
Molecular Dynamics Simulation of Kir6.2 Variants Reveals Potential Association with Diabetes Mellitus

 , , , ,
, , , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Assessment of the Deleterious Impact of nsSNPs on the KCNJ11 Gene Encodes the Kir6.2 Protein
2.2. Characterizing Disease-Associated nsSNPs
2.3. The Prediction of Protein Stability Consequences
2.4. Predicting Pathogenicity and Functional Outcomes of the KCNJ11 Gene nsSNPs
2.5. Analysis of Sequence Conservation Patterns
2.6. Predict and Analyze the Structural and Functional Properties of Kir6.2 Using the HOPE Server
2.7. MD Simulation of Kir6.2 Protein and Its Mutants
2.7.1. Temperature, Pressure, and Density
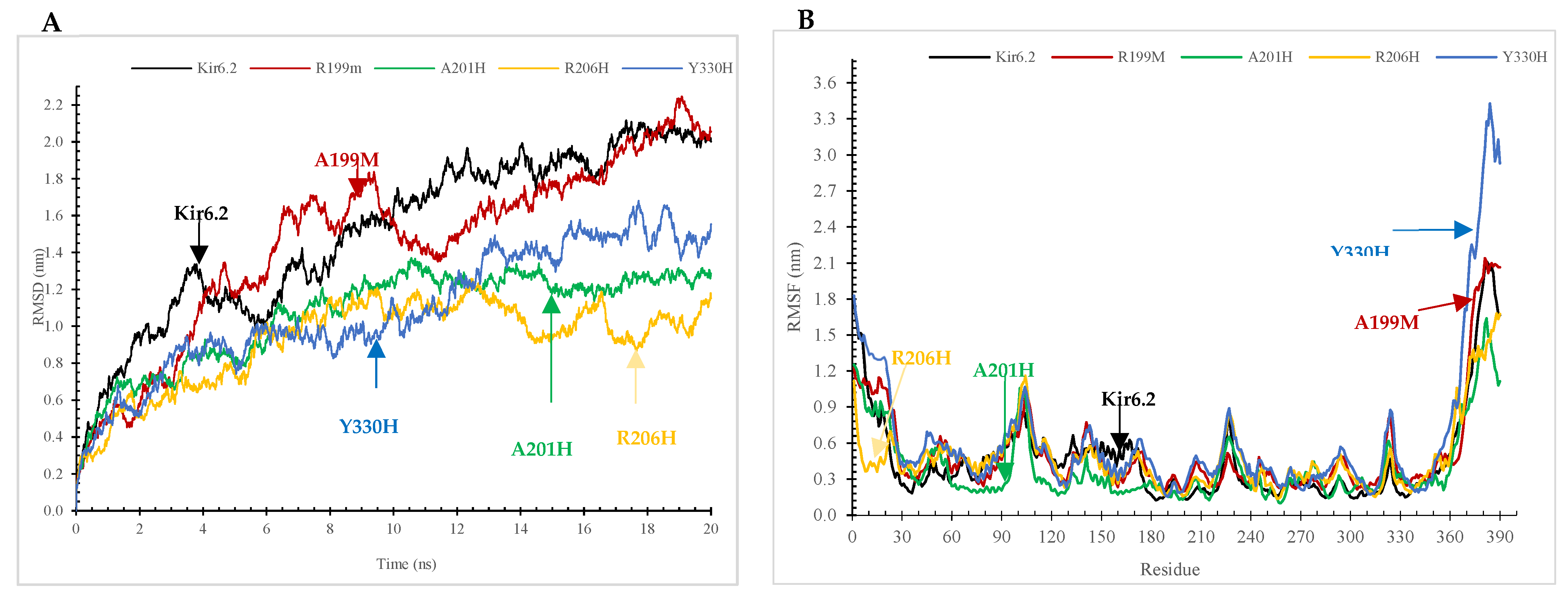
2.7.2. The Root-Mean-Square Deviation (RMSD) and Root-Mean-Square Fluctuation (RMSF) Analyses
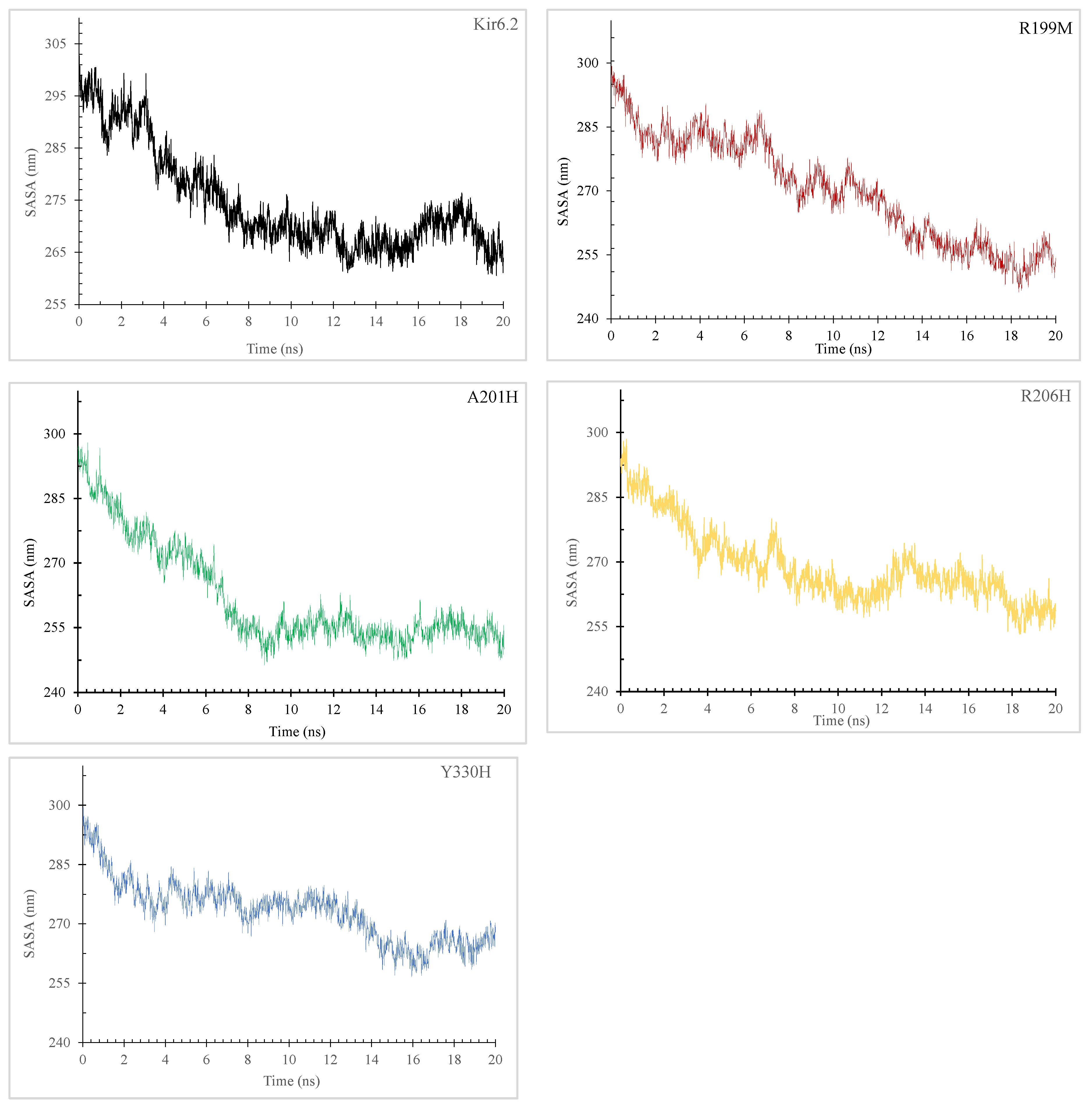
2.7.3. Radius of Gyration (Rg) and Solvent-Accessible Surface Area (SASA)
2.7.4. Dynamic Hydrogen Bonding (HB) Changes in the Kir6.2 Protein Due to Genetic Variations
3. Discussion
4. Conclusions
5. Methodology
5.1. Access to the Database
5.2. Anticipating the Impact of nsSNPs on the Functionality of the KNCJ11 Protein
5.3. Prediction of nsSNP Disease Associations on Kir6.2 Protein
5.4. Predicting Effects of nsSNPs on Kir6.2 Protein Stability
5.5. Prediction of Conserved Residues
5.6. MutPred
5.7. Hope
5.8. PyoMol
5.9. MD Simulations for Kir6.2 Protein and Its Variants
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Sameer, A.S.; Banday, M.Z.; Nissar, S. Pathophysiology of Diabetes: An Overview. Avicenna J. Med. 2020, 10, 174–188. [Google Scholar]
- Son, J.; Accili, D. Reversing Pancreatic Beta-Cell Dedifferentiation in the Treatment of Type 2 Diabetes. Exp. Mol. Med. 2023, 55, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef]
- Oliveira, S.C.; Neves, J.S.; Perez, A.; Carvalho, D. Maturity-Onset Diabetes of the Young: From a Molecular Basis Perspective toward the Clinical Phenotype and Proper Management. Endocrinol. Diabetes Nutr. 2020, 67, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Timsit, J.; Saint-Martin, C.; Dubois-Laforgue, D.; Bellanne-Chantelot, C. Searching for Maturity-Onset Diabetes of the Young (Mody): When and What for? Can. J. Diabetes 2016, 40, 455–461. [Google Scholar] [CrossRef]
- Shimomura, K.; Maejima, Y. K(Atp) Channel Mutations and Neonatal Diabetes. Intern. Med. 2017, 56, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Haghvirdizadeh, P.; Mohamed, Z.; Abdullah, N.A.; Haghvirdizadeh, P.; Haerian, M.S.; Haerian, B.S. Kcnj11: Genetic Polymorphisms and Risk of Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 908152. [Google Scholar] [CrossRef]
- Clement, A.; Guo, S.; Jansen-Olesen, I.; Christensen, S.L. Atp-Sensitive Potassium Channels in Migraine: Translational Findings and Therapeutic Potential. Cells 2022, 11, 2406. [Google Scholar] [CrossRef]
- Huang, Y.; Hu, D.; Huang, C.; Nichols, C.G. Genetic Discovery of Atp-Sensitive K(+) Channels in Cardiovascular Diseases. Circ. Arrhythmia Electrophysiol. 2019, 12, E007322. [Google Scholar] [CrossRef]
