-glucopyranosyl)benzothiazoles 153 and 2-(β-d-glucopyranosyl)-benzimidazoles 151 (Scheme 24). Acylation of the tetrazoles 147, either by acetic or trifluoroacetic anhydride, gave 5-(β-d-glucopyranosyl)-2-methyl- and -2-trifluoromethyl-1,3,4-oxadiazoles 148, respectively. Removal of the protecting groups furnished inhibitors 147 (R=H), 149, 152, and 154 exhibiting inhibitor constants in the micromolar range. The tetrazole 147 (R=H) ring of slightly acidic character was unfavorable for the binding of this compound to the GP enzyme. The neutral aglycones in 149 (Ki = 212 μM) and 154 (Ki = 229 μM) result in moderate inhibitors. The most efficient inhibitor was benzimidazole 152 (Ki = 11 μM) [50].

Synthesis of 5-(β-d-glucopyranosyl)tetrazoles 147, 5-(β-d-glucopyranosyl)-1,3,4-oxadiazoles 149, 2-(β-d-glucopyranosyl)benzothiazoles 154 and 2-(β-d-glucopyranosyl)-benzimidazoles 152 [50]
In 2005, Chrysina et al. examined inhibitors with enhanced affinity for glycogen phosphorylase that might control hyperglycemia in type 2 diabetes [51]. Three analogs of β-d-glucopyranose: 2-(β-d-glucopyranosyl)-5-methyl-1,3,4-oxadiazole 155, 2-(β-d-glucopyranosyl)-benzothiazole 156 and 2-(β-d-glucopyranosyl)-benzimidazole 157 were examined (Fig. 12). The compounds showed competitive inhibition with Ki values of 145.2 μM, 76 μM and 8.6 μM, respectively. In order to establish the mechanism of this inhibition, crystallographic studies were carried out and the structures of GPb in complex with the three analogs were determined. The complex structures revealed that the inhibitors can be accommodated in the catalytic site of T-state GPb with very little change of the tertiary structure [51].

2-(β-d-glucopyranosyl)-5-methyl-1,3,4-oxadiazole 155, 2-(β-d-glucopyranosyl)-benzothiazole 156 and 2-(β-d-glucopyranosyl)-benzimidazole 157 [51]
In 2010, Kang and coworkers designed and synthesized pyridazinyl and thiazolyl derivative of C-glycosides [52]. They wanted to check if replacement of the phenyl ring with the corresponding heterocyclic ring could improve the GLT2 inhibitor. As shown in Scheme 25, the lithiated thiazolylglucoside 158 was converted to 5-chlorothiazolylglucoside or 5-bromothiazolylglucoside 159 by electrophilic halogenation using CCl4 and CBr4, respectively. Lithiation of 5-bromothiazole intermediate 159 was performed by treatment of LDA, and the resulting anion underwent a metal–halogen exchange reaction so that a bromine atom moved to a new position on the thiazole ring. The lithiated intermediate 160 was subjected to coupling with aldehydes to produce the desired products 161. The same conditions were applied to 5-chlorothiazole 159. The chlorine atom did not move to the 4-position but maintained the original position. The coupling reactions of 5-chlorothiazole intermediate 159 with aldehydes produced 163. Both debenzylation and reduction were concurrently performed to prepare the final products 162 and 164 (Scheme 25). Introduction of the pyridazine ring at the anomeric carbon of d-glucopyranose was carried out in a stereoselective fashion [52]. Cyclization from γ-keto ester 165 to dihydropyridazinone 166 was accomplished with hydrazine monohydrate (Scheme 26). Dihydropyridazinone 166 was oxidized to pyridazinone 167 using bromine under acetic acid. Pyridazinone 167 was converted to 6-chloro-5-benzylpyridazine 168 by treatment with POCl3. Final removal of the four benzyl groups to produce the target compound 169 was accomplished with application TMSI (Scheme 26).

Synthesis of benzylthiazolyl-C-glucosides 162 and 164 [52]

Synthesis of benzylpyridazinyl-C-glucosides 169 [52]
Biological activities of the compounds 162, 164 (Scheme 25) and 169 (Scheme 26) were evaluated by in vitro SGLT2 inhibition assay. While dapagliflozin (Scheme 18) shows highly potent inhibitory activity against human hSGLT2, it was discovered that neither pyridazinyl nor thiazolyl analogs improved hSGLT2 inhibition [52].
In 2010, Handlon and coworkers described a method of obtaining C-linked heterocyclic glucosides that could inhibit human SGLT2 [53]. The authors used the bromo heterocycles 170 and the glucal boronate 171 to obtain a series of benzisothiazole- and indolizine-β-d-glucopyranosides 174 (Scheme 27). The key step of the reactions was a palladium-catalyzed cross-coupling leading to intermediates 172. Subsequent hydroboration–oxidation reactions followed by an acidic deprotection of the sugar rings in the molecules of 173 provided the final products 174. The substrates 170 were obtained in three various ways, depending on their heterocyclic cores. The compounds were evaluated for their human SGLT1 and SGLT2 inhibition potential by monitoring the suppression of the uptake of 14C-labeled α-methyl-d-glucopyranoside by COS-7 cells, which transiently expressed human SGLT2 or SGLT1, using BacMam technology [54]. The authors focused mostly on the influence of the character of the substituents R1 and R2 and the basicity of the aromatic core on the inhibition potential of the compounds. It was found that their oral absorptions were good enough to avoid a transformation into the corresponding pro-drugs prior to the intake. Finally, the compound 174 (X=C, Y=S, Z=N, R1=t-Bu, R2=H) was found to be an inhibitor of SGLT2 with an IC50 of 10 nM [53].

Synthesis of benzisothiazole- and indolizine-β-d-glucopyranosides 174 as SGLT2 inhibitors [53]
In 2012, Yao et al. based on previous research into usage of N-indolylxylosides as SGLT2 inhibitors [55] and knowledge about metabolic stability of the C-glycosidic bond, synthesized the C-indolylxylosides as a result of a five-step synthesis. It is noteworthy that their SAR studies disclosed the key role of two substituents in the indole moiety. The presence of both a distal p-cyclopropylphenyl group and substituent in 7-position is necessary to achieve potent inhibitory activity. Using 2,3,4-tri-O-benzyl-d-xylonolactone 175 and diverse 3-bromo-1-tosyl-1H-indoles 176 as a starting material in lithium halogen exchange reaction, a variety of lactols 177 were received. Reduction with trietylsilane and boron trifluoride etherate gave C-linked β-xylosides 178 (Scheme 28). During heating of the previously obtained compounds 178 over KOH in THF/EtOH, detosylation took place, providing free indoles 179. Benzyl ether groups of 179 were removed under hydrogenolysis to furnish 180. Xylopyranosyl indoles 180 underwent N-alkylation with p-cyclopropylbenzyl bromide and gave the final products 181. Evaluation of biological activity demonstrated that from among C-indolylxylosides, compound 181 (R=F) turned out to be the strongest and metabolically stable SGLT2 and SGLT1 inhibitor. In compliance with SAR studies bearing two groups most significant for inhibition activity, it exhibits an SGLT2 EC50 value of 47 nM and SGLT1 EC50 value of 282 nM. Moreover pharmacokinetic studies showed that molecule 181 (R=7-F) is metabolically stable after intravenous and oral administration to rats [55].

Synthesis of C-indolylxylosides 181 bearing p-cyklopropylbenzyl group [55]
In 2012 Li et al. designed and synthesized analogs of SGLT2 inhibitors containing the 1,2,3-triazole motif [56]. Substituted 1,2,3-triazole is a very important building block for more complex bioactive compounds, such as tazobactam, antiviral, anti-HIV, antibacterial, and antiallergic agents [56]. The C-glucosides with triazole aglycone were constructed by click chemistry. The synthesis of the key alkyne intermediate is outlined in Scheme 29. Alkyne 187 was obtained from 2,3,4,6-tetra-O-benzyl-d-glucopyranose 182 in five steps. 2,3,4,6-Tetra-O-benzyl-d-(+)-glucono-1,5-lactone 183 was prepared by Swern oxidation of benzyl protected d-glucopyranose 182. Trimethylsilylacetylene was deprotonated with n-BuLi and treated with lactone 183 to provide ketose 184. The free hydroxyl group was reduced and the trimethylsilyl group was removed easily by stirring in a mixture of NaOH, methanol, and dichloromethane, yielding the benzyl-protected alkyne 186. Alkyne 186 and azides were used directly to construct triazole aglycon by click chemistry. Compound 186 was transformed into the acetyl-protected form 187. Triazoles 188 were then synthesized through CuAAC with the corresponding azides (Scheme 29). Finally, the acetyl protecting groups were removed to give the triazole-linked C-glycosides compounds 189. Most of the synthesized compounds demonstrated increased urinary glucose excretion in SD rats, but they increased urine volume to a lesser degree than that of dapagliflozin [56].

Synthesis of C-glucosides with triazole aglycone 189 [56]
In 2013, Bokor et al. elaborated a new method for the synthesis of 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazoles [57]. The starting compound was O-perbenzoylated β-d-glucopyranosyl formimidate 190, which reacted with tosylhydrazide to give tosylamidrazone 191 (Scheme 30). 3-(β-d-Glucopyranosyl)-5-substituted-1,2,4-triazoles 194 were prepared by acylation of O-perbenzoylated N1-tosyl-C-β-d-glucopyranosyl formamidrazone 191 and subsequent removal of the protecting groups. The best inhibitor was 3-(β-d-glucopyranosyl)-5-(2-naphthyl)-1,2,4-triazole 194 (Ki = 0.41 μM against rabbit muscle glycogen phosphorylase b).

Synthesis of 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazoles 194 [57]
In 2013, Sakamaki and coworkers described the synthesis and structure–activity relationship of thiophene-C-glucosides [58]. The synthetic route to thiophene-C-glucosides 199 is shown in Scheme 31, based on the reaction of aryl halide 196 with glucal-boronate ester 195. Coupling reaction with using dichlorobis (triphenylphosphine) palladium between aglycones 195 and glucal-boronate 196 gave 197, followed by stereoselective hydroboration and oxidation in alkaline conditions yielded 198 with the desired β-configuration. O-silyl groups of 198 were deprotected with tetra-n-butylammonium fluoride (TBAF) to afford thiophene-C-glucosides 199 (Scheme 31). The human hSGLT2 inhibitory activities and rat urinary glucose excretion (UGE) effects of 199 were evaluated. As a result, they showed good hSGLT2 inhibitory activities. In particular, the chlorothiophene derivative 199 showed remarkable inhibitory activity against hSGLT2 (IC50 = 4.0 nM) [58].

Synthesis of thiophene-C-glucosides 199 [58]
In 2014, Somsak et al. synthesized derivatives of d-xylose with aglycones of the most efficient glucose-derived inhibitors of glycogen phosphorylase to explore the specificity of the enzyme towards the structure of the sugar part of the molecules [59]. 2-(β-d-Xylopyranosyl)benzimidazole 204 (Scheme 32) and 3-substituted-5-(β-d-xylopyranosyl)-1,2,4-triazoles 209 (Scheme 33) were obtained in multistep procedures from O-perbenzoylated β-d-xylopyranosyl cyanide 200.

Synthesis of 2-(β-d-xylopyranosyl)benzimidazole 204 [59]

Synthesis of 3-(β-d-xylopyranosyl)-5-substituted-1,2,4-triazoles 209 [59]
Cycloadditions of nitrile-oxides and O-peracetylated exo-xylal 212 obtained from the corresponding β-d-xylopyranosyl cyanide 210 furnished xylopyranosylidene-spiro-isoxazoline derivatives 214 (Scheme 34) [59].

Synthesis of xylopyranosylidene-spiro-isoxazoline derivatives 214 [59]
Oxidative ring closure of O-peracetylated β-d-xylopyranosyl-thiohydroximates prepared from 1-thio-β-d-xylopyranose 215 and nitrile-oxides gave xylopyranosylidene-spiro-oxathiazoles 217 and 218 (Scheme 35) [59]. The fully deprotected test compounds 204, 209, 214, 219, and 220 were assayed against rabbit muscle glycogen phosphorylase b. Evaluation showed very weak inhibition for 3-(2-naphthyl)-5-(β-d-xylopyranosyl)-1,2,4-triazole 209 only, while all other compounds proved ineffective in a concentration of 625 μM. Observations showed that the aglycones rendering their glucose derivatives to nanomolar inhibitors are not yet capable of completely overriding the effect of losing the side chain of the glucose moiety [59].

