
Abstract
Emerging evidence suggests that NLRP3 inflammasome was associated with various kinds of immunological diseases including colitis. However, there are few drugs targeting inflammasomes in the treatment of colitis. Several flavonoids have been found to affect the inflammasome pathway, but the mechanism is still confusing. Here we report that VI-16, a synthetic flavonoid compound, exerts potent anti-inflammatory effects on macrophages in DSS-induced colitis mice, which intervened in the activation of NLRP3 inflammasome without affecting intestinal epithelial cells. However, the protection of VI-16 against DSS-induced colitis was dependent on NLRP3 expression in hematopoietic cells. Furthermore, this inhibitory effect of VI-16 was found to be at least partially achieved by decreasing the mitochondrial ROS generation without affecting autophagy. Further studies confirm that VI-16 inhibits the binding of Txnip to NLRP3 by reducing oxidative stress and ultimately inhibits NLRP3 inflammasome. This demonstrates the ability of VI-16 to inhibit the NLRP3 inflammasome activation and its potential use in the treatment of inflammatory bowel disease.
Similar content being viewed by others


Introduction
Ulcerative colitis is an idiopathic inflammatory bowel disease characterized by chronic and recurrent inflammation. The exact cause of ulcerative colitis has not been determined yet, but it seems to be associated with genetic, immune, and environmental factors.1 The gastrointestinal tract is typically colonized with microbiota, which needs to interact with the host immune system to achieve a homeostasis of intestinal health.2 Failure to maintain homeostasis between host and microbes may result in spontaneous colitis or increased sensitivity to colitis.3,4,5,6
Innate immune system is an important defense mechanism of the body against microbes in non-specific ways. As an important part of the host innate immune system, inflammasomes are involved in such inflammatory gut diseases.7 Inflammasomes is a protein complex composed of pattern recognition receptors, such as NLRP1, NLRP3, NLRC4, or AIM2, adapter protein ASC and effector pro-caspase-1. Activation of inflammasome causes auto-cleavage of caspase-1, which mediates the maturation of IL-1β and IL -18.8 In order to avoid unnecessary inflammation damage to host cells, inflammasomes are strictly suppressed during the resting stage and only activated under appropriate trigger conditions.9 On the contrary, dysregulation of inflammasome signaling has been implicated in the pathogenesis of several diseases, including cancer, infectious diseases, and autoimmune diseases.10,11,12
In a variety of inflammasomes, NLRP3 inflammasome is activated by a variety of stimuli, such as bacterial toxins, danger-associated molecular patterns (DAMPs), and pathogen-associated molecular patterns (PAMPs), making NLRP3 the most implicated inflammasome in clinical.13 Activation of NLRP3 inflammasome has been believed to be associated with a variety of diseases, such as ulcerative colitis, endotoxic shock, Alzheimer’s disease, obesity, atherosclerosis, and gout, suggesting that agents inhibiting NLRP3 inflammasome activation may have therapeutic potential to treat specific inflammatory disorders.14,15,16,17,18
Flavonoids are secondary metabolites of plants. There are more than 5000 flavonoids in nature, some of which have anti-inflammatory effects.19 Previous findings have shown that flavonoids inhibit NLRP3 inflammasome, but the molecular mechanism is very confusing. To date, it is not clear whether flavonoids inhibit inflammasome assembly or inflammasome components expression or caspase-1 enzyme activity.20,21,22 To elucidate the molecular mechanism of flavonoids inhibiting inflammasome, we screened out a synthetic flavonoid compound VI-16, which inhibited NLRP3 inflammasome and explored its molecular mechanism. Our results suggest that flavonoid VI-16 inhibits Txnip-dependent NLRP3 inflammasome activation by relieving oxidative stress to attenuates DSS-induced experimental colitis.
Results
VI-16 relieved DSS-induced experimental colitis symptoms
To seek for the flavonoid compounds with anti-inflammatory activities in vitro, we screened a series of synthetic flavonoid derivatives by assaying their inhibitory activities of IL-1β in differentiated THP-1 cells activated by LPS and ATP. Initially, three kinds of synthetic flavonoid derivatives, including VI-, LW-, and LL-, were examined at 10 μM in differentiated THP-1 cells (Fig. 1a). Among them, VI-14, VI-16, LW-216, and LL-402 inhibited the secretion of IL-1β. However, 10 μM VI-14, LW-216, and LL-402 showed different inhibition effects on cell survival. To further confirm the inhibitory effect of these flavonoid on the secretion of IL-1β, we adjusted the concentration of these compounds to ensure that they did not affect the survival of THP-1 cells (Fig. 1b). As shown in Fig. 1c, VI-16 showed the highest inhibitory activity on IL-1β secretion, which was comparable to the inhibitory activity of Isoliquiritigenin or MCC950, two NLRP3 inhibitors. Moreover, mature IL-1β (p17) in the supernatants were significantly suppressed by 10 μM VI-16 in LPS-primed differentiated THP-1 cells (Fig. 1d). Therefore, we selected VI-16 for further research in DSS-induced experimental colitis. It is well known that the rapid decline in body weight is an important characteristic in DSS-induced experimental colitis mice. Compared with vehicle-treated group, VI-16 at 40 and 80 mg/kg significantly attenuated the loss of body weight during the disease progression (Fig. 1e). DSS typically causes colonic shortening while such change was also improved by 40 and 80 mg/kg of VI-16 (Fig. 1f, g). These results suggested that VI-16 successfully ameliorated DSS-induced colitis.

VI-16 attenuated DSS-induced experimental colitis. a Chemical structure of VI-14, VI-16, LW-216, and LL-402. b Effect of certain concentration flavonoids, including VI-14, VI-16, LW-216, LL-402, Isoliquiritigenin and MCC950, on the surviving fraction in THP-1 cell were measured by MTT assay. c ELISA of IL-1β in supernatants of differentiated THP-1 cells. Data are presented as mean ± SD. **P < 0.01 compared with mock group; ##P < 0.01 compared with ATP group. d Immunoblot analysis of IL-1β in supernatants (SN) and extracts (Input) of LPS-primed differentiated THP-1 cells. e Body weight changes of each group (n = 8 per group) in DSS-induced colitis mice. f The length of colons and g macroscopic appearances from each group of mice were measured. Data are presented as mean ± SD. **P < 0.01 compared with normal group; #P < 0.05, ##P < 0.01 compared with DSS group
VI-16 attenuated DSS-induced colon injury
Histological analysis showed loss of crypts, infiltration of mononuclear cells, and severe mucosal damage in the colon specimens of colitis mice. Strikingly, VI-16-treated mice exhibited less inflammatory cells infiltration and intact colonic architecture without mucosal damage (Fig. 2a). VI-16 significantly decreased histological colon damage score compared to the colitis mice (Fig. 2b). The myeloperoxidase (MPO) activity in colons from VI-16-treated mice was also lower than that of vehicle-treated group (Fig. 2c). To further investigate the mechanism of protection from colitis by VI-16, we used CD11b+ as an indicator to monitor inflammation process and further determine the beneficial effect of VI-16. We observed a number of CD11b+ inflammatory cells accumulated at the mucosa of the lesion site in colons of DSS-induced colitis mice. In contrast, few CD11b+ inflammatory cells were detected in either VI-16-treated or untreated colon tissues (Fig. 2d). Thus, treatment of VI-16 alleviated the severity of colon injury.