- Walczewska-Szewc, K.; Nowak, W. Spacial Models of Malfunctioned Protein Complexes Help to Elucidate Signal Transduction Critical for Insulin Release. Biosystems 2019, 177, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Walczewska-Szewc, K.; Nowak, W. Structural Determinants of Insulin Release: Disordered N-Terminal Tail of Kir6.2 Affects Potassium Channel Dynamics through Interactions with Sulfonylurea Binding Region in a Sur1 Partner. J. Phys. Chem. B 2020, 124, 6198–6211. [Google Scholar] [CrossRef] [PubMed]
- Martin Gm Patton, B.L.; Shyng, S.L. K(Atp) Channels in Focus: Progress toward a Structural Understanding of Ligand Regulation. Curr. Opin. Struct. Biol. 2023, 79, 102541. [Google Scholar]
- Capener, C.E.; Proks, P.; Ashcroft, F.M.; Sansom, M.S. Filter Flexibility in a Mammalian K Channel: Models and Simulations of Kir6.2 Mutants. Biophys. J. 2003, 84, 2345–2356. [Google Scholar] [CrossRef]
- Brundl, M.; Pellikan, S.; Stary-Weinzinger, A. Simulating Pip(2)-Induced Gating Transitions in Kir6.2 Channels. Front. Mol. Biosci. 2021, 8, 711975. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Garon, A.; Wieder, M.; Houtman, M.J.C.; Zangerl-Plessl, E.M.; Langer, T.; Van Der Heyden, M.A.G.; Stary-Weinzinger, A. Computational Identification of Novel Kir6 Channel Inhibitors. Front. Pharmacol. 2019, 10, 549. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Xie, L.; Chao, C.; Kang, Y.; Lin, X.; Qin, T.; Xie, H.; Feng, Z.P.; Gaisano, H.Y. Phosphatidylinositol 4,5-Biphosphate (Pip2) Modulates Interaction of Syntaxin-1a with Sulfonylurea Receptor 1 to Regulate Pancreatic Beta-Cell Atp-Sensitive Potassium Channels. J. Biol. Chem. 2014, 289, 6028–6040. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Tarasov, A.I.; Craig, T.J.; Sansom, M.S.; Ashcroft, F.M. Identification of the Pip2-Binding Site on Kir6.2 by Molecular Modelling and Functional Analysis. Embo J. 2007, 26, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.F.; Kurata, H.T.; Hund, T.J.; Cunha, S.R.; Koval, O.M.; Wright, P.J.; Christensen, M.; Anderson, M.E.; Nichols, C.G.; Mohler, P.J. Dual Role of K Atp Channel C-Terminal Motif in Membrane Targeting and Metabolic Regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 16669–16674. [Google Scholar] [CrossRef]
- Zhao, C.; Mackinnon, R. Molecular Structure of an Open Human K(Atp) Channel. Proc. Natl. Acad. Sci. USA 2021, 118, e2112267118. [Google Scholar] [CrossRef]
- Castro, L.; Noelia, M.; Vidal-Jorge, M.; Sanchez-Ortiz, D.; Gandara, D.; Martinez-Saez, E.; Cicuendez, M.; Poca, M.A.; Simard, J.M.; Sahuquillo, J. Kir6.2, the Pore-Forming Subunit of Atp-Sensitive K(+) Channels, Is Overexpressed in Human Posttraumatic Brain Contusions. J. Neurotrauma 2019, 36, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Szeto, V.; Chen, N.H.; Sun, H.S.; Feng, Z.P. The Role of K(Atp) Channels in Cerebral Ischemic Stroke and Diabetes. Acta Pharmacol. Sin. 2018, 39, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Walczewska-Szewc, K.; Nowak, W. Structural Insights into Atp-Sensitive Potassium Channel Mechanics: A Role of Intrinsically Disordered Regions. J. Chem. Inf. Model. 2023, 63, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.X.; Ding, D.; Wang, M.; Kang, Y.; Zeng, X.; Chen, L. Ligand Binding and Conformational Changes of Sur1 Subunit in Pancreatic Atp-Sensitive Potassium Channels. Protein Cell 2018, 9, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, X.; Cui, J.; Zhao, M.; Yao, H. A Novel Mutation Kcnj11 R136c Caused Kcnj11-Mody. Diabetol. Metab. Syndr. 