
Abstract
The solvent-free selective hydrogenation of nitroaromatics to azoxy compounds is highly important, yet challenging. Herein, we report an efficient strategy to construct individually dispersed Co atoms decorated on niobium pentaoxide nanomeshes with unique geometric and electronic properties. The use of this supported Co single atom catalysts in the selective hydrogenation of nitrobenzene to azoxybenzene results in high catalytic activity and selectivity, with 99% selectivity and 99% conversion within 0.5 h. Remarkably, it delivers an exceptionally high turnover frequency of 40377 h–1, which is amongst similar state-of-the-art catalysts. In addition, it demonstrates remarkable recyclability, reaction scalability, and wide substrate scope. Density functional theory calculations reveal that the catalytic activity and selectivity are significantly promoted by the unique electronic properties and strong electronic metal-support interaction in Co1/Nb2O5. The absence of precious metals, toxic solvents, and reagents makes this catalyst more appealing for synthesizing azoxy compounds from nitroaromatics. Our findings suggest the great potential of this strategy to access single atom catalysts with boosted activity and selectivity, thus offering blueprints for the design of nanomaterials for organocatalysis.
Similar content being viewed by others

Introduction
Selective hydrogenation of nitrobenzene is an important reaction used to generate valued chemicals including nitrosobenzene, phenylhydroxylamine, aniline, azobenzene, and azoxybenzene1,2. Of these, azoxybenzene and its derivatives are a class of compounds that have many potential applications in dyes, pharmaceuticals, polymerization inhibitors, and food additives3. However, the complex reduction steps and low selectivity to azoxybenzene make this reaction challenging4,5. Therefore, the design of a highly selective catalyst with moderate catalytic reduction abilities towards azoxybenzene is appealing. Among most of the metals, palladium, iridium, and rhodium are typically used as active catalysts for selective hydrogenation reactions6,7,8. However, the high cost has greatly restricted the wide use of these precious metal-based catalysts. Moreover, strong bases or expensive organic reducing agents are typically employed in the reaction9. Therefore, the development of sustainable, environmentally benign, and low-cost catalysts for the selective hydrogenation of nitroaromatics to azoxy compounds is highly desirable.
Selectivity and activity in catalysis are important for efficiently producing commodity chemicals, fine chemicals, and pharmaceuticals10. Both factors are determined by the adsorption characteristics and activation ability of catalytically active sites toward reactants, intermediates, and products. These in turn are influenced by the geometric and electronic properties of these sites11,12. In homogeneous catalysis, the properties of these sites may be effectively tuned by steric and electronic structures; however, fine-tuning the selectivity in heterogeneous catalysis is challenging10,13,14. In addition to the catalytic activity-selectivity relationship, solvent waste removal also poses a formidable challenge for green chemical synthesis and energy consumption15. The use of solvent-free mechanical methods (grinding or milling) for reagents mixing and activation holds many advantages, such as shortened reaction periods, mild reaction conditions, and excellent selectivity to the target products16,17,18. Therefore, there is a strong incentive to construct highly active and selective catalyst systems that do not require the use of solvent to efficiently hydrogenate nitroaromatics to the corresponding azoxy compounds using H219,20.
Recent years have witnessed the fast development of single atom catalysts (SACs) with unique coordination environments, high atom utilization, and appealing catalytic efficacy in a number of chemical reactions14,21,22. Notably, SACs possess almost 100% atomic dispersion and high activity for each active metal sites22,23. In addition, they can elegantly bridge heterogeneous and homogeneous catalysis to endow exceptional activity, selectively, and stability24,25. The catalytic performance of SACs can be improved by adjusting the coordination environment and electronic properties of metal active sites, which are generally influenced by synthetic methods and support materials26,27,28,29,30. Modulating the electron coupling between metals and supports can effectively regulate the electronic structure of metal sites for improved catalytic efficacy31,32. Niobium pentaoxide (Nb2O5) is an important catalyst support material due to its high stability, moderate acidity, and excellent C–O and C–C bond cleavage ability33,34. Nb2O5-supported metal catalysts have found applications in a variety of catalytic reactions, including hydrodeoxygenation, C–O bond activation, Caromatic–C bonds cleavage, and aldol condensation35,36,37. However, the use of Nb2O5 as a support material for decoration of isolated non-precious metal atoms in organocatalysis has been rarely reported.
Herein, we report a facile and reliable strategy to synthesize atomically dispersed Co atoms anchored on niobium pentaoxide (Co1/Nb2O5) nanomeshes. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM), X-ray absorption spectroscopy (XAFS), and X-ray photoelectron spectroscopy (XPS) confirm the Co atoms in Co1/Nb2O5 are atomically dispersed and positively charged. This non-precious metal-based catalyst delivers exceptional catalytic efficacy on solvent-free selective hydrogenation of nitroaromatics to azoxy compounds under base-free and solvent-free conditions (Fig. 1). The as-prepared Co1/Nb2O5 has the potential to bypass the limitations of previously reported catalysts and enable rapid and efficient access to a diverse range of azoxy compounds from functionalized nitroaromatics. Theoretical studies reveal that the unique electronic structure and strong electronic metal-support coupling effect between Co atoms with support atoms in close proximity are beneficial for the efficient activation of reactants.