VI-16 reduced colon damage in DSS-induced colitis mice. a H&E stains of serial sections of colons in DSS-induced colitis mice following VI-16 treatment. b Colon histological damage score and clinical evaluation in DSS-induced colitis mice. c MPO activities in the colonic tissues were detected. d Sections of colon tissues were immunostained with DAPI (blue) and anti CD11b-FITC (green) and observed by confocal laser-scanning microscope. Results are presented as images of colon tissues (left) and quantification of CD11b+ cells (right). Representative images are shown. Scale bars, 100 µm. Data are presented as mean ± SD. **P < 0.01 compared with normal group; #P < 0.05, ##P < 0.01 compared with DSS group
VI-16 specifically suppressed IL-1β maturation in colonic macrophages rather than intestinal epithelial cells in a NLRP3-dependent manner
As previously reported, the occurrence of colitis is associated with secretion of IL-1β by colonic macrophages in a NLRP3-dependent manner.16 We examined the regulation of VI-16 on the secretion of IL-1β and IL-18 in DSS-induced experimental colitis mice. Indeed, treatment of VI-16 was able to inhibit IL-1β and IL-18 secretion in the serum after DSS challenge (Fig. 3a). Further, mature IL-1β expression was investigated in colonic macrophages of different group mice. Consistently, mature IL-1β was significantly suppressed by IV-16 in DSS-induced colitis mice colonic macrophages (Fig. 3b). Accordingly, many previous reports have demonstrated that the secretion of IL-18 caused by the activation of NLRP3 inflammasome in epithelial cells is critical for maintaining the intestinal barrier and Nlrp3 deficiency aggravated DSS-induced colitis.23 Therefore, we next examined whether the inhibition of mature IL-1β expression by IV-16 also acted on intestinal epithelial cells. As shown in Fig. 3c, VI-16 had no significant effect on the secretion of IL-1β and IL-18. In addition, VI-16 did not interfere with the expression of mature IL-1β in DSS-induced colitis mice intestinal epithelial cells (Fig. 3d). Next, we asked if VI-16 inhibition of IL-1β maturation could be dependent on Nlrp3 in colon macrophages. To address this hypothesis, we generated four groups of Nlrp3 bone marrow chimeras. In agreement with our previous results, mature IL-1β was suppressed by IV-16 in wild-type mice transplanted with wild-type bone marrow. In addition, IV-16 also inhibited mature IL-1β expression in NLRP3−/− mice receiving wild-type bone marrow (Fig. 3e). While VI-16 had no significant effect on the mature IL-1β expression in intestinal epithelial cells (Fig. 3e). Together these results indicated that VI-16 selectively suppressed IL-1β maturation in colonic macrophages without affecting intestinal epithelial cells in a NLRP3-dependent manner.

VI-16 inhibited the activation of caspase-1 in colonic macrophages rather than in intestinal epithelial cells. a Production of IL-1β and IL-18 in serum of DSS-induced colitis mice were determined by ELISA. b Immunoblot analysis of IL-1β expression in colonic macrophages. c Production of IL-1β and IL-18 in intestinal epithelial cells were determined by ELISA. d Immunoblot analysis of IL-1β expression in intestinal epithelial cells. e Bone marrow chimeras were generated with wild-type (WT) and NLRP3−/− mice. Immunoblot analysis of IL-1β expression in colonic macrophages and intestinal epithelial cells of DSS-treated bone marrow chimeras mice. Data are presented as mean ± SD. **P < 0.01 compared with normal group; #P < 0.05, ##P < 0.01 compared with DSS group
VI-16 relieved DSS-induced experimental colitis dependent on Nlrp3 expression in hematopoietic cells
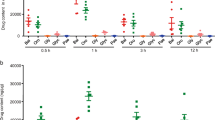
To further confirm that the relief of VI-16 from DSS-induced colitis depends on the intervention of NLRP3 inflammasome, we verified it in NLRP3−/− mice. Monitoring the changes in the weight of mice, VI-16 could not reduce the weight loss caused by DSS in NLRP3−/− mice, while NLRP3−/− mice showed more severe body weight loss than wild-type mice. (Fig. 4a). Similarly, VI-16 did not improve DSS-induced colonic shortening in NLRP3−/− mice (Fig. 4b). In the histological analysis, the loss of crypt and infiltration of inflammatory cells were still present in the colon tissues of VI-16-treated NLRP3−/− mice (Fig. 4c). This suggests that VI-16 lost anti-inflammatory function in NLRP3−/− mice. Using bone marrow chimeras, we now show that IV-16 still ameliorated the body weight loss and colonic shortening in wild-type mice reconstituted with NLRP3−/− bone marrow. Correspondingly, VI-16 could not reduce the body weight loss and colonic shortening caused by DSS in wild-type mice or NLRP3−/− mice receiving NLRP3−/− bone marrow (Fig. 4d, e). The marked improvement in the clinical manifestation of colitis was confirmed by less signs of severe histopathology in H&E-stained sections of the lamina propria in wild-type bone marrow mice after VI-16 treatment. In contrast, NLRP3−/− bone marrow mice presented with extensive crypt destruction and edema regardless of the Nlrp3 status of the mouse (Fig. 4f). Overall, these results suggest that Nlrp3 expression in hematopoietic cells is critical for protection against DSS-induced colitis by VI-16.