2021, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Madani Ha Fawzy, N.; Afif, A.; Abdelghaffar, S.; Gohar, N. Study of Kcnj11 Gene Mutations in Association with Monogenic Diabetes of Infancy and Response to Sulfonylurea Treatment in a Cohort Study in Egypt. Acta Endocrinol. 2016, 12, 157–160. [Google Scholar]
- Vedovato, N.; Cliff, E.; Proks, P.; Poovazhagi, V.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Ashcroft, F.M. Neonatal Diabetes Caused by a Homozygous Kcnj11 Mutation Demonstrates That Tiny Changes in Atp Sensitivity Markedly Affect Diabetes Risk. Diabetologia 2016, 59, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, P.; Girard, C.; Molnes, J.; Njolstad, P.R.; Ashcroft, F.M. Kir6.2 Mutations Causing Neonatal Diabetes Provide New Insights into Kir6.2-Sur1 Interactions. Embo J. 2005, 24, 2318–2330. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef]
- Galaviz, K.I.; Narayan, K.M.V.; Lobelo, F.; Weber, M.B. Lifestyle and the Prevention of Type 2 Diabetes: A Status Report. Am. J. Lifestyle Med. 2018, 12, 4–20. [Google Scholar] [CrossRef]
- Moazzam-Jazi, M.; Najd-Hassan-Bonab, L.; Masjoudi, S.; Tohidi, M.; Hedayati, M.; Azizi, F.; Daneshpour, M.S. Risk of Type 2 Diabetes and Kcnj11 Gene Polymorphisms: A Nested Case-Control Study and Meta-Analysis. Sci. Rep. 2022, 12, 20709. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for Mody Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.M. Maturity-Onset Diabetes of the Young: Update and Perspectives on Diagnosis and Treatment. Yeungnam Univ. J. Med. 2020, 37, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M. Pancreatic Beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Aftabuddin, M.; Kundu, S. Hydrophobic, Hydrophilic, and Charged Amino Acid Networks within Protein. Biophys. J. 2007, 93, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Strub, C.; Alies, C.; Lougarre, A.; Ladurantie, C.; Czaplicki, J.; Fournier, D. Mutation of Exposed Hydrophobic Amino Acids to Arginine to Increase Protein Stability. BMC Biochem. 2004, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Zervou, M.I.; Andreou, A.; Matalliotakis, M.; Spandidos, D.A.; Goulielmos, G.N.; Eliopoulos, E.E. Association of the Dnase1l3 Rs35677470 Polymorphism with Systemic Lupus Erythematosus, Rheumatoid Arthritis and Systemic Sclerosis: Structural Biological Insights. Mol. Med. Rep. 2020, 22, 4492–4498. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Saha, R.P.; Samanta, U.; Chakrabarti, P. Geometry of Interaction of the Histidine Ring with Other Planar and Basic Residues. J. Proteome Res. 2003, 2, 255–263. [Google Scholar] [CrossRef]
- Shin, S.J.; Lee, S.J.; Kim, S.K. Frequency of Gnas R201h Substitution Mutation in Polyostotic Fibrous Dysplasia: Pyrosequencing Analysis in Tissue Samples with or without Decalcification. Sci. Rep. 2017, 7, 2836. [Google Scholar] [CrossRef]
- Marusiak, A.A.; Stephenson, N.L.; Baik, H.; Trotter, E.W.; Li, Y.; Blyth, K.; Mason, S.; Chapman, P.; Puto La Read, J.A.; Read, J.A.; et al. Recurrent Mlk4 Loss-of-Function Mutations Suppress Jnk Signaling to Promote Colon Tumorigenesis. Cancer Res. 2016, 76, 724–735. [Google Scholar] [CrossRef]