Synthesis of xylopyranosylidene-spiro-oxathiazoles 219 and 220 [59]
Investigations on the inhibitory and binding properties of different monosaccharides indicated the superior effectiveness of d-glucose [60, 61]. Changes in the sugar configuration as well as removal or replacement of substituents of the glucose moiety proved detrimental for the inhibition. Therefore, in 2015 Bokor et al. elaborated synthetic methods for D-glucal attached to oxadiazoles by a C–C bond [62]. For the preparation of the target compounds 226, two main routes were used; the functionalized glucal 226 was made by the formation of the heterocycle in the final stage (Scheme 36) or the 1,2-double bond can be introduced into a preformed C-glucopyranosyl heterocycle 227 (Scheme 37). Introduction of the double bond was effected by either DBU induced elimination of benzoic acid from O-perbenzoylated glucopyranosyl precursors 221 (X=H) or Zn/N-methylimidazole mediated reductive elimination from the 1-bromoglucopyranosyl starting compounds 221 (X=Br) (Scheme 36). Test compounds 226 were obtained by Zemplen debenzoylation. Unfortunately, none of these showed significant inhibition of rabbit muscle glycogen phosphorylase b, indicating that the binding of the aglycones was not strong enough to override the detrimental effects of the changes in the sugar parts of the molecules [62].

First method of compounds 226 synthesis [62]

Second method of compounds 226 synthesis [62]
In 2016, Bokor et al. designed various C-glucopyranosyl-1,2,4-triazolones as potential inhibitors of GP [63]. Syntheses of these compounds were performed with O-perbenzoylated glucose derivatives 228, 230, and 233 as precursors (Scheme 38). Boiling a solution of carbamoyl-C-β-d-glucopyranosyl formamidrazone 228 in m-xylene gave 3-β-d-glucopyranosyl-1,2,4-triazol-5-one 229. Cyclization of 230 in boiling DMF produced the expected triazolone 231. Reaction of tosyl-C-β-d-glucopyranosyl formamidrazone 233 with ethyl chloroformate furnished 3-β-d-glucopyranosyl-1-tosyl-1,2,4-triazol-5-one 234 (Scheme 38). In situ prepared β-d-glucopyranosylcarbonyl isocyanate 237 was transformed by PhNHNHBoc into 3-β-d-glucopyranosyl-1-phenyl-1,2,4-triazol-5-one 240, while the analogous 1-(2-naphthyl) derivative 243 was obtained from the unsubstituted triazolone 242 by naphthalene-2-boronic acid in a Cu(II) catalyzed N-arylation (Scheme 39). Test compounds were prepared by Zemplen deacylation. The new glucose derivatives had weak or no inhibition of rabbit muscle glycogen phosphorylase b. The best inhibitor was 3-β-d-glucopyranosyl-1-(2-naphthyl)-1,2,4-triazol-5-one 244 (Ki = 80 μM) (Scheme 39) [63].

Syntheses of C-glucopyranosyl-1,2,4-triazolones 229, 232, and 235 [63]

Synthesis of 3-β-D-glucopyranosyl-1-phenyl-1,2,4-triazol-5-one 241 and 1-(2-naphthyl) derivative 244 [63]
Glucose-based spiro-isoxazolines can be considered as anti-hyperglycemic agents against type 2 diabetes through GP inhibition. In 2016, d-glucopyranosylidene-spiro-isoxazolines 252 were prepared by 1,3-dipolar cycloaddition of nitrile oxides 249 generated in situ to methylene exo-glucals 250 (Scheme 40) [64]. Reagents 249 were generated by reaction of a sodium hypochlorite 246 and oximes 245. Appropriate oximes 245 reacted also with NCS 247 and aryl α-chloroaldoximes 248 were prepared. Hydrochloric acid elimination in the presence of NEt3 afforded reactive nitrile oxides 249. O-unprotected spiro-isooxazolines 252 were evaluated as GP inhibitors and exhibited IC50 values ranging from 1 to 800 μM. The tetra-O-acetylated spiro-isoxazoline 251 bearing 2-naphthyl residue shoved a much lower value compared to that of the O-unprotected analog 252 [64]. The 2-naphthyl substituted glucopyranosylidene-spiro-isoxazoline 252 was the best compound identified in this study (GPb Ki = 0.63 μM).

Synthesis of spiro-isooxazolines 252 by 1,3-dipolar cycloaddition of nitrile oxides 249 to exo-glucals 250 [64]
Syntheses of a series of C-glucopyranosyl pyrroles, indole, and an improved preparation of C-glucopyranosyl imidazoles allowed in 2016 Kantsadi et al. to study and compare their inhibitory efficiency against GP [65]. C-β-D-Glucopyranosyl pyrrole derivatives 258, 260, and 262 were prepared in the reactions of pyrrole 254, 2-aryl-pyroles 255, and 3-aryl-pyrroles 256 with O-peracetylated β-d-glucopyranosyl trichloroacetimidate 253 (Scheme 41). (2β-d-Glucopyranosyl) indole 267 was obtained by a cross-coupling of O-perbenzylated β-d-glucopyranosyl acetylene 263 with N-tosyl-2-iodoaniline 264 followed by spontaneous ring closure (Scheme 42) [65]. An improved synthesis of O-perbenzoylated 2-(β-d-glucopyranosyl) imidazoles 270 was achieved by reacting C-glucopyranosyl formimidates 268 with α-aminoketones 269 (Scheme 43) [65]. The deprotected compounds were assayed with isoforms of glycogen phosphorylase to show no activity of the pyrroles 258, 260, 262, and indole 267 against rabbit muscle GPb [65]. The imidazoles 271 proved to be the best-known glucose-derived inhibitors of not only the muscle enzymes (both a and b) but also of the pharmacologically relevant human liver hlGPa (Ki = 156 and 26 nM for the phenyl and 2-naphthyl derivatives, respectively). An X-ray crystallographic study of the rmGPb-imidazole complexes revealed structural features of the strong binding, and also allowed explaining the absence of inhibition for the pyrrole and indole derivatives [65].

Synthesis of C-β-d-glucopyranosyl pyrrole derivatives 258, 260, and 262 [65]

Synthesis of 2-(β-d-glucopyranosyl) indole 267 [65]

Synthesis of 2-(β-d-glucopyranosyl) imidazoles 271 [65]
Figure 13 shows the structure of the best inhibitors from heteroaromatic C-glycoside derivatives described in Sect. 4.2. Again, the analysis of the structure of the best inhibitors leads to the conclusion that the highest activity is ensured by the presence of glucose as a structural element. Only in the case of compounds 181 and 209 is it xylose. It can also be seen that the high inhibitor activity is guaranteed by the presence of a structural element such as a five-membered heteroaromatic ring containing two or three nitrogen atoms. The high activity of the inhibitor is also ensured by the presence of such heteroaromatic rings as: 1,3,4-oxadiazole (compound 149), benzisothiazole (compound 174), indole (compound 181), thiophene (compound 199), and isoxazoline (compound 252). Again, presents a distal p-cyclopropylphenyl group (compound 181) is necessary to achieve potent inhibitory activity. Also, the glycone and aglycone spiro combination (compounds 144) ensures high inhibitor activity.

Inhibitory properties of the best inhibitors from the heteroaromatic C-glycosyl derivatives described in Sect. 4.2
4.3 Other C-Glycosyl Derivatives
In 2009, Bisht and coworkers described the synthesis of aryl butenoyl C-glycosides 277 by aldol condensation of peracetylated glycosyl acetones 275 with aromatic aldehydes followed by deacetylation with methanolic NaOMe (Scheme 44) [66]. β-C-Glycosidic ketones 274 were prepared in one step directly from the unprotected sugar 272 and pentane-2,4-dione 273 under aqueous conditions by Knoevenagel condensation [67]. Compounds 275 on aldol reaction with different aldehydes under ambient reaction conditions resulted in (E)-4-aryl-1-(glycopyranosyl)-but-3-en-2-ones 276 [68]. Prepared C-glycosides 277 were evaluated for their α-glucosidase, glucose-6-phosphatse, and glycogen phosphorylase enzyme inhibitory activities in vitro and in vivo. Three of the compounds 277 (Ar=2-naphthyl, phenyl, 3,4-dimethoxyphenyl) showed potent enzyme inhibitory activities as compared to standard drugs such as acarbose and metformin. These C-glycosides caused a significant decline in the hyperglycemia of the diabetic rats post sucrose-load [66].

Synthesis of (E)-4-aryl-1-(β-d-glucopyranosyl)-but-3-en-2-ones 277 [66]
Another approach for the inhibition of GP could take advantage of multivalency. In 2009, Cecioni and coworkers examined influence multivalency for the inhibition of GP [69]. They synthesized two distinct trivalent inhibitors of GP through Cu(I)-assisted 1,3-dipolar cycloaddition and by formation of a trisoxadiazole derivative. The perbenzoylated glucosyl cyanide 278 was reacted with hydroxylamine hydrochloride in pyridine to afford the desired amidoxime 279 (Scheme 45). The formation of the O-acyl-amidoxime 280 was achieved with 4-pentynoic acid in the presence of EDCI/HOBt as coupling agents. The use of thermal activation combined with TBAF catalysis provided the cyclic oxadiazole 281. The alkyne-terminated oxadiazole 281 was then engaged in a Huisgen’s Cu(I)-catalyzed 1,3-dipolar cycloaddition reaction under microwaves activation with benzyl azide to afford 1,4-disubstituted 1,2,3-triazole 282. Debenzoylation of compound 282 afforded hydroxylated GP inhibitor candidate 283. The reaction of 1,3,5-tris(azidomethyl)benzene with the alkyne derivative 281 under microwave activation and Cu(I) catalysis afforded the cycloadduct 284. The saponification of the benzoate ester 284 provided the fully hydroxylated macromolecule 285. Also, a more condensed trifunctional macromolecule was prepared in which the C-glucosyl-oxadiazole moiety was directly attached to a benzene ring. A biological study of the inhibiting properties of these trivalent inhibitors of GP have shown that the valency of the molecules influences slightly the inhibition of the enzyme, whereas the presence of a spacer arm between the core and the pharmacophore moieties does not. Authors reassumed that multivalent inhibitors were always superior to their monovalent counterparts [69].

Synthesis of inhibitors of GP 283 and 285 through Cu(I)-assisted 1,3-dipolar cycloaddition [69]
In 2010, Kakinuma et al. gave considerable attention to 5-thioglucose—derived SGLT2 inhibitors, which have a sulfur atom in place of the oxygen atom in the glucose ring [70]. It is known that 5′-thio-N-acetyllactosamine is 200 times more resistant to digestion by β-galactosidase [71], and methyl α-5′-thiomaltoside is not hydrolyzed at all by glucoamylase [72]. In previous articles [73, 74], it was also described that O-aryl 5-thio-β-glucoside is a SGLT inhibitor. Kakinuma et al. developed a synthetic strategy for preparing C-phenyl 1-thio-d-glucitol derivatives 290, as it is outlined in Scheme 46 [70]. Compounds 288 were obtained by adding thiolactone 287 to Grignard reagents prepared from compounds 286 and magnesium powder. The hydroxyl group of 288 was reduced β-stereoselectively to afford compounds 289. Finally, the benzyl ether of compounds 289 was removed by catalytic hydrogenation with palladium hydroxide under a hydrogen atmosphere or, alternatively, compounds 290 were obtained by removal of the benzyl group using Lewis acid conditions to prevent reduction of the chloride (Scheme 46). (1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol 290 (R1=OMe, R2=Me, R3=OEt) exhibited potent SGLT2 inhibition activity (IC50 = 2.26 nM) [70]. Since 2014, this 1-thio-d-glucitol 290 has been known as luseogliflozin, and it is an orally active SGLT2 inhibitor developed by Taisho Pharmaceutical for the treatment of patients with type 2 diabetes mellitus [75].

Synthesis of C-phenyl 1-thio-d-glucitols 290 selective SGLT2 inhibitors [70]
An approach to controlling blood glucose levels in individuals with type 2 diabetes is to target R-amylases and intestinal glucosidases using R-glucosidase inhibitors acarbose and miglitol. One of the intestinal glucosidases targeted is the N-terminal catalytic domain of maltase-glucoamylase (ntMGAM), one of the four intestinal glycoside hydrolase 31 enzyme activities responsible for the hydrolysis of terminal starch products into glucose [76]. In 2010, Sim and coworkers presented the X-ray crystallographic studies of ntMGAM in complex with a new class of R-glucosidase inhibitors derived from natural extracts of Salacia reticulata, a plant used traditionally in Ayurvedic medicine for the treatment of type 2 diabetes [76]. In extracts, active compounds were: salacinol 291, kotalanol 292, and de-O-sulfonated kotalanol 293 (Fig. 14). This study revealed that kotalanol 293 is the most potent ntMGAM inhibitor reported to date (Ki = 0.03 μM), some 2000-fold better than the compounds currently used in the clinic, and highlights the potential of the salacinol class of inhibitors as future drug candidates [76].