Nlrp3 in hematopoetic cells is critical for protection against DSS-induced injury by VI-16. a Body weight changes of each group (n = 6 per group) were recorded in DSS-treated wild-type and NLRP3−/− mice following 80 mg/kg VI-16 treatment. b The length of colons from each group of mice were measured. c H&E stains of serial sections of colons induced by DSS in wild-type and NLRP3−/− mice following 80 mg/kg VI-16 treatment. d–f Bone marrow chimeras were generated with wild-type (WT) and NLRP3−/− mice. Mice (n = 6 per group) were treated with 3% DSS for 7 days, followed by regular drinking water for 3 days. d Body weight changes, e colon length, and f histopathological changes in colon tissue were examined. Data are presented as mean ± SD. *P < 0.05, **P < 0.01
VI-16 inhibited the activation of NLRP3 inflammasome in vitro
IL-1β and IL-18 were processed as an inactive precursor, pro-IL-1β and pro-IL-18, which has to be cleaved by cleaved caspase-1 to produce the mature form. Caspase-1 cleavage depends on activation of inflammasome. To investigate the function of VI-16 in inflammasome activation, we analyzed the secretion of IL-1β in LPS-primed differentiated THP-1 cells and primary bone marrow derived macrophages (BMDMs) that were treated with 10 μM VI-16 before treatment with ATP, a specific NLRP3 inflammasome activator. VI-16 suppressed IL-1β secretion in macrophages in response to treatment with ATP. In contrast, VI-16 had no effect on the activation of IL-1β secretion in response to treatment with poly (dA:dT), an AIM2 inflammasome activator, or muramyldipeptide (MDP), an NLRP1 inflammasome activator, or flagellin, an NLR family CARD domain-containing protein 4 (NLRC4) inflammasome activator (Fig. 5a). Moreover, mature IL-1β in the supernatants and cleaved caspase-1 in the lysates were significantly suppressed by VI-16 in LPS-primed differentiated THP-1 cells and primary BMDMs in response to ATP, without affecting in response to poly (dA:dT), MDP, or flagellin (Fig. 5b). Moreover, pre-treatment with VI-16 concentration-dependently suppressed IL-1β secretion in response to NLRP3 inflammasome activators, including ATP, nigericin and monosodium urate crystals (MSU), in LPS-primed macrophages (Fig. 5c). Consistently, mature IL-1β in the supernatants and cleaved caspase-1 in the lysates were significantly suppressed by VI-16 in LPS-primed macrophages in response to ATP, nigericin, and MSU (Fig. 5d). Together these results indicated that VI-16 suppressed the activation of NLRP3 inflammasome in vitro.

VI-16 inhibited caspase-1 activation and IL-1β maturation by interrupting NLRP3 inflammasome activation in vitro. a ELISA of IL-1β in supernatants of LPS-primed differentiated THP-1 cells and BMDMs treated with 10 μM of VI-16, followed by incubation with ATP (5 mM) for 1 h, MDP (200 ng /ml) or flagellin (200 ng /ml) for 6 h, or transfection of poly(dA:dT). Data are presented as mean ± SD. **P < 0.01. b Immunoblot analysis of IL-1β in supernatants (SN) and caspase-1 in extracts (Input) of LPS-primed differentiated THP-1 cells and BMDMs treated as above. c ELISA of IL-1β in supernatants of LPS-primed differentiated THP-1 cells and BMDMs treated with 2.5, 5, or 10 μM of VI-16, followed by incubation with ATP (5 mM) for 1 h, nigericin (4 μM) for 3 h or MSU (150 µg/ml) for 6 h. Data are presented as mean ± SD. **P < 0.01. #P < 0.05, ##P < 0.01 compared with stimulation group. d Immunoblot analysis of IL-1β in supernatants (SN) and caspase-1 in extracts (Input) of LPS-primed differentiated THP-1 cells treated as above
VI-16 suppressed NLRP3 inflammasome assembly and reduced mitochondrial reactive oxygen species generation
Next, we sought to determine the mechanism how VI-16 inhibited the activation of NLRP3 inflammasome (Fig. 6a). Immunoprecipitation analysis revealed that NLRP3 inflammasome formation was interrupted by VI-16. Similarly, VI-16 suppressed colocalization of ASC and caspase-1 in response to ATP (Fig. 6b). This suggests that VI-16 inhibited the assembly of NLRP3 inflammasome. Activation of NLRP3 inflammasome assembly is dependent on the generation of mitochondrial reactive oxygen species (mtROS).24 Next, we analyzed generation of mtROS in LPS-primed differentiated THP-1 cells pretreated with VI-16 before ATP treatment. As shown in Fig. 6c, d, VI-16 reduced mtROS generation in concentration-dependently manner. To avoid excessive activation of NLRP3 inflammasome, ROS-generating mitochondria are constantly removed by mitophagy, a specialized process of autophagy.25 Therefore, we analyzed autophagy activity after VI-16 treatment. Transfection of a plasmid, expressing GFP-LC3 (pcDNA3.1-GFP-LC3), into THP-1 cells was used as a tool to monitor autophagy activity. Immunofluroscence analysis showed that VI-16 treatment did not affect autophagy activity (Fig. 6e). Furthermore, there was no significant change in the expression of p62 and LC3-II after VI-16 treatment (Fig. 6f). These results suggested that VI-16 reduced mitochondrial ROS generation without affecting the autophagy activity.

VI-16 inhibited NLRP3 inflammasome assembly and reduced mitochondrial reactive oxygen species generation. LPS-primed differentiated THP-1 cells treated with indicated concentration of VI-16, followed by incubation with ATP (5 mM) for 1 h. a Immunoblot analysis of ASC immunoprecipitates in LPS-primed differentiated THP-1 cells, probed for NLRP3, pro-caspase-1, and ASC. b Confocal microscopy of LPS-primed differentiated THP-1 cells, immunostained for ASC (green) and caspase-1 (red). Scale bars, 10 µm. Data are representative of at least three experiments. c MitoSOX was measured by flow cytometry in LPS-primed differentiated THP-1 cells. d Histograms represented the geometric mean mitoSOX fluorescence. e Induction of GFP + dots in LPS-primed differentiated THP-1 cells expressing GFP-LC3. Results are presented as quantification of GFP-LC3+ dots (top) and images of individual cells (bottom). Scale bars, 10 µm. f Immunoblot analysis of p62 and LC3 expression in LPS-primed differentiated THP-1 cells. Data are presented as mean ± SD. **P < 0.01 compared with control group; ##P < 0.01 compared with ATP group
VI-16 inhibited the dissociation of Txnip from Trx-1 and the association of Txnip with NLRP3 by enhancing the antioxidant defenses
Excessive mtROS production causes Trx-1 to dissociate from Txnip, which subsequently binds to NLRP3 and promotes NLRP3 inflammasome activation. We first analyzed the effect of VI-16 on the expression of Txnip and Trx-1. Western blots and real-time PCR analysis showed that VI-16 treatment did not affect Txnip and Trx-1 protein and mRNA expression (Fig. 7a, b). Next, immunofluroscence analysis showed that VI-16 inhibited the colocalization of NLRP3 and Txnip (Fig. 7c). Immunoprecipitation analysis revealed that VI-16 suppressed the dissociation of Txnip from Trx-1 and binding to NLRP3 in response to ATP, nigericin, and MSU, respectively (Fig. 7d). Txnip has been identified as a negative regulator of the Trx-1 reductase activity, that Txnip was directly interacted with the redox-active domain of Trx-1.26 Subsequently, we investigated the effect of VI-16 on the antioxidant defense. As shown in Fig. 7e, VI-16 decreased the level of reactive oxygen species (ROS). Taken together, VI-16 inhibited the dissociation of Txnip from Trx-1 and binding to NLRP3 by enhancing the antioxidant defenses.