a Oxidation of anilines into aromatic azoxy compounds. b Reduction of nitroarenes to aromatic azoxy compounds. c Hydrogenation of nitroarenes to aromatic azoxy compounds with noble metal catalyst in the presence of base. d This work: Direct hydrogenation of nitroarenes into aromatic azoxy compounds with non-noble metal catalysts under base-free and solvent-free conditions.
Results
Synthesis and characterization of atomically dispersed Co catalyst
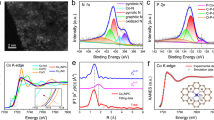
An efficient two-step strategy (Fig. 2a) incorporating incipient wetness impregnation and microwave irradiation procedures was developed to create individually dispersed cobalt atoms over niobium pentaoxide nanomeshes (Co1/Nb2O5). Typically, Nb2O5 was prepared by reacting ammonium niobate oxalate hydrate, melamine, and ammonium chloride in ethanol, followed by a calcination step in air. Electron microscopy characterization demonstrates that the as-prepared Nb2O5 possesses a nanomesh structure (Supplementary Figs. 1 and 2). Subsequently, the as-prepared Nb2O5 was homogeneously mixed with cobalt acetate aqueous solution by an incipient wetness impregnation approach (Co2+@Nb2O5). After drying by an infrared lamp, the dried powder was microwave-treated at 800 W for 10 s to obtain Co1/Nb2O5 with cobalt loading of 0.42 wt% and cobalt dispersion of 97% (Supplementary Table 1). This suggests that the vast majority of Co species were in the form of isolated Co atoms in Co1/Nb2O5. Similar nanomesh morphologies between Co1/Nb2O5 and Nb2O5 are observed by electron microscopy characterizations (Fig. 2b–d and Supplementary Fig. 3). An average height of ~6 nm is observed for Co1/Nb2O5 as measured by atomic force microscope (AFM) imaging (Fig. 2d inset). Aberration-corrected HAADF-STEM imaging as shown in Fig. 2e displays lattice fringe spacings of 0.393 and 0.315 nm which correspond to the (001) and (180) facets of Nb2O5. The corresponding ring-like selected area electron diffraction (SAED) pattern is shown in Fig. 2e inset and agrees well with the previous reports38,39. A magnified AC HAADF-STEM image reveals that the isolated Co atoms are distributed over the Nb2O5 surface (Fig. 2f). Note that the cobalt atom has a lower Z contrast relative to niobium atoms. Additionally, the enlarged area provides preliminary evidence for the presence of isolated Co atoms over the Nb2O5 surface (Fig. 2f inset). Energy-dispersive X-ray spectroscopy (EDS) analysis demonstrates the homogeneously distributed O, Co, and Nb over Nb2O5 surface (Fig. 2g). By contrast, a 5.12 wt% Co NPs-containing catalyst (Co NPs/Nb2O5, Co dispersion of 39%) was created with large metallic Co species (Supplementary Fig. 4 and Table 1).

a Schematic illustration of the synthesis strategy. b SEM image. c TEM image. d HR-TEM image (the inset is an AFM image showing the height profile). e AC HAADF-STEM image (the inset is SAED pattern). f AC HAADF-STEM image at high magnification. The red, blue, and purple atoms represent O, Nb, and Co, respectively. g EDS mapping images.