Structures of R-glucosidase inhibitors from Salacia reticulata: salacinol 291, kotalanol 292, and de-O-sulfonated kotalanol 293 [76]
In 2011, Wang et al. synthesized triazolyl phenylalanine and tyrosine-aryl C-glycoside hybrids via microwave-assisted Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition [77]. Successive enzymatic assay identified the synthesized glycoconjugates as novel PTP1B inhibitors with low micromole-ranged inhibitory activity and at least several-fold selectivity over other homologous PTPs tested. As shown in Scheme 47, the azido phenylalaninyl or tyrosinyl derivatives 297 were synthesized according to the literature [78]. For the synthesis of the O-propynyl C-glycoside 296, the known C-glucosyl 1,4-dimethoxybenzene 294 was first regioselectively silylated on its 6-position with TBDMSCl followed by full O-benzylation with NaH and BnBr. Then, the TBS group was desilylated with AcCl to give the free 6-OH, which was propargylated in the presence of NaH and propargyl bromide. Huisgen [3 + 2] cycloaddition between the azides 297 and the sugar alkyne 296 was catalyzed by sodium ascorbate and CuSO4·5H2O yielding the click adducts 298 (Scheme 47). The saponification with LiOH led to the carboxylic acids 299. The following hydrogenolysis gave the fully deprotected amino acid-sugar hybrid 300. Benzyl groups on glucosyl moiety of compounds were found crucial for PTP1B inhibition. The biological assay identified the glycoconjugates that contain the carboxylic acid and benzyl moieties as more active PTP1B inhibitors compared to their ester and debenzylated counterparts [77].

Synthesis of triazolyl phenylalanine and tyrosine-aryl C-glycoside hybrids 300 via microwave-assisted Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition [77]
In 2011, Kim et al. designed and synthesized novel macrocyclic C-aryl glucoside SGLT2 inhibitors [79]. Two different synthetic routes of macrocyclization were adopted. Alkylation of alcohol 301 with (5-bromopentyloxy)(tert-butyl)-diphenylsilane 302 in the presence of sodium hydride in DMF produced 303 (Scheme 48). Desilylation of 303 with TBAF gave alcohol 304. Removal of the allyl group was carried out using NaBH4 in the presence of tetrakis(triphenylphosphine) palladium(0) to give phenol 305 in quantitative yield. The primary alcohol of 305 was transformed into the corresponding iodide 306 by action of iodine, triphenylphosphine, and imidazole in benzene. The iodide 306 underwent macrocyclization to 307 under conditions of potassium carbonate and 18-crown-6 in DMF. Removal of the benzyl groups on the carbohydrate moiety proceeded with either BCl3 in methylene chloride or hydrogenolysis on Pd/C in MeOH and THF to produce the target compound 308 (Scheme 48). Among the compounds tested, [1, 7] dioxacyclopentadecine macrocycles 308 possessing ethoxyphenyl at the distal ring showed the best in vitro inhibitory activity (IC50 = 0.778 nM) against human hSGLT2 [79].

Synthesis of macrocyclic C-aryl glucoside SGLT2 inhibitor 308 [79]
In 2012, Ohtake et al. discovered a novel class of inhibitors, which have an O-spiroketal C-arylglucoside scaffold [80]. Compound 312 (R1=Et)—tofogliflozin (Scheme 49) is a selective SGLT2 inhibitor that is one of the inhibitors for the treatment of type 2 diabetes. Ohtake et al. worked on synthesis of tofogliflozin using the computational modeling and comparing other pharmacophore models that were derived from earlier inhibitors. O-Spiroketal C-arylglucosides 312 were prepared from 309 through two pathways, as outlined in Scheme 49. Compounds 311 were synthesized from the aldehyde 310, which could be obtained by oxidation of 309, followed by addition of Grignard reagents or lithiated benzenes and reduction. Compounds 314 were prepared utilizing the Suzuki coupling reactions. After debenzylation of 309 using boron trichloride, benzyl alcohol moiety was selectively chlorinated by treatment of chlorotrimethylsilane with dimethyl sulfoxide. Four hydroxyl groups of the resulting benzyl chloride were acetylated to afford 313. Suzuki coupling reactions of 313 with the corresponding 4-substituted phenylboronic acids gave 314. Deprotections (debenzylation for 311 or deacetylation for 314) afforded the test compounds 312. Two products 312 (R=Et or iPr) were submitted to clinical trials. Both products showed a similar degree of increase in renal glucose excretion after oral dosing [80]. However, the next clinical trials turned out the 312 (R=Et) is much better, because it had more desirable profiles in oral bioavailability and renal excretion than 314 (R=iPr).

Synthesis of O-spiroketal C-arylglucoside scaffold 312 [80]
The discovery of structurally distinct SGLT2 inhibitors has been mainly focused on the modification of the aglycones, while modification to the glucose residue is less known. Therefore, in 2016 Yan and coworkers decided to examine a series of C-aryl glucosides containing dioxa-bicycle for inhibition activity against hSGLT2 [81]. Ertugliflozin [82], bearing a unique dioxa-bicycle in place of the glucose residue of dapagliflozin, is distinct from other inhibitors and shows even better SGLT2 inhibitory activity, which is currently under phase III clinical trial. The synthesis of dioxa-bicycle C-aryl glucoside 327 is outlined in Scheme 50. Allylation of 315 in the presence of boron trifluoride etherate formed compound 316, which was converted to ether 318. Conversion of the allyl intermediate 318 to aldehyde 320 using a Pd-catalyzed double-bond migration and next reaction with K2Os2O4 and sodium periodate was made. Aldehyde 320 was then reduced to the alcohol 321, which was then protected as the methoxymethyl ether 322. Deprotection of TBSO ether gave alcohol 323, and the primary hydroxyl group of 323 was subjected to iodination using Ph3P, imidazole and iodine to give 324, which upon elimination using DBU in toluene furnished 325. Sharpless dihydroxylation and acid-promoted one-pot MOM removal followed by stereoselective intramolecular trapping of the putative oxonium ion intermediate gave compound 326. Hydrogenolysis of the benzyl-protecting groups yielded target compound 327 (Scheme 50). The target compound 327 was subsequently subjected to biological evaluation as novel C-aryl glucoside SGLT2 inhibitor. Compound 327 showed good inhibitory activity against hSGLT2 IC50 = 714 nM [81].

Synthesis of ertugliflozin 327 [81]
According to our group interest of synthesis of C-glycosyl derivatives we also occupied in the synthesis of some derivatives. In 2013, we developed a convenient and efficient procedure for the preparation of fused uracils - pyrano[2,3-d]pyrimidines with sugar moiety [83]. The reaction sequence was: Knoevenagel condensation of unprotected sugars 328 and 1,3-dimethylbarbituric acid 329 in water, acetylation of C-glycosides 330 and hetero-Diels–Alder reaction (Scheme 51). O-Acetylated 1,3-dimethyl-2,4,6-trioxo-pyrimidin-5-ylidene derivatives 331 were used as new heterodienes in the synthesis of pyrano[2,3-d]pyrimidines 333 and 334 containing a sugar moiety. Solvent-free hetero-Diels–Alder cycloadditions of O-acetylated pyrimidin-5-ylidene alditols 331 with enol ethers 332 were investigated at room temperature. New, enantiomerically pure cis and trans diastereoisomers of pyrano[2,3-d]pyrimidines 333 with alditol moiety were obtained. The same pyrimidin-5-ylidene alditols 331 underwent conjugate Michael addition-cyclizations with malononitrile 335 at room temperature to afford optically active uracils 336—diastereoisomers of pyrano[2,3-d]pyrimidine-6-carbonitriles with a sugar moiety (Scheme 52) [83]. None of the C-glycosyl derivatives of pyrano[2,3-d]pyrimidines presented in Schemes 51 and 52 have been evaluated for their pharmacological activity as inhibitors in treatment of type 2 diabetes mellitus.

Synthesis of pyrano[2,3-d]pyrimidines 333 and 334 with sugar moiety [83]

Synthesis of pyrano[2,3-d]pyrimidine-6-carbonitriles 336 with a sugar moiety [83]
Our group also described a convenient and efficient method for the synthesis of chromeno[2,3-d]pyrimidine-2,4-diones containing different sugar moieties [84]. Dimedone enamines were used as dienophiles in hetero-Diels–Alder reactions. The cycloaddition reactions of O-acetylated 1,3-dimethyl-2,4,6-trioxo-pyrimidin-5-ylidene alditols 337, representing a 1-oxa-1,3-butadiene system, with dimedone enamines 338 afforded only one enantiomerically pure cis diastereoisomer of chromeno[2,3-d]pyrimidine-2,4-diones 339 in each reaction (Scheme 53). Analysis of NMR spectra allowed the determination that prepared fused uracils containing amino and enol functional groups exist as a mixture of the neutral form (NF) and zwitterions—dipolar ions (DI). By this simple hetero-Diels–Alder reaction, we can introduce into fused uracil systems such important for biological interaction groups as: different sugar moieties, enol moiety, and different amino groups. The prepared fused uracils contain both amine and enol functional groups, so share amphiprotic properties, and they are zwitterions in solid state [84]. None of the C-glycosyl derivatives of chromeno[2,3-d]pyrimidines 339 have been examined as inhibitors in the treatment of type 2 diabetes.

Synthesis of C-glycosides-chromeno[2,3-d]pyrimidines 339 containing different sugar moieties [84]
Figure 15 presents inhibitory properties of the best inhibitors from the other C-glycosyl derivatives described in Sect. 4.3. The variety of active inhibitor structures is in this case greater than in the above-mentioned groups of glycosides. For example, aryl butenoyl C-glycosides 277 can cause a significant decline in the hyperglycemia of the diabetic rats post sucrose-load. 5-Thioglucose—derived inhibitor 290, which has a sulfur atom in place of the oxygen atom in the glucose ring, is known as luseogliflozin and it is an orally active SGLT2 inhibitor for the treatment of patients with type 2 diabetes mellitus. The activity of inhibitors may also be influenced by multivalency. On the basis of tests of trivalent inhibitors activity (for example compound 285), it was found that valency of the molecules influences slightly the inhibition of the enzyme, whereas the presence of a spacer arm between the core and the pharmacophore moieties does not. Multivalent inhibitors were always superior to their monovalent counterparts. In turn, the natural C-glycoside kotalanol 293, which structures comprise a 1,4-anhydro-4-thio-d-arabinitol core and polyhydroxylated acyclic chain, is the most potent ntMGAM inhibitor reported to date (Ki = 0.03 μM) and highlights the potential of the salacinol class of inhibitors as future drug candidates. Active inhibitors can also be glycoconjugates, such as triazolyl phenylalanine and tyrosine-aryl C-glycoside hybrids (compound 299). Biological assay identified the glycoconjugates 299 that contain the carboxylic acid and benzyl moieties as more active PTP1B inhibitors compared to their ester and debenzylated counterparts. Also, macrocyclic C-glycosides may be active inhibitors. For example, [1, 7] dioxacyclopentadecine macrocycle 308 possessing ethoxyphenyl at the distal ring showed the best in vitro inhibitory activity (IC50 = 0.778 nM) against human hSGLT2. Compound 312—tofogliflozin represents a novel class of SGLT2 inhibitors, which have an O-spiroketal C-arylglucoside scaffold. In turn, ertugliflozin 327 contains a unique dioxa-bicycle in place of the glucose residue of dapagliflozin, and is distinct from other inhibitors, and shows even better SGLT2 inhibitory activity.