VI-16 inhibited the dissociation of Txnip from Trx-1 and to binding to NLRP3 by improving the antioxidant capacity. The protein expression (a) and mRNA levels (b) of Txnip and Trx-1 were detected in LPS-primed differentiated THP-1 cells treated with indicated concentration of VI-16, followed by incubation with ATP (5 mM) for 1 h. c Confocal microscopy of LPS-primed differentiated THP-1 cells, immunostained for NLRP3 (green) and Txnip (red). Scale bars, 10 µm. d Immunoblot analysis of Txnip immunoprecipitates in LPS-primed differentiated THP-1 cells, followed by incubation with ATP (5 mM) for 1 h, nigericin (4 μM) for 3 h or MSU (150 µg/ml) for 6 h, then probed for NLRP3, Trx-1, and Txnip. e Levels of the ROS were measured in LPS-primed differentiated THP-1 cells. Data are presented as mean ± SD. *P < 0.05, **P < 0.01 compared with control group; #P < 0.05, ##P < 0.01 compared with ATP group
VI-16 suppressed Txnip-dependent NLRP3 inflammasome activation by reducing oxidative stress
In order to clarify the molecular mechanism of VI-16 inhibiting NLRP3 inflammasome, we validated the effect of Txnip on the activation of NLRP3 inflammasome by transfection of siRNA. As shown in Fig. 8a, knockdown Txnip expression reduced expression of mature IL-1β in the supernatants and cleaved caspase-1 in the lysates after treatment with ATP, nigericin and MSU. As the association of Txnip and Trx-1 prevents the activation of NLRP3 inflammasome, knockdown Trx-1 expression promoted IL-1β secretion. However, VI-16 did not inhibit the secretion of IL-1β after knockdown Trx-1 expression (Fig. 8b, c). This suggests that VI-16 suppresses NLRP3 inflammasome activation depending on the regulation of Txnip on Trx-1 antioxidant capacity.

VI-16 inhibited Txnip mediated NLRP3 inflammasome activation by resisting oxidative stress. a Immunoblot analysis of IL-1β in supernatants (SN) and caspase-1, Txnip in extracts (Input) of LPS-primed differentiated THP-1 cells transfected with siRNA with scrambled sequence or siRNA specific for Txnip. b Immunoblot analysis of differentiated THP-1 cells transfected with siRNA with scrambled sequence (−) or siRNA specific for Trx-1. c ELISA of IL-1β in supernatants of LPS-primed differentiated THP-1 cells, transfected with Txnip siRNA, treated with 10 μM of VI-16, followed by treating for 1 h with ATP (5 mM), 3 h with nigericin (4 μM) and 6 h with MSU (150 µg/ml). Data are presented as mean ± SD. *P < 0.05, **P < 0.01 compared with scramble+DMSO group. d Level of ROS was measured in LPS-primed differentiated THP-1 cells treated with 5 mM of NAC for different time, followed by treating for 1 h with ATP (5 mM). e Immunoblot analysis of Txnip immunoprecipitates in LPS-primed differentiated THP-1 cells treated with 5 mM of NAC for different time, probed for Trx-1, NLRP3, and Txnip. f Immunoblot analysis of IL-1β in supernatants (SN) and extracts (Input) of LPS-primed differentiated THP-1 cells treated with 5 mM of NAC for different time. g Level of ROS was measured in LPS-primed differentiated THP-1 cells treated with 10 μM of VI-16, followed by treating for 6 h with H2O2 (10 mM) or rotenone (10 mM). Data are presented as mean ± SD. *P < 0.05. h Immunoblot analysis of IL-1β in supernatants (SN) and caspase-1 in extracts (Input) of LPS-primed differentiated THP-1 cells treated with 10 μM of VI-16, followed by treating for 6 h with H2O2 (10 mM) or rotenone (10 mM)
Subsequently, we investigated the effect of ROS scavenger N-acetyl-L-cysteine (NAC) on NLRP3 inflammasome. The addition of NAC to THP-1 macrophage resulted in much more ROS being cleared (Fig. 8d). Simultaneously, Txnip began to associate with Trx-1. Completed association was evident 1 h after stimulation, at which time the dissociation of Txnip from NLRP3 was observed (Fig. 8e). Consistently, the expression of mature IL-1β in the supernatants was decreased by NAC (Fig. 8f). This indicates that scavenging oxidative stress inhibited Txnip-dependent NLRP3 inflammasome activation.
Additional support for the idea that VI-16 inhibits NLRP3 inflammasome by reducing oxidative stress was provided by experiments using H2O2 or Rotenone, which resulted in more mature IL-1β and caspase-1 in LPS-primed differentiated THP-1 cells. As shown in Fig. 8g, the generation of ROS, increased by H2O2 or Rotenone, was suppressed by VI-16. Similarly, VI-16 reduced expression of mature IL-1β in the supernatants and cleaved caspase-1 in the lysates in response to H2O2 or Rotenone (Fig. 8h). These data confirmed that VI-16 inhibited NLRP3 inflammasome activation which was induced by oxidative stress.
Downregulation of Txnip expression by DSS in intestinal epithelial cells is the basis for VI-16 specific inhibition of NLRP3 inflammasome in colonic macrophages
In our study, VI-16 specific inhibition of NLRP3 inflammasome in colonic macrophages rather than in intestinal epithelial cells was a puzzling result. Therefore, we first detected the expression of Txnip protein in intestinal epithelial cells in DSS-induced colitis mice at different time points. The expression of Txnip at protein and mRNA levels was significantly downregulated by DSS in intestinal epithelial cells (Fig. 9a, b). Similarly, DSS also downregulated Txnip expression at protein and mRNA levels in intestinal epithelial cells of NLRP3−/− mice (Fig. 9c, d). However, VI-16 had no significant effect on Txnip expression at protein and mRNA levels in colonic macrophages (Fig. 9e, f). Therefore, VI-16 selectively inhibited Txnip-dependent NLRP3 inflammasome activation in colonic macrophages rather than in intestinal epithelial cells.