- De Franco, E.; Saint-Martin, C.; Brusgaard, K.; Knight Johnson, A.E.; Aguilar-Bryan, L.; Bowman, P.; Arnoux, J.B.; Larsen, A.R.; Sanyoura, M.; Greeley, S.A.W.; et al. Update of Variants Identified in the Pancreatic Beta-Cell K(Atp) Channel Genes Kcnj11 and Abcc8 in Individuals with Congenital Hyperinsulinism and Diabetes. Hum. Mutat. 2020, 41, 884–905. [Google Scholar] [CrossRef]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P.J.; Ashcroft, F.M. New Insights into K(Atp) Channel Gene Mutations and Neonatal Diabetes Mellitus. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Sagen, J.V.; Raeder, H.; Hathout, E.; Shehadeh, N.; Gudmundsson, K.; Baevre, H.; Abuelo, D.; Phornphutkul, C.; Molnes, J.; Bell, G.I.; et al. Permanent Neonatal Diabetes Due to Mutations in Kcnj11 Encoding Kir6.2: Patient Characteristics and Initial Response to Sulfonylurea Therapy. Diabetes 2004, 53, 2713–2718. [Google Scholar] [CrossRef]
- Vaxillaire, M.; Populaire, C.; Busiah, K.; Cave, H.; Gloyn, A.L.; Hattersley At Czernichow, P.; Froguel, P.; Polak, M. Kir6.2 Mutations Are a Common Cause of Permanent Neonatal Diabetes in a Large Cohort of French Patients. Diabetes 2004, 53, 2719–2722. [Google Scholar] [CrossRef]
- Ghahremanian, S.; Rashidi Mm Raeisi, K.; Toghraie, D. Molecular Dynamics Simulation Approach for Discovering Potential Inhibitors against SARS-CoV-2: A Structural Review. J. Mol. Liq. 2022, 354, 118901. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, D.; Perodeau, J.; Nieuwkoop, A.J.; Oschkinat, H. Mas Nmr Detection of Hydrogen Bonds for Protein Secondary Structure Characterization. J. Biomol. Nmr. 2020, 74, 247–256. [Google Scholar] [CrossRef]
- Prabantu Vm Naveenkumar, N.; Srinivasan, N. Influence of Disease-Causing Mutations on Protein Structural Networks. Front. Mol. Biosci. 2020, 7, 620554. [Google Scholar]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive Characterization of Protein-Protein Interactions Perturbed by Disease Mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef]
- Razban, R.M.; Shakhnovich, E.I. Effects of Single Mutations on Protein Stability Are Gaussian Distributed. Biophys. J. 2020, 118, 2872–2878. [Google Scholar] [CrossRef]
- Alzahrani, O.R.; Mir, R.; Alatwi, H.E.; Hawsawi, Y.M.; Alharbi, A.A.; Alessa, A.H.; Albalawi, E.S.; Elfaki, I.; Alalawi, Y.; Moharam, L.; et al. Potential Impact of Pi3k-Akt Signaling Pathway Genes, Klf-14, Mdm4, Mirnas 27a, Mirna-196a Genetic Alterations in the Predisposition and Progression of Breast Cancer Patients. Cancers 2023, 15, 1281. [Google Scholar] [CrossRef]
- Lind, L. Genome-Wide Association Study of the Metabolic Syndrome in UK Biobank. Metab. Syndr. Relat. Disord. 2019, 17, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Elfaki, I.; Mir, R.; Almutairi, F.M.; Duhier, F.M.A. Cytochrome P450: Polymorphisms and Roles in Cancer, Diabetes and Atherosclerosis. Asian Pac. J. Cancer Prev. 2018, 19, 2057–2070. [Google Scholar] [PubMed]