X-ray diffraction (XRD) patterns of samples are exhibited in Fig. 3a. Typical diffraction peaks of Nb2O5 (JCPDS No. 30-0873) agree well with SAED patterns as shown in Fig. 2e. Moreover, there are no evident metallic cobalt peaks found in Co1/Nb2O5, suggesting that these Co species are highly dispersed on the Nb2O5 surface, even for Co NPs/Nb2O5. Raman spectra are shown in Fig. 3b. The overlapped Raman signals are categorized into three band groups40,41: a high-wavenumber (νHi, 485 cm−1 ~ 808 cm−1), a mid-wavenumber (νMid, 175 ~ 370 cm−1), and a low-wavenumber band group (νLo, 93 ~ 172 cm−1). The peaks in the 100 ~ 400 cm–1 region are associated with typical bending modes of Nb–O–Nb linkages41. After Co deposition, the intensities of peaks at 120 cm–1 in Co1/Nb2O5 and Co NPs/Nb2O5 are enhanced, while the peaks at 230 cm–1 are weakened. This suggests the formation of Co–O–Nb linkages and disordering of the bending modes in Nb2O541. In addition, the initially high-intensity peak at 690 cm–1 is reduced significantly due to the Co deposition partially distorting the Nb2O5 structure41,42. X-ray photoelectron spectroscopy (XPS) was conducted to understand the electronic properties of samples (Fig. 3c and Supplementary Fig. 5). Compared with the Co NPs/Nb2O5 sample (Fig. 3c), the Co 2p3/2 peak of Co1/Nb2O5 is located at 781.1 eV, agreeing well with assignment as a positively charged Co species43,44. The O 1 s and Nb 3d spectra of Co1/Nb2O5 and Co NPs/Nb2O5 are shown in Supplementary Fig. 5c. We observe surface oxygen vacancies in both samples, indicating the presence of electronic coupling interaction between the introduced Co species and the Nb2O5 support45. The Nb 3d spectra of both samples can be fitted to two peaks for Nb 3d5/2 and Nb 3d3/2, respectively. The position of the Nb 3d5/2 peak of Co1/Nb2O5 is centered at 207.1 eV, which is slightly lower than that of Co NPs/Nb2O5 (207.3 eV), suggesting a relatively larger concentration of Nb4+ species and oxygen vacancies over the former46. Nonetheless, the chemical shift suggests that only a small portion of the niobium present in either sample is in the form of Nb4+ species. In situ CO-diffuse reflectance infrared Fourier transform spectroscopy (CO-DRIFTS) was performed to investigate atomic structures of Co species in Co1/Nb2O5 and Co NPs/Nb2O5 (Fig. 3d). The broad bands around 2171 cm−1 and 2115 cm−1 are ascribed to residual gas-phase CO molecules47,48. For Co1/Nb2O5, a pair of CO adsorption peaks were observed at 2089 cm−1 and 2067 cm−1, respectively, which could be assigned to linearly adsorbed CO on positively charged Co species49,50. The absence of other peaks excludes the presence of Co multiatomic species in Co1/Nb2O5. In the case of Co NPs/Nb2O5, two peaks appeared at 2015 cm−1 and 1984 cm−1, respectively, which are associated with the CO adsorbing on metallic Co in linear and bridge form (Supplementary Fig. 6)51. Electron paramagnetic resonance (EPR) spectra (Supplementary Fig. 7) demonstrate a sharp signal at a g value of 2.003 for Co1/Nb2O5, which is associated with the coordinatively unsaturated Co species in the catalyst45. H2-temperature-programmed reduction (H2-TPR) results are shown in Supplementary Fig. 8. Two peaks at 508 °C and 588 °C are observed for Nb2O5, corresponding to surface Nb–O–Nb and interior Nb–O–Nb sites46. After introducing Co species, only two peaks at 341 °C and 484 °C are observed, which are associated with the reduction of Co–O–Nb and interior Nb–O–Nb species, respectively. This suggests Nb2O5 can serve as a heterogeneous support for anchoring the isolated Co atoms and restricting them from agglomeration.

a XRD patterns. b Raman spectra. c XPS Co 2p spectra. d In situ CO-DRIFTS of Co1/Nb2O5. e O K-edge spectra. f XANES spectra at Co K-edge. g Fourier transformed k3-weighted Co K-edge of EXAFS spectra. h EXAFS fitting in R space (inset is the model of Co1–O4). i 3D contour WT-EXAFS plot.
To understand the chemical state of Co species in the catalysts, Co L2,3-edge near-edge X-ray absorption fine structure (NEXAFS) results were collected with synchrotron-based soft X-ray radiation. As shown in Supplementary Fig. 9, the peak of the Co L-edge spectrum in Co1/Nb2O5 displays a positive shift of 0.9 eV relative to that of Co NPs/Nb2O5, indicating a higher valence state of Co in Co1/Nb2O552,53. This is in good agreement with the XPS results as discussed in Fig. 3c. O K-edge NEXAFS spectra (Fig. 3e) exhibit a lowered 4dt2g peak intensity for O–Nb coupling after deposition of Co species in Nb2O5. These results, in conjunction with the XPS O 1 s spectra, demonstrate that the Co1/Nb2O5 and Co NPs/Nb2O5 interfaces facilitate the formation of oxygen vacancies in these catalysts54. Synchrotron radiation-based ultraviolet photoemission spectroscopy (UPS) was performed to study the electronic state of Co1/Nb2O5 and Co NPs/Nb2O5. As displayed in Supplementary Fig. 10, the values of valence band maxima for Co1/Nb2O5 and Co NPs/Nb2O5 are determined to be 2.27 eV and 2.56 eV, respectively. This indicates a change in the electron arrangement of Co 3d orbitals, which is associated with the metal-support interaction and the coordination environment. Because the valence electrons near the Fermi level contribute greatly to the d states, the change in the valence band signifies the movement of the d band center. Fourier-transform infrared (FT-IR) spectra of samples are shown in Supplementary Fig. 11 and signals typical of Nb–O–Nb and Nb=O are observed. N2 adsorption/desorption isotherms (Supplementary Fig. 12 and Table 1) show the specific surface area of Nb2O5, Co1/Nb2O5, and Co NPs/Nb2O5 are determined to be 49.8, 56.7, and 80.0 m2/g, respectively. TGA/DSC results show similar results for Co1/Nb2O5 and Co NPs/Nb2O5 (Supplementary Fig. 13). This implies that the incorporation of Co species into Nb2O5 did not significantly affect the material properties.