DSS down-regulated Txnip expression in intestinal epithelial cells without in colonic macrophages. The a protein expression and b mRNA levels of was detected in intestinal epithelial cells extracts of DSS-induced wild-type mice following DSS treatment. The c protein expression and d mRNA levels of was detected in intestinal epithelial cells extracts of DSS-induced NLRP3−/− mice following DSS treatment. The e protein expression and f mRNA levels of was detected in colonic macrophages extracts of DSS-induced wild-type mice following DSS treatment. Data are presented as mean ± SD. *P < 0.05, **P < 0.01 compared with 0 day group
Discussion
In this study, VI-16 has emerged from many flavonoids due to its inhibitory activity on IL-1β secretion. To assess the anti-inflammatory activity of VI-16, we investigated the roles of VI-16 in DSS-induced colitis. The results showed that VI-16 attenuated the severity of colitis by reducing intestinal mucosal damage, the infiltration of inflammatory cells and inhibiting excessive secretion of inflammatory cytokines in wild-type mice, while it lost the function of alleviating DSS-induced colitis in NLRP3−/− bone marrow mice. In the development of colitis, NLRP3 inflammasome was activated excessively and persistently, which was seem to be a potential target for therapeutics of IBD.16 Then, activation of NLRP3 inflammasome was inhibited after VI-16 treatment in colonic macrophages rather that in intestinal epithelial cells. Meanwhile, VI-16 suppressed the generation of mtROS without affecting the autophagy activity. Further studies show that VI-16 inhibited the dissociation of Txnip from Trx-1 and binding to NLRP3 by reducing oxidative stress and ultimately inhibited NLRP3 inflammasome. Together these data indicated that VI-16 attenuated DSS-induced colitis through reducing oxidative stress to suppress Txnip-dependent NLRP3 inflammasome activation in colonic macrophages.
Flavonoids have good anti-inflammatory activity, but the mechanism is complex. Studies have shown that the anti-inflammatory mechanisms of some flavonoids are associated with pro-inflammatory enzymes, such as cyclooxygenase-2, nitric oxide synthase and lipoxygenase, and inflammatory cytokines, such as IL-1β, IL-6, and TNF-α.27,28,29 Flavonoids also interfere with the activation of transcription factors, including nuclear factor-kappa B (NF-κB), activator protein 1 (AP-1), signal transducer, and transcription activation (STAT) and peroxisome proliferator-activated receptor (PPAR) family.29,30,31,32 Therefore, it is certain that flavonoids have anti-inflammatory effects in vivo and in vitro. More importantly, flavonoids showed the activity of inhibiting NLRP3 inflammasome. But the specific molecular mechanism remains confusing. In order to further study the anti-inflammatory mechanism of flavonoid compounds, we first screened the anti-inflammatory activity of flavonoids. The concentration of flavonoids tested initially was related to pharmacology, since concentrations higher than 10 μM flavonoid cannot easily reach in the blood stream. Previous pharmacokinetic studies have shown that the blood concentrations of flavonoids and their metabolites after oral intake are relatively low (<10 μM).33 Therefore, the active flavonoid compound 10 μM is considered ineffective in our initial experiment because they may not show any inhibitory action of NLRP3 inflammasome activation in vivo.
Ulcerative colitis is a chronic inflammatory disease that not only impairs the quality of life of patients, but also contributes to the occurrence of colon cancer.34 In general, the treatment of inflammatory bowel disease depends on the severity of the disease. Most of the time may require immunosuppressive agents, such as prednisone, TNF inhibition, azathioprine, and methotrexate, to control symptoms.35 However, these immunosuppressants also have potential side effects, including steroid dependence36 and severe infection.37 Therefore, there is an urgent need for new strategies with fewer side effects. Inflammasome is a cytoplasmic protein complex, which is becoming more and more recognized by its clinical importance in autoimmune, infectious, and metabolic diseases.38 When inflammasome is activated and assembled, it converts inactive pro-caspase-1 into cleaved caspase-1. Among the several isoforms of caspases, caspase-1 is known to mediate inflammation, cellular necrosis and pyroptosis. Cleaved caspase-1 cleaves pro-IL-1β and pro-IL-18 to produce active forms. The mature IL-1β and IL-18 are released from the cells during the inflammatory response. Therefore, blocking the activation of NLRP3 inflammasome in macrophages is a new strategy to interrupt inflammatory and immunological response. However, other studies have confirmed that the activation of inflammasome in intestinal epithelial cells leads to IL-18 secretion, which contributes to the realization of intestinal epithelial barrier function.23 Colitis was more severe in NLRP3−/− mice than in wild-type mice. Therefore, agents that selectively inhibit the activation of NLRP3 inflammasome in colonic macrophages without affecting intestinal epithelial cells has the potential to treat ulcerative colitis. In our study, VI-16 acted on colon macrophages rather than intestinal epithelial cells depending on the expression of Txnip. This was because DSS reduced the expression of Txnip in intestinal epithelial cells. It had been confirmed that inflammatory cells expressing Txnip were abundant in the lamina propria and submucosa of the colon in ulcerative colitis patients, which is the result of increased inflammatory cells infiltrating into the colon. However, Epithelial cells expressing the Txnip transcript were obviously decreased in the colonic mucosa of UC patients compared to normal controls.39 This is consistent with our findings. Thus, the down-regulation of Txnip in intestinal epithelial cells may be related to the pathogenesis of inflammatory bowel disease and colitis-associated carcinogenesis, but further studies are still needed.
Mitochondrial function plays an important role in regulating inflammation, since mitochondria are the main intracellular source of ROS (90%).40 Many studies have shown that mtROS is an important factor in activating NLRP3 inflammasome by promoting the dissociation of Txnip from Trx-1 and binding to NLRP3.41 Reducing oxidative stress is the preferred solution to suppress NLRP3 inflammasome and inflammation, which has been confirmed by NAC addition in our study. Autophagy is the main way to eliminate damaged mitochondria and reduce mtROS.42 Therefore, induction of autophagy has become an important way to control inflammation.43 In our study, VI-16 significantly reduced the production of mtROS without affecting autophagy, suggesting that VI-16 has other ways of regulating mtROS. Previous studies have demonstrated that Trx-1, which is converted from the reduced form to the oxidized form, can resist oxidative stress, while Txnip can only be bind to the reduced form Trx-1.26 When ROS is antagonised by the reduced form of Trx-1, Trx-1 protein itself forms disulfide bonds, so that Txnip is released and binds to NLRP3 to activate NLRP3. The effect of VI-16 on reducing oxidative stress ensures the existence of more reductive form Trx-1, which has been confirmed by transfection of Trx-1 siRNA. At the same time, other stimulations that cause oxidative stress, such as hydrogen peroxide or rotenone, similarly activated NLRP3 inflammasome. Correspondingly, VI-16 also inhibited NLRP3 inflammasome activation in response to hydrogen peroxide or rotenone. It is confirmed that VI-16 alleviate the activation of NLRP3 inflammasome induced by oxidative stress (Fig. 10).

The possible mechanism of VI-16 inhibits NLRP3 inflammasome in colonic macrophages. VI-16 intervened in the association of Txnip with NLRP3 to inhibit NLRP3 inflammasome activation by reducing oxidative stress
In conclusion, the present study demonstrated that VI-16 selectively suppress Txnip-dependent NLRP3 inflammasome activation by relieving oxidative stress in colonic macrophages to attenuates DSS-induced colitis. This implies that flavonoid VI-16 may be a potential treatment for colitis. However, the integrated safety assessment and optimization of treatment options in clinical application of VI-16 deserve further study.