- Elfaki, I.; Mir, R.; Abu-Duhier, F.M.; Jha, C.K.; Ahmad Al-Alawy, A.I.; Babakr, A.T.; Habib, S.A.E. Analysis of the Potential Association of Drug-Metabolizing Enzymes Cyp2c9*3 and Cyp2c19*3 Gene Variations with Type 2 Diabetes: A Case-Control Study. Curr. Drug Metab. 2020, 21, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Pipatpolkai, T.; Corey, R.A.; Proks, P.; Ashcroft, F.M.; Stansfeld, P.J. Evaluating Inositol Phospholipid Interactions with Inward Rectifier Potassium Channels and Characterising Their Role in Disease. Commun. Chem. 2020, 3, 147. [Google Scholar] [CrossRef] [PubMed]
- Gerasimavicius, L.; Livesey, B.J.; Marsh, J.A. Loss-of-Function, Gain-of-Function and Dominant-Negative Mutations Have Profoundly Different Effects on Protein Structure. Nat. Commun. 2022, 13, 3895. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Anishchenko, I.; Bordin, N.; Sillitoe, I.; Velankar, S.; Baker, D.; Orengo, C. Characterizing and Explaining the Impact of Disease-Associated Mutations in Proteins without Known Structures or Structural Homologs. Brief Bioinform. 2022, 23, bbac187. [Google Scholar] [CrossRef] [PubMed]
- Elfaki, I.; Knitsch, A.; Matena, A.; Bayer, P. Identification and Characterization of Peptides That Bind the Ppiase Domain of Parvulin17. J. Pept. Sci. 2013, 19, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using Polyphen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Patch Am Ellard, S. Using Sift and Polyphen to Predict Loss-of-Function and Gain-of-Function Mutations. Genet. Test. Mol. Biomarkers 2010, 14, 533–537. [Google Scholar] [CrossRef]
- Johnson, A.D.; Handsaker, R.E.; Pulit, S.L.; Nizzari, M.M.; O‘donnell, C.J.; De Bakker, P.I. Snap: A Web-Based Tool for Identification and Annotation of Proxy Snps Using Hapmap. Bioinformatics 2008, 24, 2938–2939. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. Panther Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive Api. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R.; Ws-Snps, G.O. A Web Server for Predicting the Deleterious Effect of Human Protein Variants Using Functional Annotation. BMC Genom. 2013, 14, S6. [Google Scholar] [CrossRef] [PubMed]
- Akhoundi, F.; Parvaneh, N.; Modjtaba, E.B. In Silico Analysis of Deleterious Single Nucleotide Polymorphisms in Human Bub1 Mitotic Checkpoint Serine/Threonine Kinase B Gene. Meta Gene 2016, 9, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, H.V.; Babu, P.R.; Subbiah, U. Determination of Deleterious Single-Nucleotide Polymorphisms of Human Lyz C Gene: An In Silico Study. J. Genet. Eng. Biotechnol. 2022, 20, 92. [Google Scholar]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional Annotations Improve the Predictive Score of Human Disease-Related Mutations in Proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, C.; Lv, H.; Zhang, M.; Li, X. In Silico Analysis and High-Risk Pathogenic Phenotype Predictions of Non-Synonymous Single Nucleotide Polymorphisms in Human Crystallin Beta A4 Gene Associated with Congenital Cataract. PLoS ONE 2020, 15, E0227859. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Consurf 2016: An Improved Methodology to Estimate and Visualize Evolutionary Conservation in Macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the Molecular and Phenotypic Impact of Amino Acid Variants with Mutpred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein Structure Analysis of Mutations Causing Inheritable Diseases. An E-Science Approach with Life Scientist Friendly Interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. Charmm General Force Field: A Force Field for Drug-like Molecules Compatible with the Charmm All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SIFT | Polyphen | SNAP2 | PANTHER | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variant ID | Alleles | nsSNP | Predication | Score | Score | Predication | Predication | Accuracy | Predication | Pdel |
| rs74339576 | C>A | R301L | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs74339576 | C>G | R301P | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs74339576 | C>T | R301H | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs80356621 | T>C | K170R | deleterious | 0 | 0.997 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs80356622 | C>G | K170N | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs80356624 | C>A | R201L | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs80356624 | C>T | R201H | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs80356625 | G>A | R201C | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs104894237 | G>A | P254L | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs104894248 | T>C | H259R | deleterious | 0 | 0.998 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs377091338 | G>A | R301C | deleterious | 0 | 1 | probably damaging | Effect | 85% | probably damaging | 0.85 |
| rs377091338 | G>C | R301G | deleterious | 0.01 | 1 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs387906783 | A>G | F60S | deleterious | 0.2 | 1 | probably damaging | Effect | 85% | probably damaging | 0.89 |
| rs587783672 | C>T | E227K | deleterious | 0 | 1 | probably damaging | Effect | 85% | probably damaging | 0.85 |
| rs587783675 | A>G | Y330H | deleterious | 0.04 | 1 | probably damaging | Effect | 85% | probably damaging | 0.85 |
| rs750778014 | C>A | R192L | deleterious | 0 | 1 | probably damaging | Effect | 91% | probably damaging | 0.85 |
| rs761575495 | G>A | T302I | deleterious | 0.01 | 1 | probably damaging | Effect | 85% | probably damaging | 0.57 |
| rs761575495 | G>T | T302N | deleterious | 0.01 | 1 | probably damaging | Effect | 85% | probably damaging | 0.57 |
| rs775204908 | G>A | R206C | deleterious | 0 | 1 | probably damaging | Effect | 85% | probably damaging | 0.85 |
| rs797045637 | C>A | G289V | deleterious | 0.01 | 1 | probably damaging | Effect | 91% | probably damaging | 0.89 |
| rs1174593640 | C>T | E292K | deleterious | 0 | 0.997 | probably damaging | Effect | 91% | probably damaging | 0.85 |
| rs1404429785 | C>T | G156R | deleterious | 0 | 1 | probably damaging | Effect | 91% | probably damaging | 0.89 |
| rs1437510576 | C>A | S265I | deleterious | 0 | 1 | probably damaging | Effect | 91% | probably damaging | 0.89 |
| rs1554901747 | C>A | R206L | deleterious | 0 | 1 | probably damaging | Effect | 91% | probably damaging | 0.85 |
| rs2133379125 | A>G | F315S | deleterious | 0 | 1 | probably damaging | Effect | 85% | probably damaging | 0.89 |