The atomic dispersion and coordination information of Co species in Co1/Nb2O5 were examined by synchrotron-radiation X-ray absorption fine structure spectroscopy (XAFS). As disclosed in Fig. 3f, the pre-edge of Co K-edge in Co1/Nb2O5 is located between those of CoO and Co3O4, and closer to that of CoO. This indicates the valence state of Co species is between Co2+ and Co3+, though closer to Co2+. The positively charged Co species result from the strong charge transfer between atomically dispersed Co species and the Nb2O5 support55. In the k-space of the extended X-ray absorption fine structure spectra (EXAFS), Co1/Nb2O5 displays a different pattern compared with Co foil (Supplementary Fig. 14), implying they possess different coordination structures. Figure 3g shows the Fourier-transformed k3-weighted EXAFS spectra of Co in Co1/Nb2O5 together with other reference samples. The Co foil reference exhibits a dominant peak at 2.19 Å that is associated with Co–Co scattering in the first coordination sphere. CoO displays two peaks at 1.67 Å and 2.62 Å that can be indexed to Co–O and Co–Co scattering, respectively. Co3O4 exhibits two peaks at 1.52 Å and 2.49 Å, also corresponding to Co–O and Co–Co scattering, respectively. As for Co1/Nb2O5, there is only one prominent peak centered at 1.43 Å and this is assigned to Co–O scattering in the first shell. These results confirm the atomic distribution of Co species in Co1/Nb2O5. The EXAFS fitting results (Fig. 3h, Supplementary Fig. 15 and Table 2) reveal that the Co atom is encircled by oxygen atoms with an average coordination number of 4.6. Wavelet transform (WT) results (Fig. 3i and Supplementary Fig. 16) confirm the presence of Co‒O bond in Co1/Nb2O5, in line with the Co K-edge FT-EXAFS analysis. Together, these findings demonstrate an atomic dispersion of Co species over Nb2O5 support.
Evaluation of catalytic performance
The catalytic performance of Co1/Nb2O5 in the selective hydrogenation of nitrobenzene to azoxybenzene was initially evaluated under 1 atm of H2 at 20 °C with addition of solvent (Supplementary Table 3). A series of solvents were screened and the use of tetrahydrofuran/H2O (4:1, v:v) mixed solvents secured the optimum reaction conditions (Supplementary Table 4). We observe low catalytic activity in the case of Nb2O5, implying the Co species are essential for the catalytic performance (Supplementary Table 5). With Co1/Nb2O5, the reaction proceeds smoothly to give the desired azoxybenzene with high conversion (99%) and selectivity (99%) within 1.5 h (Supplementary Fig. 17 and Table 5). No side product of aniline is detected for this reaction. Various Co salts, including Co(NO3)2, Co(Ac)2, CoCl2, and CoPc, are unable to efficiently catalyze this transformation, resulting in low catalytic activity (Supplementary Table 5). Accordingly, an extremely high turnover frequency (TOF) value of 11524 h−1 is noted for Co1/Nb2O5 compared with the control samples. For Co NPs/Nb2O5 (Supplementary Fig. 18 and Table 5), aniline is identified as the main product with a high selectivity of 99%, together with trace amounts of azoxybenzene and azobenzene.
Next, we sought to investigate the catalytic efficacy of Co1/Nb2O5 in catalyzing this reaction under solvent-free conditions. We observed an exceptionally high catalytic activity of Co1/Nb2O5, with maximized atomic utilization. The reaction yielded azoxybenzene with excellent conversion (99%) and exclusive selectivity (99%) within merely 0.5 h (Fig. 4a and Supplementary Fig. 19). Additionally, the 99% selectivity remains unchanged following a further 9.5 h reaction, emphasizing excellent catalyst performance. This implies that the undesired side reaction was effectively constrained in the absence of Co–Co bonds. In the case of Co NPs/Nb2O5, aniline is the main product (Fig. 4b). This might result from the aggregated Co species over Nb2O5 with multiple Co–Co bonds. Catalysts consisting of Co single atoms anchored on other oxides using the same synthetic method with O–coordination structures were also prepared, being Co1/MgO and Co1/V2O5, respectively (Supplementary Figs. 20 and 21, and Table 2). A nitrogen-doped carbon support with Co single atoms (Co1/N-C) was fabricated to represent different coordination environments, for example, Co–N coordination (Supplementary Fig. 22 and Table 2). Remarkably, an exceptional TOF value of 40377 h−1 for Co1/Nb2O5 was determined, significantly higher than other control samples (Fig. 4c). Poor catalytic activity and selectivity of Co1/MgO, Co1/V2O5, and Co1/N-C are observed (Supplementary Fig. 23). This might result from the unique coordination environments, electronic structures, and electronic metal-support interactions of catalytically active Co sites in Co1/Nb2O5 that influence the reaction pathways and energy barriers. Under these mild conditions, Co1/Nb2O5 also demonstrates superior catalytic performance compared with previously reported catalysts (Supplementary Table 6).