Materials and methods
Reagents and antibodies
VI-14 (C24H29NO5, MW: 411.49) and VI-16 (C25H29NO5, MW: 423.50) were obtained from Z.L. (China Pharmaceutical University, China). LW-216 (C30H31NO5, MW: 485.57) and LL-402 (C26H29F3N2O5, MW: 506.51) were obtained from Z.L. (China Pharmaceutical University, China). Compounds were dissolved in dimethylsulfoxide (DMSO) as a stock solution, stored at −20 °C, and freshly diluted with medium to the final concentration in vitro study. In vivo study, VI-16 was prepared as intragastric administration (0.5% sodium carboxyl methyl cellulose (CMC)) by Dr. Xue Ke from College of Pharmacy, China Pharmaceutical University. The DSS-treated group of mice were administered 0.5% CMC as vehicle.
LPS (E. coli: Serotype O55:B5), DMSO, phorbol 12-myristate 13-acetate (PMA), adenosine triphosphate (ATP), and Nigericin were purchased from Sigma–Aldrich (St. Louis, USA). Dextran sulfate sodium (DSS, molecular weight 36–50 kDa) was purchased from MP Biomedicals Inc. (Irvine, USA). Isoliquiritigenin, MCC950, MDP, flagellin (Salmonella typhimurium), and MSU were purchased from InvivoGen (San Diego, USA).
Dye DAPI was purchased from Invitrogen (Carlsbad, USA). Paraformaldehyde (PFA) was purchased from Yonghua Chemical Technology (Jiangsu) Co. Ltd. (Changshu, China). Triton X-100 was purchased from Shanghai Chao Rui Biotech. Co. Ltd. (Shanghai, China). BSA was purchased from Roche Diagnosis Ltd. (Shanghai, China).
Myeloperoxidase (MPO) activity assay kit was purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). ELISA kits for mouse IL-1β, IL-18 and human IL-1β, IL-18 were purchased from Boster Biotech Co. Ltd. (Wuhan, China).
Primary antibodies against caspase-1, ASC, Txnip, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Antibodies to Trx-1 was from Bioworld (OH, USA) and antibodies against IL-1β was purchased from Cell Signaling Technology (Danvers, MA); Antibodies to NLRP3 was obtained from Abcam (Cambridge, UK). IRDyeTM800 conjugated secondary antibodies were obtained from Rockland Inc. (Philadelphia, USA). FITC-anti-CD11b/ M1/70 and APC-anti-MHC II/M5-114.15.2 were purchased from eBioscience (San Diego, USA). PE-anti-F4/80/BM8 was from Biolegend (San Diego, CA). Alexa Fluor 488 donkey anti-rabbit IgG, Alexa Fluor 594 donkey anti-mouse IgG were obtained from Invitrogen (CA, USA).
Cell culture
Human acute monocytic leukemia THP-1 cells were obtained from CBCAS (Cell Bank of the Chinese Academic of Sciences, Shanghai, China). THP-1 cells were cultured in RPMI-1640 medium (Gibco, Carlsbad, USA), supplemented with 10% (v/v) fetal bovine serum (Gibco, Carlsbad, USA) and 0.05 mM 2-mercaptoethanol, 100 U/ml benzyl penicillin and 100 mg/ml streptomycin. Cells were cultured in a humidified environment with 5% CO2 at 37 °C. Differentiation of THP-1 cells was induced by 0.5 mM phorbol 12-myristate 13-acetate (PMA) for 3 h. The differentiated cells were washed three times with PBS and treated with 500 ng/ml LPS in the absence or presence of VI-16.
Bone marrow derived macrophages (BMDMs) were isolated from C57BL/6 mice and precursor cells were cultured in complete DMEM medium (Gibco, Carlsbad, USA) supplemented with 30% L929 cell-conditioned medium, as the source of macrophage colony-stimulating factor. Cells were harvested and seeded on cell culture dishes (60 × 15 mm). Exchanging culture medium every 3 days, adherent macrophages were obtained within about one week. Then, the cells were washed three times with PBS and treated with 500 mg/ml LPS in the absence or presence of VI-16.
Animals
C57BL/6 J mice (male, 6–8 weeks old, weighing 18–22 g) were supplied Shanghai Laboratory Animal Center, China Academy of Sciences. NLRP3 knockout (NLRP3−/−) mice (male, 6–8 weeks old, weighing 18–22 g) were obtained from Gang Hu’s laboratory. The mice were manipulated in accordance with the local Ethical Committee guidelines. Experimental protocols were in accordance with National Institutes of Health regulations and approved by the Institutional Animal Care and Use Committee. Throughout the acclimatization and study periods, all animals had access to food and water ad libitum and were maintained on a 12 h light/dark cycle (21 ± 2 °C with a relative humidity of 45 ± 10%).
DSS-induced colitis and design of drug treatment
Colitis was induced by administration of DSS in drinking water. The mice received either drinking regular water (control) or 3% (w/v) DSS drinking water (model) for 7 days and thereafter provided with regular water for 3 days. The mice were randomly assigned to normal, DSS-treated, VI-16 (20, 40, or 80 mg/kg)-treated groups. VI-16 was given intragastrically from day 1 to day 10, respectively.
Macroscopic assessment and histological analysis of colonic lesions
After DSS-induced colitis, the mice were sacrificed and colons were removed, opened longitudinally, and washed with phosphate-buffered saline (PBS). The pieces of colonic tissue were fixed in 10% neutral-buffered formalin and routinely paraffin embedded and processed. The histological analysis was performed as previously described.44
Immunofluorescence of colon tissues
The CD11b+ cells infiltration analysis was performed on paraffin-embedded colon tissue sections. Briefly, the sections were deparaffinized, rehydrated, and washed in PBS. After treating with 3% hydrogen peroxide, blocking with 3% bovine serum albumin (BSA), the sections were incubated for 1 h at room temperature with anti-CD11b+ (1:100). The slides were then counter-stained with DAPI for 30 min. The reaction was stopped by thorough washing in water for 5 min. Images were acquired by confocal laser-scanning microscope (Olympus, NY, USA). Settings for image acquisition were identical for control and experimental tissues.
Cytokine quantification by enzyme-linked immunoassay
Colons from mice in each group were homogenated with lysis buffer to extract total protein. The homogenate was centrifuged at 12,000 × g at 4 °C for 15 min. The amount of total extracted protein was determined by BCA protein assay kit (Thermo, MA, USA). The amounts of IL-1β and IL-18 were measured by ELISA kit. IL-1βand IL-18 production in supernatant differentiated THP-1 cells and BMDM were measured by ELISA kits according to the manufacturers’ recommendations.