| rs80356621 | T>C | K170R | deleterious | 0 | 0.997 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs80356622 | C>G | K170N | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.85 |
| rs80356624 | C>A | R201L | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs80356624 | C>T | R201H | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs80356625 | G>A | R201C | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs764444072 | C>G | A87P | deleterious | 0 | 0.998 | probably damaging | Effect | 85% | probably damaging | 0.57 |
| rs770553801 | A>G | F60L | deleterious | 0.02 | 1 | probably damaging | Effect | 85% | probably damaging | 0.89 |
| rs780511484 | G>A | R192C | deleterious | 0 | 1 | probably damaging | Effect | 85% | probably damaging | 0.85 |
| rs1435239409 | T>C | K170E | deleterious | 0 | 1 | probably damaging | Effect | 85% | probably damaging | 0.85 |
| rs1474444717 | A>C | W68G | deleterious | 0 | 1 | probably damaging | Effect | 91% | probably damaging | 0.89 |
| rs1591694925 | T>G | Y304S | deleterious | 0 | 1 | probably damaging | Effect | 95% | probably damaging | 0.89 |
| rs1591694946 | T>G | T302P | deleterious | 0.01 | 1 | probably damaging | Effect | 91% | probably damaging | 0.57 |
| rs1953581831 | A>C | M199R | deleterious | 0 | 0.997 | probably damaging | Effect | 85% | probably damaging | 0.85 |
| rs1953584995 | C>T | G165D | deleterious | 0 | 1 | probably damaging | Effect | 91% | probably damaging | 0.89 |
| rs1953585779 | T>C | Q152R | deleterious | 0 | 0.997 | probably damaging | Effect | 91% | probably damaging | 0.85 |
| rs1953586783 | C>G | G134A | deleterious | 0 | 0.998 | probably damaging | Effect | 91% | probably damaging | 0.89 |
| PhD-SNP | SNP&Go | |||||
|---|---|---|---|---|---|---|
| Variant ID | Alleles | nsSNP | Prediction | Score | Predication | Score |
| rs74339576 | C>A | R301L | Disease | 1 | Disease | 10 |
| rs80356622 | C>G | K170N | Disease | 0 | Disease | 9 |
| rs80356624 | C>A | R201L | Disease | 3 | Disease | 10 |
| rs80356624 | C>T | R201H | Disease | 5 | Disease | 9 |
| rs80356625 | G>A | R201C | Disease | 3 | Disease | 9 |
| rs104894237 | G>A | P254L | Disease | 0 | Disease | 9 |
| rs104894248 | T>C | H259R | Disease | 2 | Disease | 9 |
| rs587783672 | C>T | E227K | Disease | 7 | Disease | 9 |
| rs587783675 | A>G | Y330H | Disease | 4 | Disease | 9 |
| rs775204908 | G>A | R206C | Disease | 1 | Disease | 9 |
| rs797045637 | C>A | G289V | Disease | 2 | Disease | 9 |
| rs1404429785 | C>T | G156R | Disease | 2 | Disease | 9 |
| rs2133379125 | A>G | F315S | Disease | 4 | Disease | 9 |
| rs80356622 | C>G | K170N | Disease | 0 | Disease | 9 |
| rs80356624 | C>A | R201L | Disease | 3 | Disease | 10 |
| rs80356624 | C>T | R201H | Disease | 5 | Disease | 9 |
| rs80356625 | G>A | R201C | Disease | 3 | Disease | 9 |
| rs764444072 | C>G | A87P | Disease | 3 | Disease | 8 |
| rs780511484 | G>A | R192C | Disease | 3 | Disease | 9 |
| rs1435239409 | T>C | K170E | Disease | 0 | Disease | 9 |
| rs1591694946 | T>G | T302P | Disease | 2 | Disease | 9 |
| rs1953581831 | A>C | M199R | Disease | 5 | Disease | 9 |
| rs1953584995 | C>T | G165D | Disease | 4 | Disease | 9 |
| rs1953585779 | T>C | Q152R | Disease | 5 | Disease | 9 |
| Variant ID | Alleles | nsSNP | I-Mutant | MUpro |
|---|---|---|---|---|