Conversion and selectivity of (a) Co1/Nb2O5 and (b) Co NPs/Nb2O5. c The corresponding TOF values of different samples. d Arrhenius plots and Ea values. e Recycling results (Reaction time: 5 min). f The proposed reaction routes. In situ DRIFTS spectra were recorded during the hydrogenation of nitrobenzene over (g) Co1/Nb2O5 and (h) Co NPs/Nb2O5. (NB nitrobenzene; NSB nitrosobenzene; PHA phenylhydroxylamine; AB azobenzene; AOB azoxybenzene; AN aniline).
Kinetic studies were performed to gain more insights into the origin of the catalytic activity of Co1/Nb2O5 based on initial conversion rates of nitrobenzene at different temperatures (Fig. 4d and Supplementary Fig. 24). Compared with Co NPs/Nb2O5, a lowered activation energy (Ea) of 39 kJ mol−1 was determined for Co1/Nb2O5 which suggests an enhanced catalytic activity. This offers evidence that the functionalization of a moderate amount of atomically dispersed Co atoms over Nb2O5 can significantly boost the catalytic activity. After 10 cycles of repetitive use, this Co1/Nb2O5 catalyst exhibits admirable stability without noticeable activity degradation (Fig. 4e and Supplementary Table 7). The crystalline structure and morphology of recycled Co1/Nb2O5 catalyst do not show any evident differences (Supplementary Fig. 25). Importantly, the EXAFS results of spent Co1/Nb2O5 catalyst (Supplementary Fig. 26 and Table 2) demonstrate that the dispersion and coordination environment of Co atoms are unchanged after 10 cycles. In addition, Co L-edge and O K-edge NEXAFS (Supplementary Fig. 27) reveal that no substantial electronic structural changes were observed. These results imply a strong metal-support interaction in the Co1/Nb2O5 catalyst with high stability.
H2 dissociation ability over catalysts plays a crucial role in the hydrogenation reactions56,57,58. H2-temperature-programmed desorption (H2-TPD) measurements were initially performed and the results (Supplementary Fig. 28) show that Co NPs/Nb2O5 exhibits a higher intensity of desorption peaks over Co1/Nb2O5 and Nb2O5, implying the existence of a higher amount of active sites and greater H2 adsorption capacity (Supplementary Table 1). The desorption temperature of Co1/Nb2O5 is slightly smaller than that of Co NPs/Nb2O5, but higher than that of Nb2O5. Based on Kyriakou’s work59, the H2 dissociation barrier will be lower on the active sites once the corresponding binding energy of dissociated H atoms is higher. Therefore, the higher H2 desorption temperature of Co NPs/Nb2O5 suggests that it favors the activation and dissociation of H2 more efficiently than Co1/Nb2O5 and Nb2O5. The H2 dissociation activity of the samples was further evaluated using an H2-D2 exchange experiment (Supplementary Fig. 29). The HD formation rate follows the order Co NPs/Nb2O5 > Co1/Nb2O5 > Nb2O5. Co NPs/Nb2O5 achieved a higher HD formation rate than those of Co1/Nb2O5 and Nb2O5, suggesting that the addition of Co species could considerably enhance the H2 dissociation activity and subsequently promote the hydrogenation reactions. Although Co NPs/Nb2O5 exhibits excellent nitrobenzene conversion, it exhibits extremely poor azoxybenzene selectivity. This implies that the difference in H2 dissociation activity might not be the only reason affecting the overall catalytic performance of the catalyst. More discussion on the origin of the selectivity difference can be found in the kinetics simulations of key hydrogenation steps in the Mechanism investigation section.
Based on these experimental findings, we assume that the Co1/Nb2O5 follows the steps from 1-2-3, while the Co NPs/Nb2O5 take the steps from 1-2-6 under the reaction conditions (Fig. 4f). To confirm this point, the selective hydrogenation of nitrobenzene was examined on Co1/Nb2O5 and Co NPs/Nb2O5 by in situ DRIFTS. In the case of Co1/Nb2O5 (Fig. 4g), the spectra show a slow consumption of nitrobenzene (IR bands at 1552, 1543, and 1349 cm−1)60 and the appearance of azoxybenzene with rapidly increased IR bands9,61,62 at 1576 and 1437 cm−1. The intermediate species of phenylhydroxylamine (1496 and 1302 cm−1)9 and nitrosobenzene (1507 and 1119 cm−1)9,61 were also observed. For Co NPs/Nb2O5 (Fig. 4h), the IR bands of nitrobenzene decrease gradually, while the bands of aniline (1623, 1614, 1606, and 1375 cm−1)9,61 increase, indicating H2 introduction favors the formation of aniline. In addition, we also observed the key reaction intermediate of nitrosobenzene and phenylhydroxylamine. These IR results are in excellent agreement with the experimental results. To validate the utility of the Co1/Nb2O5 catalyst in large-scale organocatalysis, we scaled up the reaction (50-fold) to the gram scale and established optimized conditions (Supplementary Fig. 30). The results show the catalytic performance of Co1/Nb2O5 was nearly identical to the lab-scale to give azoxybenzene, underscoring the potential of reaction scale tolerance.