Inhibitory properties of the best inhibitors from the other C-glycosyl derivatives described in Sect. 4.3
5 Directions of the Latest Research in 2018–2019
In order to show the latest research directions, papers from 2018 to 2019 are described in a separate Sect. 5. When searching for articles on subject this publication, it was noted that the number of articles describing the synthesis of new inhibitors decreased, and at the same time the number of publications depicting the extraction of active natural O-glycosides from plant materials increased. This trend applies in particular to 2018 and 2019. Sect. 5 presents articles on the latest modifications of the C-glycosides structure aimed at increasing their effectiveness as inhibitors. Suitable modifications were made to previously tested C-glycosides, in the aglycone structure as well as in the sugar molecule structure. Next, the latest proposals concerning the structure of N-glycosides as antidiabetic agents were presented and finally the structures of active O-glycosides isolated from plants were presented.
In 2018, Kerru et al. published a review article about current antidiabetic agents and their molecular targets [85]. In this review, authors described the use of heterocyclic scaffolds, which have been evaluated for their biological response as inhibitors against their respective antidiabetic molecular targets over the 5-year period from 2012 to 2017. Investigation reveals a diverse target set which includes protein tyrosine phosphatase 1 B (PTP1B), dipeptidly peptidase-4 (DPP-4), free fatty acid receptors 1 (FFAR1), G protein-coupled receptors (GPCR), peroxisome proliferator activated receptor-g (PPARg), sodium glucose co-transporter-2 (SGLT2), α-glucosidase, aldose reductase, glycogen phosphorylase (GP), fructose-1,6-bisphosphatase (FBPase), glucagon receptor (GCGr), and phosphoenolpyruvate carboxykinase (PEPCK). The article presents the structures of various active heterocyclic compounds, of which glycoside derivatives constitute a small group.
Kun and coworkers examined 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazoles, which have been revealed as an effective scaffold for the development of potent glycogen phosphorylase inhibitors [86]. The potency of these compounds is very sensitive to the nature of the alkyl/aryl 5-substituent. Authors have chosen for synthesis nine predicted candidates after in silico screening of 2335 new analogues. The compounds 349 were prepared in O-perbenzoylated forms by either ring transformation of 5-β-d-glucopyranosyl tetrazole 340 by N-benzyl-arenecarboximidoyl chlorides, ring closure of C-(β-d-glucopyranosyl)formamidrazone 341 with aroyl chlorides, or that of N-(β-d-glucopyranosylcarbonyl)arenethiocarboxamides 347 by hydrazine, followed by deprotections (Scheme 54). Five compounds had Ki’s < 10 μM (349 a–e) with potent low μM inhibitors (rmGPa, hlGPa) and three of these (349a–c) on the submicromolar range for rmGPa [86]. The 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazoles described by Kun et al. are predicted to have drug-like potential with only permeability flagged as a potential issue to efficacy.

Synthesis of 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazoles 349 [86]. The collected yields for individual synthesis steps are not given in the article
In 2018, Kyriakis et al. studied the inhibitory effect of different groups, in size and hydrophobicity, at the para position of 3-(β-d-glucopyranosyl)-5-phenyl-1, 2, 4-triazoles 350 in hlGP by kinetics and X-ray crystallography (Fig. 16) [87]. The most bioactive compound was the one with an amine substituent to show a Ki value of 0.43 μM. The best C-β-d glucopyranosyl triazole GP inhibitor reported thus far is the compound which has a 2-napthyl group and displays a Ki value of 0.172 μM for the liver enzyme. Structural studies have revealed the physicochemical diversity of the β-pocket providing information for future rational inhibitor design studies. Comparison of the Ki values of the inhibitors studied and their structural mode of binding revealed that the addition of the para group led to significant increments in potency only when this group exploited hydrophilic or hydrophobic interactions within the β-pocket [87].

Structure of 3-(β-d-glucopyranosyl)-5-phenyl-1, 2, 4-triazoles 350 [87]
Szennyes et al. performed a systematic study on the preparation of 2-C-(β-d-glucopyranosyl)pyrimidines [88]. Pinner-type cyclization of O-perbenzylated C-(β-d-glucopyranosyl)formamidine 351 with β-ketoesters, dimethyl malonate, and β-diketone-derived α,β-unsaturated β-chloroketones followed by catalytic hydrogenation resulted in various substituted 2-C-(β-d-glucopyranosyl)-pyrimidin-4(3H)-ones 354 (Scheme 55), and 2-C-(β-d-glucopyranosyl)-4,6-disubstituted-pyrimidines, respectively, in moderate to good yields. These pyrimidine derivatives were also obtained by ring closure of the unprotected C-(β-d-glucopyranosyl)formamidine 352 with the same 1,3-dielectrophiles (Scheme 55). A continuous one-pot three-step procedure starting from O-peracylated d-glycopyranosyl cyanides was also elaborated to give pyrimidines with various sugar configurations in overall yields (25–94%). These synthetic routes represent the first expansible method to obtain the target compounds. The C-glycopyranosyl pyrimidines showed moderate inhibition against α-glucosidase and β-galactosidase enzymes, and no activity against glycogen phosphorylase [88].

Synthesis of 2-C-(β-d-glucopyranosyl)-pyrimidin-4(3H)-ones 354 [88]
Kuo et al. identified a good-in-class SGLT1/SGLT2 dual inhibitor to improve blood glucose control in type 2 diabetes [89]. The synthesis of benzocyclobutane-C-glycosides 362 is described in Scheme 56. Lithium-halogen exchange of compound 355 with n-BuLi, followed by the aldol condensation with compound 356 formed 357. Reduction of compound 357 with TFA/Et3SiH gave 358. Treatment of 358 with n-BuLi, followed by the condensation with lactone 359, resulted in the formation of lactol 360. Removal of the 1-OH group with BF3.Et2O/Et3SiH provided compound 361. Deprotection of the benzyl-protecting groups with BCl3/penta-methylbenzene gave benzocyclobutane-C-glycosides 362. The biological experiments were carried out on mice, rats, dogs, and monkeys. The best inhibitor 362 (R1=H, R2=Cl) displayed very high inhibitory potency at both SGLT1 (IC50 = 45 nM) and SGLT2 (IC50 = 1 nM). New compounds have high in vivo efficacies in different animal model species [89].

Synthesis of benzocyclobutane-C-glycosides 362 [89]. The collected yields for individual synthesis steps are not given in the article
In 2019, Kuroda and co-authors reported a discovery of an SGLT1 inhibitor C-phenyl d-glucitol derivative 378 (R1=OMe, R2=H, R3=N,N-dimethylethylenediamine) (Scheme 57 and 58), with a glucose-lowering effect at a dose of 0.3 mg/kg (p.o.) in Sprague–Dawley (SD) rats [90]. The authors’ aim was to obtain a derivative that excretes mostly in bile to avoid retention of the drug in kidneys. The way of achieving this was imparting greater lipophilicity into the molecule by balancing ClogP and topological polar surface area (TPSA) together with the absorbability. The inhibitor 378 was obtained in a multi-step synthesis in which as staring materials were used: 3-isopropylphenol 363, lactone 366, and the compounds 370 and 371 (Schemes 57 and 58). Compound 378 (R1=OMe, R2=H, R3=N,N-dimethylethylenediamine) showed hSGLT1 IC50 = 29 nM, hSGLT2 IC50 = 20 nM, ClogP = 3.66, and TPSA = 161 Å2. The authors concluded that the compound 378 could potentially be useful as a therapeutic agent for patients with T2DM [90].

The synthesis of potent, low-absorbable SGLT1 inhibitor [90] (part 1)

The synthesis of potent, low-absorbable SGLT1 inhibitor 378 [90] (part 2)
In the next two articles discussed in the review, attempts were made to synthesize active inhibitors by modifying the sugar part of the C-glycoside. In 2018, Yuan and coworkers published an article which concerned the synthesis of 27 aryl C-glycosides bearing a C=N/C–N linkage at the glucosyl C6 position [91]. All of these compounds were tested for their inhibitory activity against sodium-dependent glucose co-transporter 2 (SGLT2). Among all obtained oxime ether derivatives 385, oxime (R=H) showed the best in vitro inhibitory activity (hSGLT2 EC50 = 46 nM, hSGLT1 EC50 = 3576 nM) and moreover no significant cytotoxicity and low human ether-à-go–go-related gene (hERG) inhibition. The mentioned oxime ether derivatives 385 were prepared starting from 5-bromo-2-chloro-benzoic acid 379 as outlined in Scheme 59. A mixture of α- and β-C-glucosides 380 was obtained in accordance with the procedure described in [41]. The next step included regioselective 6-O-silylation and per-O-acetylation of 380 to synthesize the fully protected β-C-glucoside 381. Then a reaction with boron trifluoride etherate in CH2Cl2 and oxidation with Dess–Martin periodinane were carried out to introduce the aldehyde at C6 position. At the end, a condensation of aldehyde 383 with different hydroxylamines in pyridine, followed by deacetylation under Zemplén conditions led to obtain the desired oxime ether derivatives 385 [91].

Synthetic pathway of oxime ether derivatives 385 [91]
The next article from 2018 from Sadurní et al. concerned the influence of molecular editing with fluorine at the C2 position of the pyranose ring of phlorizin analogues to concurrently direct β-selective glycosylation [92]. The authors proposed the methodology of fluorine-directed glycosylation to synthesize the selective SGLT2 inhibitors for type 2 diabetes. The mentioned phlorizin analogues were remogliflozin etabonate and dapagliflozin. These compounds, 396 and 397, containing fluorine at C2 position were prepared starting from triacetyl-d-glucal 386 as outlined in Scheme 60. A derivative 387 was obtained by the method described in Ref. [93]. The two glycosyl donors 388 and 389, required to access both target scaffolds, were formed according to the procedures presented in Scheme 60. The next steps in the synthesis of remogliflozin etabonate analogue included: glycosylation, benzyl-deprotection, and selective creation of the primary carbamate. Finally, the remogliflozin etabonate surrogate 396 was obtained. In the case of dapagliflozin surrogate 397, the next steps involved the activation of 391 by halogen-lithium exchange and addition to the donor lactone 389, reduction with triethylsilane and BF3·OEt2, benzyl-deprotection. Based on the conducted biological tests, it was found that the fluorinated dapagliflozin analogue 397 better selectively inhibited human SGLT2 over SGLT1 than remogliflozin etabonate analogue 396 [92].

Synthesis of remogliflozin etabonate and dapagliflozin analogues 396 and 397 [92]
Kun et al. extended the structure–activity relationships of β-d-glucopyranosyl azole type inhibitors and revealed the extreme sensitivity of such type of inhibitor towards the structure of the azole moiety [94]. Actually, these compounds are the best glucose analogue inhibitors of GP known to date. Their efficiency, among other factors, is due to the formation of an H-bridge between the heterocycle and the His-377 main chain carbonyl group in the active site of the enzyme [94]. 1-Aryl-4-β-d-gluco-pyranosyl-1,2,3-triazoles were prepared by copper-catalyzed azide-alkyne cycloadditions between O-perbenzylated or O-peracetylated-β-d-glucopyranosyl ethynes and aryl azides. 1-β-d-Gluco-pyranosyl-4-phenyl imidazole was obtained in a glycosylation of 4(5)-phenylimidazole with O-peracetylated α-d-glucopyranosyl bromide. C-β-d-Glucopyranosyl-N-substituted-tetrazoles were synthesized by alkylation/arylation of O-perbenzoylated 5-β-d-glucopyranosyl-tetrazole or from a 2,6-anhydroheptose tosylhydrazone and arenediazonium salts. 5-Substituted tetrazoles were glycosylated by O-peracetylated α-d-glucopyranosyl bromide 398 to give N-β-d-glucopyranosyl-C-substituted-tetrazoles 399 and 401 (Scheme 61) [94]. Standard deprotections gave test compounds that were assayed against rabbit muscle glycogen phosphorylase b. Most of the compounds proved inactive; the best inhibitor was 2-β-d-glucopyranosyl-5-phenyltetrazole 400 (R1=Ph) (IC50 600 μM).