| rs80356624 | C>T | R201H | Decrease | Decrease |
| rs80356625 | G>A | R201C | Decrease | Decrease |
| rs587783675 | A>G | Y330H | Decrease | Decrease |
| rs775204908 | G>A | R206C | Decrease | Decrease |
| rs2133379125 | A>G | F315S | Decrease | Decrease |
| rs780511484 | G>A | R192C | Decrease | Decrease |
| rs1953581831 | A>C | M199R | Decrease | Decrease |
| rs1953585779 | T>C | Q152R | Decrease | Decrease |
| MutPred | |||||
|---|---|---|---|---|---|
| Variant ID | Alleles | nsSNP | Effect | Score | Function Affected |
| rs80356624 | C>T | R201H | - | 0.914 | Loss of allosteric site at R201; altered DNA binding; altered metal binding |
| rs80356625 | G>A | R201C | - | 0.948 | Loss of allosteric site at R201; altered DNA binding |
| rs587783675 | A>G | Y330H | - | 0.868 | Loss of acetylation at K332; loss of sulfation at Y330; altered metal binding; altered stability |
| rs775204908 | G>A | R206C | - | 0.949 | Altered DNA binding |
| rs2133379125 | A>G | F315S | - | 0.946 | Loss of allosteric site at F315; altered ordered interface |
| rs780511484 | G>A | R192C | - | 0.897 | Loss of strand; altered metal binding |
| rs1953581831 | A>C | M199R | - | 0.957 | Loss of allosteric site at R201; altered DNA binding; altered stability; altered disordered interface |
| rs1953585779 | T>C | Q152R | no | 0.903 | No effect detected |
| Variant ID | Alleles | nsSNP | SIFT | PolyPhen | SNAP2 | PANTHER | PhD-SNP | SNP&Go | I-Mutant | MUPro | ConSurf | MutPred |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs1953585779 | T>C | Q152R | * | * | * | * | * | * | * | * | Cons, 9, BS | * |
| rs780511484 | G>A | R192C | * | * | * | * | * | * | * | * | Cons, 8, EF | * |
| rs1953581831 | A>C | M199R | * | * | * | * | * | * | * | * | Cons, 8, B | * |
| rs80356624 | C>T | R201H | * | * | * | * | * | * | * | * | Cons, 9, BS | * |
| rs80356625 | G>A | R201C | * | * | * | * | * | * | * | * | Cons, 9, BS | * |
| rs775204908 | G>A | R206C | * | * | * | * | * | * | * | * | Cons, 9, EF | * |
| rs2133379125 | A>G | F315S | * | * | * | * | * | * | * | * | Cons, 9, BS | * |
| rs587783675 | A>G | Y330H | * | * | * | * | * | * | * | * | Cons, 7, B | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elangeeb, M.E.; Elfaki, I.; Eleragi, A.M.S.; Ahmed, E.M.; Mir, R.; Alzahrani, S.M.; Bedaiwi, R.I.; Alharbi, Z.M.; Mir, M.M.; Ajmal, M.R.; et al. Molecular Dynamics Simulation of Kir6.2 Variants Reveals Potential Association with Diabetes Mellitus. Molecules 2024, 29, 1904. https://doi.org/10.3390/molecules29081904
Elangeeb ME, Elfaki I, Eleragi AMS, Ahmed EM, Mir R, Alzahrani SM, Bedaiwi RI, Alharbi ZM, Mir MM, Ajmal MR, et al. Molecular Dynamics Simulation of Kir6.2 Variants Reveals Potential Association with Diabetes Mellitus. Molecules. 2024; 29(8):1904. https://doi.org/10.3390/molecules29081904
Chicago/Turabian StyleElangeeb, Mohamed E., Imadeldin Elfaki, Ali M. S. Eleragi, Elsadig Mohamed Ahmed, Rashid Mir, Salem M. Alzahrani, Ruqaiah I. Bedaiwi, Zeyad M. Alharbi, Mohammad Muzaffar Mir, Mohammad Rehan Ajmal, and et al. 2024. "Molecular Dynamics Simulation of Kir6.2 Variants Reveals Potential Association with Diabetes Mellitus" Molecules 29, no. 8: 1904. https://doi.org/10.3390/molecules29081904