With the optimized reaction conditions established, the substrate scope of solvent-free selective hydrogenation of nitroaromatics to azoxy compounds was explored. As shown in Fig. 5 and Supplementary Figs. 31–46, a variety of nitroaromatics were tested and all of these substrates are well tolerated and converted efficiently to the corresponding azoxy compounds in high conversion (up to 99%) and selectivity (up to 99%). The nitroaromatics bearing either electron-withdrawing substituents (halogen group, and nitro group) or electron-donating substituents (hydroxyl group, methoxyl group, methyl group, and amino group), situated at the para-position of the aromatic ring, did not have a noticeable impact on the reaction output. Note that methyl and methoxy groups at either ortho- or meta-positions (2r, 2o, 2l, 2j) underwent smooth conversion to afford the desired products with slightly lower efficacy compared to the more sterically accessible ones at para-position (2a, 2b). This might be due to the steric hindrance effect of the substituent groups that unfavor the coupling reactions. In addition to symmetrical azoxy compounds, we also tried to explore the potential feasibility of synthesizing products with unsymmetrical compounds (Supplementary Fig. 47). To our delight, this Co1/Nb2O5 catalyst shows good reactivity and affords the desired unsymmetrical compounds in moderate yields in most cases. Together, the results demonstrate the developed Co1/Nb2O5 catalyst can efficiently catalyze solvent-free selective hydrogenation of nitroaromatics to yield azoxy compounds.

a Reaction temperatures and melting points of substrates in parentheses; b Conversion; c Selectivity.
Mechanism investigation
To explore the origin of the high catalytic performance of Co1/Nb2O5 catalyst, density functional theory (DFT) calculations were performed. The optimized geometric structures of Nb2O5, Co1/Nb2O5, and Co NPs/Nb2O5 are shown in Supplementary Figs. 48–50. Based on AC STEM and XAFS characterizations, we embedded a Co atom on the Nb2O5(001) surface to describe Co1/Nb2O5 and employed Co(111) to represent Co NPs/Nb2O5 for the calculation. Bader charge and charge density difference analysis of Co1/Nb2O5 indicate that the Co atom loses 0.83 e to the neighboring O atoms in Co1/Nb2O5 (Fig. 6a and Supplementary Fig. 51). This suggests the presence of electronic metal-support interactions and the positively charged Co atoms. In addition, there is orbital mixing between O and Co atoms based on the projected density of states (PDOS) in Co1/Nb2O5 relative to that of Nb2O5, as shown in Fig. 6b and Supplementary Fig. 52. The d band centers of Co in Co1/Nb2O5 and Co(111) were determined to be −2.95 eV and −2.09 eV, respectively (Fig. 6b, c). Figure 6d, e shows the reaction pathways and the corresponding calculated energy profiles over Co1/Nb2O5 and Co(111). The configurations of intermediates are displayed in Supplementary Figs. 53 and 54. The nitrobenzene adsorption energies (EPhNO2) for Co1/Nb2O5 and Co(111) were both calculated to be exothermic at −1.25 eV and −2.57 eV, respectively. The larger adsorption energy of Co(111) demonstrates it has a much stronger affinity to nitrobenzene, which is in good agreement with its d band center that is closer to the Femi level than Co1/Nb2O5. With Co1/Nb2O5, the adsorbed *PhNO2 can be hydrogenated to afford a *PhNO2H intermediate (−0.24 eV) and then form *PhNO (−1.61 eV). Subsequently, the *PhNO intermediate would be reduced to *PhNOH and then *PhN via two hydrogenation steps, both of which are downhill in the energy profile by −0.69 eV and −0.33 eV, respectively. Finally, *PhN is converted to Ph-NNOPh with high priority by a highly exothermic process (−2.15 eV) over *PhNH (−1.70 eV). In the case of Co(111), the conversion of *PhNO2 to *PhNO2H is uphill in the energy profile by 0.58 eV due to the strong adsorption of PhNO2. Following the formation of *PhNO2H, its further hydrogenation to *PhNO is energetically favorable (−1.86 eV), and then *PhNO undergoes two successive hydrogenation steps to generate *PhN and which is overall exothermic by −1.08 eV. By overcoming an energy barrier of merely 0.07 eV, *PhN can be easily transformed to *PhNH compared with a higher energy barrier of 1.17 eV to obtain Ph-NNOPh.

a Bader charge and charge density difference of Co1/Nb2O5. The isosurface level is 0.004 eÅ−3. The PDOS and d band centers of (b) Co1/Nb2O5 and (c) Co(111). Energy profiles of solvent-free selective hydrogenation of nitrobenzene over (d) Co1/Nb2O5 and (e) Co(111). Inset: the corresponding transition state configurations.