Synthesis of the best inhibitor 2-β- d -glucopyranosyl 5-substituted tetrazole 400 [94]
Scheme 62 presents N-glucosyl indole derivatives, which were designed and synthesized by Chu et al. in 2019 as sodium-dependent glucose co-transporter 2 inhibitors [95]. The aim of the research was to check how modifications in the sugar part of the N-glucosyl indoles will affect their inhibitory properties. The synthesis of compounds 408 and 410 started from N-glucosyl indole 404. 6-Aldehyde 406 was obtained by selective protection of the primary alcohol 404 with tert-butylchlorodimethylsilane (TBDMSCl), followed by immediate peracetylation by addition of acetic anhydride. Prepared the fully protected N-glucoside underwent desilylation under acidic conditions yielded the free primary alcohol 405. Dess–Martin periodinane (DMP) oxidation gave the desired aldehyde 406. Condensation of 406 with hydroxylamines and hydrazides followed by deacetylation gave oxime ethers 407 and N-acylhydrazones 409. Reduction products, hydroxylamine N-glucosyl indoles 408 and hydrazide N-glucosyl indoles 410, were also synthesized using sodium cyanoborohydride (NaBH3CN) under acidic conditions. Authors studied inhibitory activities (EC50) of all synthesized N-glucosyl indole derivatives 407, 408, 409, 410, which were determined by measuring the inhibition by uptake of [14C]-labeled α-methyl-d-glucopyranoside into hamster ovary cells stably expressing human hSGLT2 or hSGLT1 [95]. The compounds 407 and 409 had similar potency (EC50 = 212–286 nM) except 409 (R=OEt, EC50 = 1162 nM) and 409 (R=O-tert-Bu, EC50 = 867 nM). Compound 409 (R = Ph, EC50 = 258 nM) was the most potent inhibitor of SGLT2. The next step was the examination of hydroxylamine derivatives 408 and hydrazides 410. The products 408 and 410 are better inhibitors than substrates 407 and 409. The hydroxylamine derivatives 408 have similar potency (EC50 = 45–294 nM). Taking into account the group of hydrazides 410, the most activite for hSGLT2 was compoud 410 (R=Me, EC50 = 33 nM). Other hydrazides 410 had their power within range (EC50 = 63–1761 nM). Compound 410 (R=Me) was advanced into selectivity, pharmacokinetic, and in vivo glucosuria studies. Unfortunately, it was found that this hydrazide has poor pharmacokinetic properties and increases glucosuria in rats only at high doses [95].

Synthesis of N-glucosyl indole derivatives 408 and 410 [95]
A variety of medicinal plants and their active compounds have been used to treat diabetes and related chronic disorders since ancient times. Recently, there is a growing interest in developing natural antidiabetic drugs to manage diabetic complications, especially from plant sources. Thus, due to the return to nature, attention was again paid to the structure of natural O-glycosides with antidiabetic action.
In 2018, Rosas-Ramírez et al. postulated that resin glycosides from the morning glory family (Convolvulaceae) may be a source of phytotherapeutic agents with antihyperglycemic properties for the prophylaxis and treatment of non-insulin-dependent type 2 diabetes mellitus [96]. Twenty-seven individual resin glycosides were evaluated for their α-glucosidase inhibitory potential. Four of these compounds displayed an inhibitory activity comparable to acarbose. In Fig. 17, compound 411 presents the structure of one of the four active glycosides isolated from the morning glory family. Based on molecular modeling studies performed by docking analysis, it was predicted that the active compounds and acarbose bind to the α-1,4-glucosidase enzyme catalytic site of MAL12 from the yeast Saccharomyces cerevisiae through stable hydrogen bonds primarily with the amino acid residues HIS279 and GLN322 [96]. Docking studies with the human maltase-glucoamylase (MGAM) also identified binding modes for resin glycosides inside the catalytic site in the proximity of TYR1251. Resin glycosides could help to control postprandial glucose levels due to their inhibitory activity of α-glucosidases, which play a crucial role in the production of glucose [96].

Structures of natural O-glycosides with antidiabetic action isolated from plant sources: 411 (morning glory family) [96], 412 (seeds of Lens culinaris Medikus) [97], 413 (leaves of Stevia rebaudiana bertoni) [98], 414 (Gynostemma longipes) [99], 415 (Ficus species) [100], 416 (Leea indica) [101], 417 (Lu’an GuaPian tea- Camellia sinensis L.O. Kuntze) [102], 418 (onion solid waste) [103], 419 [104]
In 2018, Kim and coworkers isolated from the seeds of Lens culinaris Medikus (Fabaceae) three flavonol glycosides and tested them for their DPP-IV–inhibitory activity [97]. Figure 17 contains the structure of one of the three active glycosides (compound 412) isolated from the morning glory family. Dipeptidyl peptidase IV (DPP-IV) is a new target for the treatment of type 2 diabetes mellitus. DPP-IV inhibitors shorten the inactivation of glucagon-like peptide 1 (GLP-1), permitting the incretin to stimulate insulin release, thereby combating hyperglycemia. It was demonstrated for the first time that three isolated flavonol glycosides inhibited DPP-IV activity in a concentration-dependent manner in vitro bioassay system [97]. Molecular docking experiments of these compounds within the binding pocket of DPP-IV were also conducted. All investigated compounds readily fit within the active sites of DPP-IV, in low-energy conformations characterized by the flavone core structure having optimal electrostatic attractive interactions with the catalytic triad residues of DPP-IV.
The use of steviol glycosides as non-caloric sweeteners has proven to be beneficial for patients with type 2 diabetes mellitus, obesity, and metabolic syndrome [98]. Figure 17 shows the structure of one of the natural antidiabetic O-glycosides 413 isolated from Stevia leaves. Recent data also demonstrate that steviol and stevioside might act as glucocorticoid receptor (GR) agonists and thus correlate with adverse effects on metabolism. In 2018, Panagiotou et al. provided strong evidence that steviol and steviol glycosides exert GR-mediated effects in cancer Jurkat cells [98].
In 2018, Pham and coworkers examined the chemical composition of Gynostemma longipes, an ethnomedicinal plant used to treat type 2 diabetes mellitus by local communities in Vietnam [99]. Ten new dammarane triterpenes, including two hexanordammarane glycosides and five other dammarane glycosides, were isolated from a ethanolic extract of the whole G. longipes plant. The structures of the new compounds were elucidated using diverse spectroscopic methods. All of the isolates were evaluated for their stimulatory activities on glucose uptake. Compound 414 (Fig. 17) showed particularly potent stimulatory effects [99].
Deepa et al. presented in the review paper that extracts from various species of Ficus and isolated compounds significantly have enhanced insulin secretion and subsequently reduced blood glucose level [100]. O-Glycosides (for example compound 415, Fig. 17) isolated from Ficus species exhibited remarkable antidiabetic properties.
In 2018, Kekuda et al. prepared a comprehensive review about traditional uses, chemistry and pharmacological activities of Leea indica (Burm. f.) Merr.(Vitaceae) [101]. The plant L. indica is traditionally used, inter alia, as a medicine for diabetes. Hypoglycemic activity of alcoholic and hydroalcoholic extracts of L. indica leaves using glucose tolerance test and alloxan-induced diabetes model in rats, was evaluated. The extract administration significantly reduced blood glucose levels, indicating hypoglycemic activity of leaf extracts [101]. Figure 17 shows the structure of one of the natural O-glycosides (mollic acid arabinoside 416) that have been isolated from the plant L. indica.
In another paper from 2018, Hua and coworkers have proven that green tea may favorably modulate blood glucose homeostasis, and regular consumption of green tea can prevent the development of type 2 diabetes mellitus [102]. Authors described the inhibition of α-glucosidase and α-amylase by flavonoid glycosides from Lu’an GuaPian tea. The kaempferol monoglycoside showed inhibitory activity against α-glucosidase with IC50 at 40.02 ± 4.61 μM, and kaempferol diglycoside (Fig. 17, O-glycoside 417) showed α-amylase inhibition with IC50 at 0.09 ± 0.02 μM [95]. Application of Lu’an GuaPian green tea as a functional food ingredient can regulate postprandial hyperglycemia through inhibition of α-glucosidase/α-amylase by the mono and diglycosides of kaempferol [102].
Nile et al. described valorization of onion solid waste and their flavonols for assessment of cytotoxicity, enzyme inhibitory, and antioxidant activities [103]. Onion (Allium cepa L.) is rich in flavonols like quercetin and quercetin glycosides. These glycosides have been extracted and tested against enzymes of clinical importance in diabetes. The samples exhibited significant antidiabetic effects. Results indicated that OSW (onion solid waste) and flavonol glycosides are potential antidiabetic agents [103]. Figure 17 shows the structure of one of the natural O-glycosides (quercetin-3,4′-O-diglucoside QDG 418) that have been isolated from onion solid waste.
Jayachandran and coworkers have designed studies to accumulate the experimental evidence in support of antidiabetic effects of isoquercetin 419 (Fig. 17) [104]. Supplementation with isoquercetin significantly normalized blood sugar levels, insulin, and regulated the mRNA expression of insulin signaling genes and carbohydrate-metabolizing enzyme genes. The results achieved with isoquercetin are similar to that of the standard drug glibenclamide [104]. The findings suggest isoquercetin could be a possible therapeutic agent for treating diabetes mellitus in the near future.
6 Biological Action of the Glycosides Described in this Review and Patents of Marketed Drugs
At present, the normalization of glycemia in diabetes is a rather complicated and problematic issue of diabetology. Despite the growing knowledge of various chemical and biochemical aspects of diabetes mellitus, the specific molecular mechanisms leading to T2DM are still unknown. Current pharmacological treatments are symptomatic and aim at maintaining blood glucose levels close to the fasting normoglycemic range of 3.5–6 mM/l. A large number of oral antidiabetic drugs, which exert their effects through various mechanisms, aimed at eliminating three major metabolic disorders leading to hyperglycemia: dysfunction of β-cells, peripheral insulin resistance, excessive hepatic glucose production [7]. Promising direction for design of new drugs for the treatment of diabetes mellitus is the regulation of key carbohydrate metabolism enzymes: inhibition of glycogen phosphorylase (GP), sodium-dependent glucose transporters (SGLT), and protein tyrosine phosphatase (PTP).
Depending on the binding site of the enzyme molecule, GP inhibitors are divided into compounds that block the catalytic site and compounds that block the allosteric sites [105, 106]. The following groups of chemical compounds were studied as inhibitors of the catalytic site of GP: N-acyl-β-(d-glucopyranosylamine derivatives, N-β-d-glucopyranosyl ureas, 2-(d-glucopyranosyl)-5-methyl-1,3,4-oxadiazole derivatives, 2-(d-glucopyranosyl)-benzimidazoles, 3-substituted 5-β-d-glucopyranosyl-1,2,4-oxadiazoles, glucopyranosyliden-spiro-thiohydantoins. Efforts in identifying the best heterocyclic junction between the glucose and pharmacophore units were patented, taking into account 1,2,4-triazole and imidazole moieties [107].
Inhibitors of sodium-dependent glucose co-transporter 2 (SGLT2) are an attractive method of type 2 diabetes treatment because of their distinct mechanism of action, in which blood glucose levels are reduced independently of insulin secretion [108]. In healthy individuals, greater than 99% of the plasma glucose that is filtered in the kidney is reabsorbed, resulting in less than 1% of the total filtered glucose being excreted in urine [41]. This reabsorption process is mediated by two sodium-dependent glucose cotransporters (SGLTs): SGLT1, a low-capacity, high-affinity transporter and SGLT2, a high-capacity, low-affinity transporter that is expressed mainly in the kidney [41]. It is estimated that 90% of renal glucose reabsorption is facilitated by SGLT2 residing on the surface of the epithelial cells lining the S1 segment of the proximal tubule; the remaining 10% is likely mediated by SGLT1 localized on the more distal S3 segment of the proximal tubule [41]. Several selective SGLT2 inhibitors have been developed by structural modification of phlorizin, the first known SGLT inhibitor. C-Linked β-glycosides (gliflozins) are at advanced stages of development because of their metabolic stability, high oral bioavailability, and plasma exposure. Of these, dapagliflozin, canagliflozin, ipragliflozin, empagliflozin, tofogliflozin, luseogliflozin, ertugliflozin, and sotagliflozin have been approved for the treatment of type 2 diabetes mellitus recently [108]. These compounds are now approved as marketed drugs (Fig. 18). For example, inhibition of SGLT2 with Forxiga (dapagliflozin) reduces renal glucose reabsorption and thereby increases urinary glucose exretion [109]. From 2014, Forxiga is indicated by the FDA as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes and is not recommended for patients with type 1 diabetes mellitus.