Isolation of colonic macrophages from mice
Colonic macrophages were prepared as described previously.9 Briefly, entire colons from 30 to 50 mice were collected and washed with calcium and magnesium-free Hanks’ balanced salt solution (HBSS; Gibco, Carlsbad, USA). The tissues were opened longitudinally, cut into 2-cm pieces, and incubated with HBSS containing 0.015% dithiothreitol (Sigma, St. Louis, USA) for 15 min, followed by extensive washing in cold HBSS containing 5% heat-inactivated fetal bovine serum (Gibco, Carlsbad, USA) to remove epithelial cells and mucus. Colon pieces were finely minced and digested in complete (supplemented with penicillin– streptomycin, 2 mM L-glutamine, 15 mM HEPES, 50 mM β-mercaptoethanol (Sigma, St. Louis, USA), and 10% fetal bovine serum) Iscove’s Modified Dulbecco’s Media (IMDM) (Gibco, Carlsbad, USA) containing 167 mg/ml Liberase TL and 30 mg/ml DNase I (Roche, Indianapolis, IN) for 60 min at 37 °C. The samples were filtered, pelleted, and resuspended in 40% and 80% fraction of the Percoll solution (Sigma, St. Louis, USA). After centrifugation at 1000 × g for 20 min without brakes at room temperature, the viable cells from the interphase of the two different Percoll solutions were collected and colonic macrophages were sorted using FACSAria (BD Biosciences).
Isolation of intestine epithelial cells from mice
Intestinal epithelial cells were isolated using a modified protocol.45 The colon was cut into small pieces and incubated with 0.5 mM dithiothreitol and 3 mM EDTA at room temperature for 1.5 h without shaking. After gently removing the solution, PBS was added to the colon tissue. Crypts released from the colon by shaking the tubes. Cells were washed with PBS by centrifuging and then were solubilized in cell lysis buffer containing 1% Triton X-100, 10 mM Tris (pH 7.4), 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, and protease and phosphatase inhibitor cocktail (Sigma, St. Louis, USA).
Bone marrow chimeras
Bone marrow transfer was used to create NLRP3−/− chimera mice wherein the genetic deficiency of Nlrp3 was confined to either circulating cells (NLRP3−/− > WT chimera) or nonhematopoietic tissue (WT > NLRP3−/− chimera). In brief, bone marrows were collected from femur and tibia from donor mice, and a total of 8 × 106 bone marrow cells were injected into the tail veins of irradiated recipient mice. Reconstituted mice were treated with 0.2% neomycin for the first 2 weeks after transplantation. Mice were left for 6–8 weeks before DSS treatment to allow reconstitution.
Measurement of mature IL-1β
For IL-1β induction, macrophages were plated in 6-well plates. The medium was changed to Opti-MEM and cells were treated for 6 h with MSU (150 µg/ml), MDP (200 ng /ml), flagellin (200 ng /ml), rotenone (10 mM) or H2O2 (10 mM), for 3 h with nigericin (4 μM) or for 60 min with ATP (5 mM). For transfection of poly (dA:dT) into cells, Lipofectamine 2000 (4 µl/ml) was used according to the manufacturer’s protocol. Medium from each well was mixed with 2 ml methanol and 0.5 ml chloroform, vortexed, and centrifuged at 12,000 × g at 4 °C for 5 min. The upper phase of each sample was removed and 0.5 ml methanol was added. Samples were centrifuged again at 12,000 × g at 4 °C for 5 min as described above, then supernatants were removed and pellets were dried for 5 min at 50 °C. Then, 20 µl loading buffer was added to each sample, followed by boiling for 10 min before SDS-PAGE and immunoblot analysis with antibodies for the detection of mature IL-1β. Adherent cells in each well were lysed with the lysis buffer described above, followed by immunoblot analysis to determine the cellular content of various proteins.
Measurement of ROS and mtROS formation
The level of ROS was detected using fluorescent dye 2, 7-dichlorofluorescein-diacetate (DCFHDA) obtained from Beyotime (Shanghai, China). The level of mtROS was detected using fluorescent dye mitoSOX obtained from Invitrogen (CA, USA). Cells were collected and incubated with DCFH-DA or mitoSOX for 30 min at 37 °C in the dark. The fluorescence intensity was measured using flow cytometry.
Immunoprecipitation
Cells were collected and lysized with lysis buffer on ice for 1 h and centrifuged at 12,000 × g at 4 °C for 30 min. The supernatant fractions were collected and incubated with the appropriate antibody at 4 °C overnight and precipitated with protein A/G-agarose beads (Santa Cruz, CA, USA) for another 4 h at 4 °C. The beads were washed with the lysis buffer three times by centrifugation at 1000 × g at 4 °C. The immunoprecipitated proteins were separated by SDS-PAGE and western blot was performed with the indicated antibodies.
Western blot analysis
After whole cell lysates were prepared. Western blot analysis was prepared as described previously.46 Protein samples were separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked with 1% BSA at 37 °C for 1 h and incubated with indicated antibodies overnight at 4 °C, followed by IRDye800 conjugated secondary antibody for 1 h at 37 °C. Immunoreactive protein was detected with an Odyssey Scanning System (LI-COR Inc., Lincoln, Nebraska).
Cell transfection
GFP-LC3 plasmid (Addgene, MA, USA) and the siRNA targeting human Txnip, Trx-1 or control siRNA with scrambled sequence (Santa Cruz, CA, USA) were transfected using Lipofectamine 2000™ reagent (Invitrogen, CA, USA), according to the manufacturer’s instructions.
Immunofluorescence
For confocal imaging of fixed cells, differentiated THP-1 cells were used. After the appropriate treatment, cells were washed with PBS, fixed with 4% paraformaldehyde and washed again with PBS. Non-specific receptors on cells were blocked for 1 h with 3% BSA. Rabbit anti-ASC (Santa Cruz, CA, USA), mouse anti-caspase-1 (Santa Cruz, CA, USA), rabbit anti-NLRP3 (Abcam, Cambridge, UK), mouse anti-Txnip (Santa Cruz, CA, USA) were used for immunostaining. Alexa Fluor 488 donkey anti-rabbit IgG, Alexa Fluor 594 donkey anti-mouse IgG were used as secondary antibodies (Invitrogen, CA, USA). Samples were observed and captured with a confocal laser-scanning microscope (Olympus Corp., Tokyo, Japan).
Statistical analysis
The data shown in the study were obtained in at least three independent experiments and all results represent the mean ± S.E.M. Differences between the groups were assessed by one-way ANOVA test. Details of each statistical analysis used are provided in the figure legends. Differences with P values <0.05 were considered statistically significant.
References
Podolsky, D. K. Inflammatory bowel disease. N. Engl. J. Med. 347, 417–429 (2002).
Aychek, T. & Jung, S. Immunology. axis Toler. Sci. 343, 1439–1440 (2014).
Maloy, K. J. & Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K. & Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993).
Sellon, R. K. et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224–5231 (1998).
Vijay-Kumar, M. et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Investig. 117, 3909–3921 (2007).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Latz, E., Xiao, T. S. & Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 (2013).