We also performed a kinetic analysis to gain more insights into the reactivity of Co1/Nb2O5 and Co(111). It was found that the kinetic barrier (Eb) of the dissociation of an H2 molecule on Nb2O5 is 0.91 eV (Supplementary Fig. 55), consistent with the previous report (0.88 eV)63. The Eb of Co1/Nb2O5 (0.84 eV) is close to that of Nb2O5 (Supplementary Fig. 56); this may be due to the fact that H2 is physically adsorbed on Nb2O5 and Co1/Nb2O5, of which the adsorption energy EH2 is −0.17 and −0.24 eV, respectively. Specially, in both Nb2O5 and Co1/Nb2O5, H2 is preferentially located above the Nb site (Supplementary Fig. 57). The H2 above the Co1 site is also physisorbed while being more weakly bound (−0.12 eV). Therefore, H2 dissociation and PhNO2 hydrogenation may occur on the Nb2O5 and the Co1 site, respectively. Moreover, as expected, Co(111) exhibits stronger H2 adsorption than Co1/Nb2O5 and Nb2O5 (Supplementary Fig. 57), and its Eb is as low as 0.02 eV (Supplementary Fig. 58), which is in agreement with previous studies (0.03 eV)64. In the presence of *PhNO2, H2 dissociation on Co(111) is still kinetically favorable (Supplementary Fig. 59). These modeling results are consistent with our H2-TPD measurements and H2-D2 exchange experiments. Note that the small Eb of H2 dissociation on Co(111) should not affect the PhNO2 adsorption and the subsequent hydrogenation steps, given the stronger adsorption of PhNO2 compared to H2. As H2 is physisorbed, we also calculated H2 dissociation on *PhNO2 of Co1/Nb2O5 via the Eley−Rideal (ER) mechanism and found a small Eb of 0.34 eV (Supplementary Fig. 60).
We then moved our attention to the subsequent hydrogenation steps. For the first four hydrogenation steps, the largest Eb of Co1/Nb2O5 occurs in the conversion of *PhNOH to *PhN (0.84 eV), and that of Co(111) is the conversion of *PhNO to *PhNOH (0.95 eV). Specifically, Co1/Nb2O5 exhibits a favorable Eb of 0.22 eV for facilitating the further coupling of *PhN and *PhNO to *Ph-NNOPh, which is lower than that of the competing *Ph-NH formation (0.65 eV). By contrast, the hydrogenation of *PhN to *PhNH on Co(111) requires overcoming a moderate Eb of 1.02 eV, whereas the barrier for the *Ph-NNOPh generation is 1.21 eV. It should be noted that the active sites of the niobium oxide component are essential for the dehydration reaction because the Nb site of Co1/Nb2O5 exhibits favorable binding to*PhNO, which promotes the coupling of *PhNO and *PhN. In addition, we explored the influence of solvent on the reaction processes and found that the solvent effect did not change the product selectivity of Co1/Nb2O5 and Co(111) (Supplementary Figs. 61 and 62). For example, after considering the solvent effect, the Eb of Ph-NNOPh formation on Co1/Nb2O5 (0.49 eV) is still lower than that of the competing *Ph-NH formation (0.66 eV). These results are consistent with the experimental observations. The electronic coupling between Co atoms and adjacent coordinating oxygen atoms in Nb2O5 prevents the full hydrogenation of nitroarene, giving rise to a high selectivity towards azoxybenzene. Overall, DFT calculations provide solid evidence that Co1/Nb2O5, with its unique interface and electronic properties and strong electronic metal-support interactions, can significantly affect adsorption characteristics and activation ability with reactants, thus lowering the energy barriers and facilitating the formation of azoxy compounds, which ensures prominent catalytic activity and selectivity.
Discussion
In conclusion, we report on a facile strategy to access an efficient heterogeneous catalyst with atomically dispersed Co atoms over Nb2O5 nanomeshes. AC HAADF-STEM, CO-DRIFTS, XAFS, and XPS characterizations reveal that these isolated Co atoms are positively charged and coordinated with the neighboring oxygen atoms. This Co1/Nb2O5 catalyst exhibits exceptional catalytic efficiency in solvent-free hydrogenation of nitrobenzene to give azoxybenzene, superior to that of reported catalysts. In addition, Co1/Nb2O5 successfully promoted the solvent-free hydrogenation coupling of a broad range of nitroaromatics into the desired azoxy compounds with high efficiency. More importantly, excellent recyclability and reaction scale tolerance were demonstrated. Theoretical calculations elucidate that the unique electronic properties and strong electronic metal-support interactions of catalytically active Co sites in Co1/Nb2O5 have a substantial influence on the reaction pathways and energy barriers. Our findings underscore the great potential of this synthetic strategy for designing high-performance catalysts and provide insights into the structure-performance relationship for industrially important catalytic reactions.
Methods
Synthesis of Nb2O5
In a typical synthesis, 1.6 mmol of ammonium niobate oxalate hydrate, 16 mmol of melamine, and 40 mmol of ammonium chloride were dissolved in 40 ml of ethanol and stirred for 12 h. The mixture was then washed with ethanol and vacuum-dried at 80 °C. Subsequently, the dried powder was transferred to a tube furnace and heated at 550 °C in air for 4 h with a heating rate of 2.5 °C min−1. After cooling to room temperature, the white-colored Nb2O5 nanomeshes were obtained.