SGLT2 inhibitors approved as marketed drugs. Name, company, brand name, and year for approval are provided
Of particular importance to the downregulation of insulin signaling is protein tyrosine phosphatase 1B (PTP1B), which dephosphorylates the receptor (IR) on the surface of a cell [110]. Inhibition of PTP1B may represent a practical strategy for the treatment of type 2 diabetes [111]. Various strategies have been developed to design and synthesize potent and selective PTP1B inhibitors. The principal approach is based on mimicking the phosphotyrosine moiety. Many PTP inhibitors contain a quinone functionality. Series of C-glucosyl 1,4-benzo- and 1,4-naphtho-quinones were found as better PTP1B inhibitors than their analogues displaying 1,4-dimethoxybenzene or -naphthalene residues [40]. Modifications of molecules at the primary 6-position were made by transformation to either carboxylic, azido, or benzamido groups, or elongation by azide-alkyne click chemistry to triazole ring formation [112].
7 Conclusions
In this review, we disclosed studies on the preparation of glycosides that can be applied as inhibitors of glycogen phosphorylase, sodium glucose cotransporter 2, protein tyrosine phosphatase 1B and other more specific enzymes. The natural product phlorizin was identified as the first type 2 diabetes mellitus inhibitor. Because O-glycosides are usually hydrolytically unstable, many carbohydrate analogues such as N-glycosides and C-glycosides have been synthesized and used as enzyme inhibitors. In comparison to O-glycosides, the C-glycosides are structurally more stable against acidic and enzymatic cleavage due to the existence of their C–C glycosidic bond. Among glycomimetics, C-glycosyl compounds have attracted much attention. By use of structure–activity relationships, several new glycosides have been developed as inhibitors and studied in clinical trials. Some of the described glycoside-based molecules, for example dapagliflozin, canagliflozin, ipragliflozin, empagliflozin, tofogliflozin, luseogliflozin, ertugliflozin, and sotagliflozin, have been approved as marketed drugs.
Figure 19 shows the best GPb inhibitors described in this article, which were selected from Figs. 7, 9, 10, 11, 13, and 15 presenting activity of inhibitors having a specific O, N, or C-glycoside structure and from Sect. 5 describing directions of the latest research from 2018 to 2019. Below each compound there is information about what type of glycoside it represents. The compounds were ranked in order from the inhibitor with the highest activity (the lowest Ki) to the inhibitor with the lowest activity (the highest Ki). Where the Ki values were not reported in the publication, the inhibitory properties were compared based on the IC50 values. Seven of the eight inhibitors shown in Fig. 19 are derivatives of d-glucose. Changes in the sugar configuration as well as removal or replacement of substituents of the glucose moiety proved detrimental for the inhibition. The crucial role of the aglycon in the efficiency of the inhibitors is also presented in this article. Based on the analysis of the structure of compounds that are the best GPb inhibitors, it can be concluded that a high activity is ensured by the presence of the 2-naphthol group. Four of the eight compounds shown in Fig. 19 have a 2-naphthol group. Analysis of the structures of the best GPb inhibitors (Fig. 19) leads to the conclusion that the highest activity is possessed by inhibitors containing, as aglycones, heterocyclic rings with nitrogen atoms.

The best GPb inhibitors selected from the figures summarizing the activity of inhibitors discussed in a given Sects
In turn, Fig. 20 presents the best SGLT2 inhibitors. They were selected from Figs. 7, 9, 10, 11, 13, and 15 as representatives of glycosides with the structure described in the given Sects and from Sect. 5 describing directions of the latest research from 2018 to 2019. The compounds were arranged in order from the best inhibitor (the lowest IC50) to the inhibitor with lower activity (the highest IC50). The activity of inhibitors for which no IC50 values were reported in the publication was evaluated by comparing the EC50 values. Analysis of the structure of the best SGLT2 inhibitors allows the conclusion that not only glucose but also its derivatives or xylose provide inhibitory activity. Looking at the structure of active SGLT2 inhibitors from Fig. 20, it can also be seen that each of them has one or two phenyl groups in its structure. Considering the structure of the aglycone, one cannot distinguish a specific heteroaromatic system whose structure would be repeated in the structure of the inhibitors considered. However, it can be seen that in two cases there is a thiophene ring and in the other two an indole system. Eight of the ten SGLT2 inhibitors shown in Fig. 20 have a chlorine or fluorine atom in their structure.