Filardy, A. A., He, J., Bennink, J., Yewdell, J. & Kelsall, B. L. Posttranscriptional control of NLRP3 inflammasome activation in colonic macrophages. Mucosal Immunol. 9, 850–858 (2016).
Bruchard, M. et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64 (2013).
Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 (2009).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865 (2008).
De Nardo, D. & Latz, E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 32, 373–379 (2011).
Abderrazak, A. et al. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 4, 296–307 (2015).
Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Mao, K. et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 23, 201–212 (2013).
Han, J. W. et al. Anti-inflammatory effect of emodin via attenuation of NLRP3 inflammasome activation. Int. J. Mol. Sci. 16, 8102–8109 (2015).
Havsteen, B. Flavonoids, a class of natural products of high pharmacological potency. Biochem. Pharmacol. 32, 1141–1148 (1983).
Honda, H. et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 96, 1087–1100 (2014).
Tsai, P. Y. et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 51, 744–754 (2011).
White, J. B. et al. Some natural flavonoids are competitive inhibitors of Caspase-1, -3 and -7 despite their cellular toxicity. Food Chem. 131, 1453–1459 (2012).
Nowarski, R. et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163, 1444–1456 (2015).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008).
Goldman, S. J., Taylor, R., Zhang, Y. & Jin, S. Autophagy and the degradation of mitochondria. Mitochondrion 10, 309–315 (2010).
Nishiyama, A. et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 274, 21645–21650 (1999).
Chi, Y. S. et al. Effects of naturally occurring prenylated flavonoids on enzymes metabolizing arachidonic acid: cyclooxygenases and lipoxygenases. Biochem. Pharmacol. 62, 1185–1191 (2001).
Middleton, E. Jr., Kandaswami, C. & Theoharides, T. C. The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 52, 673–751 (2000).
Kim, H. P., Son, K. H., Chang, H. W. & Kang, S. S. Anti-inflammatory plant flavonoids and cellular action mechanisms. J. Pharmacol. Sci. 96, 229–245 (2004).
Lim, H., Park, H. & Kim, H. P. Effects of flavonoids on matrix metalloproteinase-13 expression of interleukin-1beta-treated articular chondrocytes and their cellular mechanisms: inhibition of c-Fos/AP-1 and JAK/STAT signaling pathways. J. Pharmacol. Sci. 116, 221–231 (2011).
Choi, M. J. et al. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: critical role of PPAR-gamma signaling pathway. Biochem. Pharmacol. 144, 120–131 (2017).
Zhou, R. J., Ye, H., Wang, F., Wang, J. L. & Xie, M. L. Apigenin inhibits d-galactosamine/LPS-induced liver injury through upregulation of hepatic Nrf-2 and PPARgamma expressions in mice. Biochem. Biophys. Res. Commun. 493, 625–630 (2017).
Rodriguez-Mateos, A. et al. Bioavailability, bioactivity and impact on health of dietary flavonoids and related compounds: an update. Arch. Toxicol. 88, 1803–1853 (2014).
Danese, S., Malesci, A. & Vetrano, S. Colitis-associated cancer: the dark side of inflammatory bowel disease. Gut 60, 1609–1610 (2011).
Rutgeerts, P., Van Assche, G. & Vermeire, S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology 126, 1593–1610 (2004).
Faubion, W. A. Jr. et al. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology 121, 255–260 (2001).
Beaugerie, L. et al. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 374, 1617–1625 (2009).
Lamkanfi, M. & Kanneganti, T. D. The inflammasome: a remote control for metabolic syndrome. Cell Res. 22, 1095–1098 (2012).
Takahashi, Y. et al. Decreased expression of thioredoxin interacting protein mRNA in inflamed colonic mucosa in patients with ulcerative colitis. Oncol. Rep. 18, 531–535 (2007).
Kowaltowski, A. J., de Souza-Pinto, N. C., Castilho, R. F. & Vercesi, A. E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 47, 333–343 (2009).
Abais, J. M. et al. Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J. Biol. Chem. 289, 27159–27168 (2014).
Zhong, Z. et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016).
Zhao, Y. et al. Small molecule GL-V9 protects against colitis-associated colorectal cancer by limiting NLRP3 inflammasome through autophagy. Oncoimmunology 7, e1375640 (2017).
Zhao, Y. et al. GL-V9, a new synthetic flavonoid derivative, ameliorates DSS-induced colitis against oxidative stress by up-regulating Trx-1 expression via activation of AMPK/FOXO3a pathway. Oncotarget 6, 26291–26307 (2015).
Wang, Y. et al. An LGG-derived protein promotes IgA production through upregulation of APRIL expression in intestinal epithelial cells. Mucosal Immunol. 10, 373–384 (2017).
Zhao, Y. et al. LYG-202 exerts antitumor effect on PI3K/Akt signaling pathway in human breast cancer cells. Apoptosis: Int. J. Program. Cell Death 20, 1253–1269 (2015).
Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu province (No. BK20180576), the National Natural Science Foundation of China (No. 81673461), the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. SKLNMZZCX201823), the Drug Innovation Major Project (2017ZX09301014, 2018ZX09711001-003-007, 2017ZX09101003-005-023), Program for Changjiang Scholars and Innovative Research Team in University (IRT1193), the Open Project of State Key Laboratory Cultivation Base for TCM Quality and Efficacy, Nanjing University of Chinese Medicine (No.TCMQ&E201704), Social Development Project of Jiangsu Provincial Science and Technology Department (BE2018711), and Double First-Class University project (CPU 2018GF11, CPU2018GF05).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Guo, Q., Zhu, Q. et al. Flavonoid VI-16 protects against DSS-induced colitis by inhibiting Txnip-dependent NLRP3 inflammasome activation in macrophages via reducing oxidative stress. Mucosal Immunol 12, 1150–1163 (2019). https://doi.org/10.1038/s41385-019-0177-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-019-0177-x
This article is cited by
-
The ‘speck’-tacular oversight of the NLRP3-pyroptosis pathway on gastrointestinal inflammatory diseases and tumorigenesis
Journal of Biomedical Science (2023)
-
Toll-like receptors and NLRP3 inflammasome-dependent pathways in Parkinson’s disease: mechanisms and therapeutic implications
Journal of Neurology (2023)
-
Melatonin, an endogenous hormone, modulates Th17 cells via the reactive-oxygen species/TXNIP/HIF-1α axis to alleviate autoimmune uveitis
Journal of Neuroinflammation (2022)
-
Diosmetin has therapeutic efficacy in colitis regulating gut microbiota, inflammation, and oxidative stress via the circ-Sirt1/Sirt1 axis
Acta Pharmacologica Sinica (2022)
-
Pectolinarigenin Suppresses LPS-Induced Inflammatory Response in Macrophages and Attenuates DSS-Induced Colitis by Modulating the NF-κB/Nrf2 Signaling Pathway
Inflammation (2022)