The best SGLT2 inhibitors selected from the figures summarizing the activity of inhibitors discussed in a given Sects
The conclusions drawn from the analysis of the structure of the best inhibitors should be taken into account when designing antidiabetic drugs. We sincerely hope that this article will stimulate further research in glycoside derivatives synthesis and will encourage scientists to design novel inhibitors in the treatment of type 2 diabetes mellitus.
Abbreviations
- Ac:
-
Acetyl
- Ar:
-
Aryl
- Asn:
-
Asparagine
- Asp:
-
Aspartic acid
- Bn:
-
Benzyl
- Boc:
-
tert-Butyloxycarbonyl
- iBu:
-
Isobutyl
- n-Bu:
-
n-Butyl
- t-Bu:
-
tert-Butyl
- Bz:
-
Benzoyl
- CAN:
-
Cerium ammonium nitrate
- CuAAC:
-
Copper-catalyzed azide–alkyne cycloaddition
- DBU:
-
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DCC:
-
N,N’-Dicyclohexylcarbodiimide
- DCM:
-
Dichloromethane
- DIAD:
-
Diisopropyl azodicarboxylate
- DMAP:
-
N,N-Dimethyl-4-aminopyridine
- DME:
-
Dimethoxyethane
- DMF:
-
Dimethylformamide
- DMSO:
-
Dimethyl sulfoxide
- EDCI:
-
N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide
- Et:
-
Ethyl
- GP:
-
Glycogen phosphorylase
- GS:
-
Glycogen synthase
- Hetaryl:
-
Heteroaromatic ring
- HMPA:
-
Hexamethylphosphoramide
- HOBt:
-
Hydroxybenzotriazole
- HPLC:
-
High-performance liquid chromatography
- IC50 :
-
Half maximal inhibitory concentration
- Ki :
-
Inhibition constant
- LDA:
-
Lithium diisopropylamide
- Me:
-
Methyl
- MOM:
-
Methoxymethyl
- MOMCl:
-
Methoxymethyl chloride
- Ms:
-
Mesyl
- NBS:
-
N-bromosuccinimide
- NCS:
-
N-chlorosuccinimide
- NMM:
-
N-Methylmorpholine
- NMO:
-
N-Methylmorpholine N-oxide
- Ph:
-
Phenyl
- iPr:
-
Isopropyl
- PTP1B:
-
Protein tyrosine phosphatase 1B
- QSAR:
-
Quantitative structure–activity relationship
- RMGP:
-
Rat muscle glycogen phosphorylase
- SAR:
-
Structure–activity relationship
- SGLT:
-
Sodium-dependent glucose cotransporter
- TBA:
-
tert-Butyl alcohol
- TBAF:
-
Tetrabutylammonium fluoride
- TBDMS:
-
tert-Butyldimethylsilyl
- TBDMSCl:
-
tert-Butyldimethylsilyl chloride
- TBS:
-
tert-Butyldimethylsilyl
- TBSCl:
-
tert-Butyldimethylsilyl chloride
- T2DM:
-
Type 2 diabetes mellitus
- TEA:
-
Triethylamine
- TFA:
-
Trifluoroacetic acid
- THF:
-
Tetrahydrofuran
- TIPS:
-
Triisopropylsilyl
- TMS:
-
Trimethylsilyl
- TMSI:
-
Trimethylsilyl iodide
- TMSOTf:
-
Trimethylsilyl trifluoromethanesulfonate
- Ts:
-
Tosyl
- Tyr:
-
Tyrosine
References
Wild S, Roglic G, Green A, Sicree R, King H (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053
Lipscombe LL, Hux JE (2007) Trends in diabetes prevalence, incidence, and mortality in Ontario, Canada 1995–2005: a population-based study. Lancet 369:750–756
Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN (2009) Hyperglycemic crises in adult patients with diabetes. Diabetes Care 32:1335–1343
Ross SA, Gulve EA, Wang M (2004) Chemistry and biochemistry of type 2 diabetes. Chem Rev 104:1255–1282
Drzewoski J, Kasznicki J, Trojanowski Z (2009) The role of metabolic memory in the natural history of diabetes mellitus. Polskie Archiwum Medycyny Wewnętrznej 119:493–499
Barford D, Johnson LN (1989) The allosteric transition of glycogen phosphorylase. Nature 340:609–616
Spasov AA, Chepljaeva NI, Vorobev ES (2016) Glycogen phosphorylase inhibitors in the regulation of carbohydrate metabolism in type 2 diabetes. Russ J Bioorganic Chem 42:133–142
Sprang SR, Acharya KR, Goldsmith EJ, Stuart DI, Varvill K, Fletterich RJ, Madsen NB, Johnson LN (1988) Structural changes in glycogen phosphorylase induced by phosphorylation. Nature 336:215–221
Sprang SR, Withers SG, Goldsmith EJ, Fletterich RJ, Madsen NB (1991) Structural basis for the activation of glycogen phosphorylase b by adenosine monophosphate. Science 254:1367–1371
Krimm I, Lancelin JM, Praly JP (2012) Binding evaluation of fragment-based scaffolds for probing allosteric enzymes. J Med Chem 55:1287–1295
Ellsworth B, Washburn WN, Sher PM, Wu G, Meng W (2001) C-Aryl glucoside SGLT2 inhibitors. PCT Int Appl. WO2001- 027128 Chem Abstr 134:281069
Toyama Y, Kobayashi Y, Noda A, Toyama I, Toyama T (2001) C-Glycosides and preparation of thereof as antidiabetic agents. US Patent 6627611 Chem Abstr 135: 304104
Somsak L, Bokor E, Czifrak K, Juhasz L, Toth M (2011) Carbohydrate derivatives and glycomimetic compounds in established and investigational therapies of type 2 diabetes. In: Zimering MB (ed) Topics in the prevention, treatment and complications of type 2 diabetes. InTech Open Access Publisher, Rijeka, pp 103–126
So SS, Karplus M (2001) Evaluation of designed ligands by a multiple screening method: application to glycogen phosphorylase inhibitors constructed with a variety of approaches. J Comput Aided Mol Des 15:613–647
So SS, Karplus M (1999) A comparative study of ligand-receptor complex binding affinity prediction methods based on glycogen phosphorylase inhibitors. J Comp Aided Mol Des 13:243–258
Bokor E, Kun S, Toth DM, Praly JP, Vidal S, Somsak L (2017) C-Glycopyranosyl arenes and hetarenes: synthetic methods and bioactivity focused on antidiabetic potential. Chem Rev 117:1687–1754
Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA (1987) Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest 79:1510–1515
Huang CS, Yin MC, Chiu LC (2011) Antihyperglycemic and antioxidative potential of Psidium guajava fruit in streptozotocin-induced diabetic rats. Food Chem Toxicol 49:2189–2195
Oha WK, Leea CH, Leea MS, Baea EY, Sohnb CB, Oha H et al (2005) Antidiabetic effects of extracts from Psidium guajava. J Ethnopharmacol 96:411–415
Eidenberger T, Selg M, Krennhuber K (2013) Inhibition of dipeptidyl peptidase activity by flavonol glycosides of guava (Psidium guajava L.): a key to the beneficial effects of guava in type II diabetes mellitus. Fitoterapia 89:74–79
Diaz-Lobo M, Garcia-Amoros J, Fita I, Velasco D, Guinovart JJ, Ferrer JC (2015) Selective photoregulation of the activity of glycogen synthase and glycogen phosphorylase, two key enzymes in glycogen metabolism. Org Biomol Chem 13:7282–7288
Tang W, Li S, Liu Y, Wu JCh, Pan MH, Huang MT, Ho ChT (2017) Anti-diabetic activities of cis- and trans-2,3,5,4′-tetrahydroxystilbene 2-O-β-glucopyranoside from Polygonum multiflorum. Mol Nutr Food Res 61:1600871
Watson KA, Mitchell EP, Johnson LN, Cruciani G, Son JC, Bichard CJF, Fleet GWJ, Oikonomakos NG, Kontou M, Zographos SE (1995) Glucose analogue inhibitors of glycogen phosphorylase: from crystallographic analysis to drug prediction using GRID force-field and GOLPE variable selection. Acta Crystallogr D51:458–472
Oikonomakos NG, Kosmopolou M, Zographos SE, Leonidas DD, Somsák L, Nagy V, Praly JP, Docsa T, Toth B, Gergely P (2002) Binding of N-acetyl-N’-β-d-glucopyranosyl urea and N-benzoyl-N-β-d-glucopyranosyl urea to glycogen phosphorylase b. Kinetic and crystallographic studies. Eur J Biochem 269:1684–1696
Gyorgydeak Z, Hadady Z, Felföldi N, Krakomperger A, Nagy V, Toth M, Brunyanszki A, Docsa T, Gergely P, Somsák L (2004) Synthesis of N-(β-d-glucopyranosyl)- and N-(2-acetamido-2-deoxy-β-d-glucopyranosyl) amides as inhibitors of glycogen phosphorylase. Bioorg Med Chem 12:4861–4870
Czifrák K, Hadady Z, Docsa T, Gergely P, Schmidt J, Wessjohannd L, Somsák L (2006) Synthesis of N-(β-d-glucopyranosyl) monoamides of dicarboxylic acids as potential inhibitors of glycogen phosphorylase. Carbohydr Res 341:947–956
Somsák L, Felföldi N, Kónya B, Huse C, Telepo K, Bokor E, Czifrak K (2008) Assessment of synthetic methods for the preparation of N-β-d-glucopyranosyl-N ‘- substituted ureas,—thioureas and related compounds. Carbohydr Res 343:2083–2093
Nagy V, Felföldi N, Kónya B, Praly JP, Docsa T, Gergely P, Chrysina ED, Tiraidis C, Kosmopoulou MN, Alexacou KM, Konstantakaki M, Leonidas DD, Zographos SE, Oikonomakos NG, Kozmon S, Tvaroška I, Somsák L (2012) N-(4-Substituted-benzoyl)-N’-(β-d-glucopyranosyl)ureas as inhibitors of glycogen phosphorylase: synthesis and evaluation by kinetic, crystallographic, and molecular modelling methods. Bioorg Med Chem 20:1801–1816
Kónya B, Docsa T, Gergely P, Somsák L (2012) Synthesis of heterocyclic N-(β-d-glucopyranosyl) carboxamides for inhibition of glycogen phosphorylase. Carbohydr Res 351:56–63
Parmenopoulou V, Kantsadi AL, Tsirkone VG, Chatzileontiadou DSM, Manta S, Zographos SE, Molfeta Ch, Archontis G, Agius L, Hayes JM, Leonidas DD, Komiotis D (2014) Structure based inhibitor design targeting glycogen phosphorylase b. Virtual screening, synthesis, biochemical and biological assessment of novel N-acyl-β-D-glucopyranosylamines. Bioorg Med Chem 22:4810–4825
Anand N, Jaiswal N, Pandey SK, Srivastava AK, Tripathi RP (2011) Application of click chemistry towards an efficient synthesis of 1,2,3-1 H-triazolyl glycohybrids as enzyme inhibitors. Carbohydr Res 346:16–25
Goyard D, Chajistamatiou AS, Sotiropoulou AI, Chrysina ED, Praly JP, Vidal S (2014) Efficient atropodiastereoselective access to 5,5′-bis-1,2,3-triazoles: studies on 1-glucosylated 5-halogeno 1,2,3-triazoles and their 5-substituted derivatives as glycogen phosphorylase inhibitors. Chem Eur J 20:5423–5432
Goyard D, Docsa T, Gergely P, Praly JP, Vidal S (2015) Synthesis of 4-amidomethyl-1-glucosyl-1,2,3-triazoles and evaluation as glycogen phosphorylase inhibitors. Carbohydr Res 402:245–251
Bai S-T, Xiong D-C, Niu Y, Wu Y-F, Ye X-S (2015) Synthesis of novel N-glycoside derivatives via CuSCN-catalyzed reactions and their SGLT2 inhibition activities. Tetrahedron 71:4909–4919
Chu K-F, Yao Ch-H, Song J-S, Chen Ch-T, Yeh T-K, Hsieh T-Ch, Huang Ch-Y, Wang M-H, Wu S-H, Chang W-E, Chao Y-S, Lee J-C (2016) N-Indolylglycosides bearing modifications at the glucose C6-position as sodium-dependent glucose co-transporter 2 inhibitors. Bioorganic Med Chem 24:2242–2250
Yao CH, Song JS, Chen CT, Yeh TK, Hung MS, Chang CC, Liu YW, Yuan MC, Hsieh CJ, Huang CY, Wang MH, Chiu CH, Hsieh TC, Wu SH, Hsiao WC, Chu KF, Tsai CH, Chao YS, Lee JC (2011) Discovery of novel N-β-d-xylosylindole derivatives as sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors for the management of hyperglycemia in diabetes. J Med Chem 54:166–178
Mamais M, Degli Esposti A, Kouloumoundra V, Gustavsson T, Monti F, Venturini A, Chrysina ED, Markovitsi D, Gimisis T (2017) A new potent inhibitor of glycogen phosphorylase reveals the basicity of the catalytic site. Chem Eur J 23:8800–8805
He L, Zhang YZ, Tanoh M, Chen GR, Praly JP, Chrysina ED, Tiraidis C, Kosmopoulou M, Leonidas DD, Oikonomakos NG (2007) In the search of glycogen phosphorylase inhibitors: synthesis of C-glycopyranosylbenzo(hydro)quinones—inhibition of and binding to glycogen phosphorylase in the crystal. Eur J Org Chem 2007(4):596–606
Moller DE (2001) New drug targets for type 2 diabetes and the metabolic syndrome. Nature 414:821
Lin L, Shen Q, Chen G-R, Xie J (2008) β-C-Glycosiduronic acids and β-C-glycosyl compounds: new PTP1B inhibitors. Bioorganic Med Chem Lett 18:6348–6351
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem 51:1145–1149
Kato E, Kawabatta J (2010) Glucose uptake enhancing activity of puerarin and the role of C-glucoside suggested from activity of related compounds. Bioorganic Med Chem Lett 20:4333–4336
Nomura S, Sakamaki S, Hongu M, Kawanishi E, Koga Y, Sakamoto T, Yamamoto Y, Ueta K, Kimata H, Nakayama K, Tsuda-Tsukimoto M (2010) Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J Med Chem 53:6355–6360
Elkinson S, Scott LJ (2013) Canagliflozin: first global approval. Drugs 73:979–988
Dietrich E, Powell J, Taylor JR (2013) Canagliflozin: a novel treatment option for type 2 diabetes. Drug Des Dev Ther 7:1399–1408
Xu B, Feng Y, Cheng H, Song Y, Lv B, Wu Y, Wang C, Li S, Xu M, Du J, Peng K, Dong J, Zhang W, Zhang T, Zhu L, Ding H, Sheng Z, Welihinda A, Roberge JY, Seed B, Chen Y (2011) C-Aryl glucosides substituted at the 4′-position as potent and selective renal sodium-dependent glucose co-transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes. Bioorganic Med Chem Lett 21:4465–4470
Zhang W, Welihinda A, Mechanic J, Ding H, Zhu L, Lu Y, Deng Z, Sheng Z, Lv B, Chen Y, Robergeb JY, Seed B, Wanga Y-X (2011) Egt1442, a potent and selective (SGLT2) inhibitor, attenuates blood glucose and HbA1c levels in db/db mice and prolongs the survival of stroke-prone rats. Pharmacol Res 63:284–293
Imamura M, Nakanishi K, Suzuki T, Ikegai K, Shiraki R, Ogiyama T, Murakami T, Kurosaki E, Noda A, Kobayashi Y, Yokota M, Koide T, Kosakai K, Ohkura Y, Takeuchi M, Tomiyama H, Ohta M (2012) Discovery of ipragliflozin (Asp1941): a novel C-glucoside with benzothiophene structure as a potent and selective sodium glucose co-transporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes mellitus. Bioorganic Med Chem 20:3263–3279
Somsák L, Kovács L, Toth M, Osz E, Szilágyi L, Györgydeák Z, Dinya Z, Docsa T, Toth B, Gergely P (2001) Synthesis of and a comparative study on the inhibition of muscle and liver glycogen phosphorylases by epimeric pairs of d-gluco- and -d-xylopyranosylidene-spiro-(thio)hydantoins and N-(d-glucopyranosyl) amides. J Med Chem 44:2843–2848
Hadady Z, Tóth M, Somsák L (2004) C-(β-d-Glucopyranosyl)heterocycles as potential glycogen phosphorylase inhibitors. Arkivoc 7:140–149
Chrysina ED, Kosmopoulou MN, Tiraidis C, Kardakaris R, Bischler N, Leonidas DD, Hadady Z, Somsak L, Docsa T, Gergely P, Oikonomakos NG (2005) Kinetic and crystallographic studies on 2-(β-d-glucopyranosyl)-5-methyl-1, 3, 4-oxadiazole, -benzothiazole, and -benzimidazole, inhibitors of muscle glycogen phosphorylase b. Evidence for a new binding site. Protein Sci 14:873–888
Kang SY, Song K-S, Lee J, Lee S-H, Lee J (2010) Synthesis of pyridazine and thiazole analogs as (SGLT2) inhibitors. Bioorganic Med Chem 18:6069–6079
Zhou H, Danger DP, Dock ST, Hawley L, Roller SG, Smith CD, Handlon AL (2010) Synthesis and SAR of benzisothiazole- and indolizine-β-d-glucopyranoside inhibitors of SGLT2. ACS Med Chem Lett 1:19–23
Condreay JP, Witherspoon SM, Clay WC, Kost TA (1999) Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc Natl Acad Sci USA 96:127–132
Yao Ch-H, Song J-S, Chen Ch-T, Yeh T-K, Hsieh T-Ch, Sz-H Wu, Huang Ch-Y, Huang Y-L, Wang M-H, Liu Y-W, Tsai Ch-H, Kumar ChR, Lee J-Ch (2012) Synthesis and biological evaluation of novel C-indolylxylosides as sodium-dependent glucose co-transporter 2 inhibitors. Eur J Med Chem 55:32–38
Li L-T, Zhou L-F, Li Y-J, Huang J, Liu R-H, Wang B, Wang P (2012) Facile synthesis of 1,2,3-triazole analogs of (SGLT2) inhibitors by ‘click chemistry’. Bioorganic Med Chem Lett 22:642–644
Bokor E, Docsa T, Gergely P, Somsak L (2013) C-Glucopyranosyl-1,2,4-triazoles as new potent inhibitors of glycogen phosphorylase. ACS Med Chem Lett 4:612–615
Sakamaki S, Kawanishi E, Koga Y, Yamamoto Y, Kuriyama C, Matsushita Y, Ueta K, Nomura S (2013) Synthesis and biological evaluation of thiophene-C-glucosides as sodium-dependent glucose cotransporter 2 inhibitors. Chem Pharm Bull 61:1037–1043
Somsak L, Bokor E, Czibere B, Czifrak K, Koppany C, Kulcsar L, Kun S, Szilagyi E, Toth M, Docsa T, Gergely P (2014) Synthesis of C-xylopyranosyl- and xylopyranosylidene-spiroheterocycles as potential inhibitors of glycogen phosphorylase. Carbohydr Res 399:38–48
Sprang SR, Goldsmith EJ, Fletterick RJ, Withers SG, Madsen NB (1982) Catalytic site of glycogen phosphorylase: structure of the T state and specificity for α-d-Glucose. Biochem 21:5364–5371
Street IP, Armstrong CR, Withers SG (1986) Hydrogen bonding and specificity. Fluorodeoxy sugars as probes of hydrogen bonding in the glycogen phosphorylase-glucose complex. Biochemistry 25:5027–6021
Bokor E, Szennyes E, Csupasz T, Toth N, Docsa T, Gergely P, Somsak L (2015) C-(2-Deoxy-d-arabino-hex-1-enopyranosyl)-oxadiazoles: synthesis of possible isomers and their evaluation as glycogen phosphorylase inhibitors. Carbohydr Res 412:71–79
Bokor E, Szeles Z, Docsa T, Gergely P, Somsák L (2016) C-Glucopyranosyl-1,2,4-triazol-5-ones: synthesis and inhibition of glycogen phosphorylase. Carbohydr Res 429:128–134
Goyard D, Konya B, Chajistamatiou AS, Chrysina ED, Leroy J, Balzarin S, Tournier M, Tousch D, Petit P, Duret C, Maurel P, Somsák L, Docsa T, Gergly P, Praly JP, Azay-Milhau J, Vidal S (2016) Glucose-derived spiro-isoxazolines are anti-hyperglycemic agents against type 2 diabetes through glycogen phosphorylase inhibition. Eur J Med Chem 108:444–454
Kantsadi AL, Bokor E, Kun S, Stravodimos GA, Chatzileontiadou DSM, Leonidas DD, Juhász-Tóth E, Szakács A, Batta G, Docsa T, Gergely P, Somsák L (2016) Synthetic, enzyme kinetic, and protein crystallographic studies of C-β-d-glucopyranosyl pyrroles and imidazoles reveal and explain low nanomolar inhibition of human liver glycogen phosphorylase. Eur J Med Chem 123:737–745
Bisht SS, Fatima S, Tamrakar AK, Rahuja N, Jaiswal N, Srivastava AK, Tripathi RP (2009) Synthetic studies in butenonyl C-glycosides: preparation of polyfunctional alkanonyl glycosides and their enzyme inhibitory activity. Bioorganic Med Chem Lett 19:2699–2703
Rodrigues F, Canac Y, Lubineau A (2000) A convenient, one-step, synthesis of β-C-glycosidic ketones in aqueous media. Chem Comm:2049–2050
Bisht SS, Pandey J, Sharma A, Tripathi RP (2008) Aldol reaction of beta-C-glycosylic ketones: synthesis of C-(E)-cinnamoyl glycosylic compounds as precursors for new biologically active C-glycosides. Carbohydr Res 343:1399–1406
Cecioni S, Argintaru OA, Docsa T, Gergely P, Praly JP, Vidal S (2009) Probing ultivalency for the inhibition of an enzyme: glycogen phosphorylase as a case study. N J Chem 33:148–156
Kakinuma H, Oi T, Hashimoto-Tsuchiya Y, Arai M, Kawakita Y, Fukasawa Y, Iida I, Hagima N, Takeuchi H, Chino Y, Asami J, Okumura-Kitajima L, Io F, Yamamoto D, Miyata N, Takahashi T, Uchida S, Yamamoto K (2010) (1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol (TS-071) is a potent, selective sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for type 2 diabetes treatment. J Med Chem 53:3247–3261
Yuasa H, Hindsgaul O, Palcic MM (1992) Chemical-enzymatic synthesis of 5′-thio-N-acetyllactosamine: the first disaccharide with sulfur in the ring of the nonreducing sugar. J Am Chem Soc 114:5891–5892
Hashimoto H, Kawanishi M, Yuasa H (1996) Synthesis of methyl 5′-thio-α-isomaltoside via an acyclic monothioacetal and its behavior toward glucoamylase. Chem Eur J 2:556–560
Sato M, Kakinuma H, Asanuma H (2004) Preparation of aryl 5-thio-β-d-glucopyranoside derivatives as remedies for diabetes. PCT Int Appl. WO2004014931, 2004, Chem Abstr 140:199631
Kumeda S, Asami J, Tomoike H, Fukuhara N, Hachiuma K, Sato M, Kakinuma H, Yamamoto K, Miyata N, Takahashi K, Nakaike S (2005) SGL0010 is a novel orally active inhibitor of sodium-dependent glucose cotransporter and enhances urinary glucose excretion. Presented at the 65th Scientific Sessions of American Diabetes Association San Diego CA June 10–14 Poster 475–P
Markham A, Elkinson S (2014) Luseogliflozin: first global approval. Drugs 74:945–950
Sim L, Jayakanthan K, Mohan S, Nasi R, Johnston BD, Pinto BM, Rose DR (2010) New glucosidase inhibitors from an Ayurvedic herbal treatment for type 2 diabetes: structures and inhibition of human intestinal maltase-glucoamylase with compounds from Salacia reticulata. Biochemistry 49:443–451
Wang J, He X, Gao L, Sheng L, Shi X, Li J, Chen G (2011) Synthesis of triazole-linked amino acid-aryl C-glycoside hybrids via click chemistry as novel PTP1B inhibitors. Chin J Chem 29:1227–1232
Goddard-Borger ED, Stick RV (2007) An efficient, inexpensive, and shelf-stable diazotransfer reagent: imidazole-1-sulfonyl azide hydrochloride. Org Lett 9:3797–3800
Kim MJ, Lee SH, Park SO, Kang H, Lee JS, Lee KN, Jung ME, Kim J, Lee J (2011) Novel macrocyclic C-aryl glucoside SGLT2 inhibitors as potential antidiabetic agents. Bioorganic Med Chem 19:5468–5479
Ohtake Y, Sato T, Kobayashi T, Nishimoto M, Taka N, Takano K, Yamamoto K (2012) Discovery of tofogliflozin, a novel C-arylglucoside with an O-spiroketal ring system, as a highly selective sodium glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem 55:7828–7840
Yan Q, Ding N, Li Y (2016) Synthesis and biological evaluation of novel dioxa-bicycle C-aryl glucosides as (SGLT2) inhibitors. Carbohydr Res 421:1–8
Mascitti V, Maurer TS, Robinson RP, Bian J, Boustany-Kari CM, Brandt T, Collman BM, Kalgutkar AS, Klenotic MK, Leininger MT et al (2011) Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 inhibitors. J Med Chem 54:2952–2960
Pałasz A, Kalinowska-Tłuścik J, Jabłoński M (2013) Application of 2,4,6-trioxo-pyrimidin-5-ylidene alditols in the synthesis of pyrano(2,3-d) pyrimidines containing a sugar moiety by hetero-Diels-Alder reactions and by conjugate Michael addition-cyclizations. Tetrahedron 69:8216–8227
Pałasz A, Cież D, Musielak B, Kalinowska-Tłuścik J (2015) Application of dimedone enamines as dienophiles: stereoselectives synthesis of amino enols of fused uracils containing a sugar moiety by hetero-Dielse-Alder reactions of barbituric acid 5-ylidene alditols with dimedone enamines. Tetrahedron 71:8911–8924
Kerru N, Singh-Pillay A, Awolade P, Singh P (2018) Current anti-diabetic agents and their molecular targets: a review. Eur J Med Chem 152:436–488
Kun S, Begum J, Kyriakis E, Stamati ECV, Barkas TA, Szennyes E, Bokor E, Szabo KE, Stravodimos GA, Sipos A, Docsa T, Gergely P, Moffatt C, Patraskaki MS, Kokolaki MC, Gkerdi A, Skamnaki VT, Leonidas DD, Somsak L, Hayes JM (2018) A multidisciplinary study of 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazole derivatives as glycogen phosphorylase inhibitors: computation, synthesis, crystallography and kinetics reveal new potent inhibitors. Eur J Med Chem 147:266–278
Kyriakis E, Solovou TGA, Kun S, Czifrák K, Szocs B, Juhász L, Bokor E, Stravodimos GA, Kantsadi AL, Chatzileontiadou DSM, Skamnaki VT, Somsák L, Leonidas DD (2018) Probing the β-pocket of the active site of human liver glycogen phosphorylase with 3-(C-β-d-glucopyranosyl)-5-(4-substituted-phenyl)-1,2,4-triazole inhibitors. Bioorganic Chem 77:485–493
Szennyes E, Bokor E, Langer P, Gyemant G, Docsa T, Siposd A, Somsak L (2018) The first general synthesis of 2-C-(β-d-glycopyranosyl)pyrimidines and their evaluation as inhibitors of some glycoenzymes. N J Chem 42:17439–17446
Kuo G-H, Gaul MD, Liang Y, Xu JZ, Du F, Hornby P, Xu G, Qi J, Wallace N, Lee S, Grant E, Murray WV, Demarest K (2018) Synthesis and biological evaluation of benzocyclobutane-C-glycosides as potent and orally active SGLT1/SGLT2 dual inhibitors. Bioorganic Med Chem Lett 28:1182–1187
Kuroda S, Kobashi Y, Oi T, Kawabe K, Shiozawa F, Okumura-Kitajima L, Sugisaki-Kitano M, Io F, Yamamoto K, Kakinuma H (2019) Discovery of potent, low-absorbable sodium-dependent glucose cotransporter 1 (SGLT1) inhibitor SGL5213 for type 2 diabetes treatment. Bioorganic Med Chem 27:394–409
Yuan MC, Yeh TK, Chen CT, Song JS, Huang YC, Hsieh TC, Huang C-Y, Huang Y-L, Wang M-H, Wu S-H, Yao C-H, Chao Y-S, Lee J-C (2018) Identification of an oxime-containing C-glucosylarene as a potential inhibitor of sodium-dependent glucose co-transporter 2. Eur J Med Chem 143:611–620
Sadurní A, Kehr G, Ahlqvist M, Wernevik J, Sjögren HP, Kankkonen C et al (2018) Fluorine-directed glycosylation enables the stereocontrolled synthesis of selective SGLT2 inhibitors for Type II diabetes. Chem Eur J 24:2832–2836
Bucher C, Gilmour R (2010) Fluorine-directed glycosylation. Angew Chem Int Ed 49:8724–8728
Kun S, Bokor E, Sipos A, Docsa T, Somsák L (2018) Synthesis of new C- and N-d-glucopyranosyl derivatives of imidazole, 1,2,3-triazole and tetrazole, and their evaluation as inhibitors of glycogen phosphorylase. Molecules 23(666):1–17
Chu K-F, Song J-S, Chen C-T, Yeh T-K, Hsieh T-C, Huang C-Y, Wang M-H, Wu S-H, Yao C-H, Chao Y-S, Lee J-C (2019) Synthesis and biological evaluation of N-glucosyl indole derivatives as sodium-dependent glucose co-transporter 2 inhibitors. Bioorganic Chem 83:520–525
Rosas-Ramírez D, Escandon-Rivera S, Pereda-Miranda R (2018) Morning glory resin glycosides as α-glucosidase inhibitors: in vitro and in silico analysis. Phytochemistry 148:39–47
Kim B-R, Young Kim H, Choi I, Kim J-B, Hyun Jin Ch, Han A-R (2018) DPP-IV Inhibitory potentials of flavonol glycosides isolated from the seeds of lens culinaris: in vitro and molecular docking analyses. Molecules 23(1998):1–10
Panagiotou Ch, Mihailidou Ch, Brauhli G, Katsarou O, Moutsatsou P (2018) Effect of steviol, steviol glycosides and stevia extract on glucocorticoid receptor signaling in normal and cancer blood cells. Mol Cell Endocrinol 460:189–199
Pham HTT, Ha TKQ, Cho HM, Lee BW, An JP, Tran VO, Oh WK (2018) Insulin mimetic activity of 3,4-seco and hexanordammarane triterpenoids isolated from gynostemma longines. J Nat Prod 81:2470–2482
Deepa P, Sowndhararajan K, Kim S, Park SJ (2018) A role of Ficus species in the management of diabetes mellitus: a review. J Ethnopharmacol 215:210–232
Kekuda PTR, Raghavendra HL, BharadwajNA Akhilesha S (2018) Traditional uses, chemistry and pharmacological activities of Leea indica (Burm. f.) Merr. (Vitaceae): a comprehensive review. Int J Green Pharm 12:S73–S80
Hua F, Zhou P, Wu H-Y, Chu G-X, Xiea Z-W, Bao G-H (2018) Inhibition of α-glucosidase and α-amylase by flavonoid glycosides from Lu’an GuaPian tea: molecular docking and interaction mechanism. Food Funct 9:4173–4183
Nile A, Nile SH, Kim DH, Keum YS, Seok PG, Sharma K (2018) Valorization of onion solid waste and their flavonols for assessment of cytotoxicity, enzyme inhibitory and antioxidant activities. Food Chem Toxicol 119:281–289
Jayachandran M, Zhang T, Ganesan K, Xu B, Chung SSM (2018) Isoquercetin ameliorates hyperglycemia and regulates key enzymes of glucose metabolism via insulin signaling pathway in streptozotocin-induced diabetic rats. Eur J Pharmacol 829:112–120
Gaboriaud KN, Skaltsounis AL (2013) Glycogen phosphorylase inhibitors: a patent review (2008–2012). Expert Opin Ther Pat 23:1017–1032
Donnier-Marechal M, Vidal S (2016) Glycogen phosphorylase inhibitors: a patent review (2013–2015). Expert Opin Ther Pat 26:199–212
Somsák L, Bokor É, Vágvölgyiné Tóth M, Juhász L, Czifrák K, Kónya B, Kun S, Páhi A, Szőcs B, Varga G, Gergely P, Docsa T, Kóder L, Nagy K (2013) Preparation of imidazolyl and triazolyl glycosides as glycogen phosphorylase inhibitors and antitumor agents. WO2013061105A2
Abdul-Ghani MA, Norton L, DeFronzo RA (2011) Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev 32:515–531
DeFronzo RA, Hompesch M, Kasichayanula S, Liu X, Hong Y, Pfister M, Morrow LA, Leslie BR, Boulton DW, Ching A, LaCreta FP, Griffen SC (2013) Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 36:3169–3176
Cohen P (2006) The twentieth century struggle to decipher insulin signalling. Nat Rev Mol Cell Biol 7:867–873
Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK (2011) Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell 147:185–198
Lin L, Shen Q, Chen G-R, Xie J (2008) Synthesis of triazole-linked β-C-glycosyl dimers as inhibitors of PTP1B. Bioorg Med Chem 16:9757–9763
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pałasz, A., Cież, D., Trzewik, B. et al. In the Search of Glycoside-Based Molecules as Antidiabetic Agents. Top Curr Chem (Z) 377, 19 (2019). https://doi.org/10.1007/s41061-019-0243-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-019-0243-6