Synthesis of Co1/Nb2O5
In a typical synthesis, 0.4 g of as-prepared Nb2O5 was subjected to an incipient wetness impregnation method with 600 µl cobalt(II) acetate ethanol solution (20 mg/ml), followed by an infrared lamp drying step (Co2+@Nb2O5). The dried powder was sealed in an argon-filled glass vial and microwave-treated at 800 W for 10 s (Microwave Oven, Galanz) to obtain Co1/Nb2O5. The metal loading in the catalyst was determined to be 0.42 wt%.
Synthesis of Co NPs/Nb2O5
The preparation method was the same as that of Co1/Nb2O5, except 1.3 ml of cobalt(II) acetate ethanol solution (100 mg/ml) was used. The metal loading in the catalyst was determined to be 5.12 wt%.
Catalytic evaluation
The solvent-free selective hydrogenation of nitroaromatics on Co1/Nb2O5 was evaluated under atmosphere pressure. Typically, 20 mmol of nitrobenzene and 100 mg of Co1/Nb2O5 (with a molar ratio of 2800:1) were introduced into 25 ml of a round bottom flask connected with a balloon filled with H2. The catalytic reaction was performed at 20 °C. After the reaction, 50 µl of the resultant mixture was added to 2 ml of ethyl acetate before centrifugation. The corresponding organic compounds were extracted and analyzed by gas chromatography (GC, Techcomp GC-7980) equipped with an HP-5 capillary column and a flame ionization detector. The qualitative analysis was performed by gas chromatography-mass spectrometry (GC-MS, 7890 and 5975 C, Agilent). The used catalyst was separated from the reaction mixture by centrifugation and washed with ethanol before being vacuum-dried at 60 °C for the next catalytic cycle.
For the reaction with solvents, 10 mg of Co1/Nb2O5, 2 mmol of nitrobenzene, and 5 ml of mixed solvents (tetrahydrofuran: H2O = 4:1, v:v) were added into a 25 ml of Schlenk glass vessel tube with a molar ratio of nitrobenzene: Co is 2800:1. The catalytic reaction was performed at 20 °C under H2 atmosphere.
The turnover frequency (TOF) values of the catalysts were determined below 10% conversion of the substrate and based on exposed Co atoms. The conversion, selectivity, yield, and TOF are defined as follows:
Data availability
The data supporting the findings of this work are available within the article and Supplementary Information. All data are available from the corresponding authors upon request.
References
Doherty, S. et al. Highly selective and solvent-dependent reduction of nitrobenzene to N-phenylhydroxylamine, azoxybenzene, and aniline catalyzed by phosphino-modified polymer immobilized ionic liquid-stabilized AuNPs. ACS Catal. 9, 4777–4791 (2019).
Formenti, D., Ferretti, F., Scharnagl, F. K. & Beller, M. Reduction of nitro compounds using 3d-non-noble metal catalysts. Chem. Rev. 119, 2611–2680 (2019).
Dai, Y. et al. Light-tuned selective photosynthesis of azo- and azoxy-aromatics using graphitic C3N4. Nat. Commun. 9, 60 (2018).
Yan, H. et al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 9, 3197 (2018).
Zhao, J.-X. et al. Selectivity regulation in Au-catalyzed nitroaromatic hydrogenation by anchoring single-site metal oxide promoters. ACS Catal. 10, 2837–2844 (2020).
Lou, Y. et al. Pocketlike active site of Rh1/MoS2 single-atom catalyst for selective crotonaldehyde hydrogenation. J. Am. Chem. Soc. 141, 19289–19295 (2019).
Ma, Y. et al. High-density and thermally stable palladium single-atom catalysts for chemoselective hydrogenations. Angew. Chem. Int. Ed. 59, 21613–21619 (2020).
Jin, H. et al. Sabatier phenomenon in hydrogenation reactions induced by single-atom density. J. Am. Chem. Soc. 145, 12023–12032 (2023).
Wu, B. et al. Ru single atoms for efficient chemoselective hydrogenation of nitrobenzene to azoxybenzene. Green Chem. 23, 4753–4761 (2021).
Chen, Z., Liu, J., Koh, M. J. & Loh, K. P. Single-atom catalysis: from simple reactions to the synthesis of complex molecules. Adv. Mater. 34, 2103882 (2022).
Guo, J. et al. Metal-organic frameworks as catalytic selectivity regulators for organic transformations. Chem. Soc. Rev. 50, 5366–5396 (2021).
Kaden, W. E., Wu, T., Kunkel, W. A. & Anderson, S. L. Electronic structure controls reactivity of size-selected Pd clusters adsorbed on TiO2 surfaces. Science 326, 826–829 (2009).
Zaera, F. Designing sites in heterogeneous catalysis: are we reaching selectivities competitive with those of homogeneous catalysts? Chem. Rev. 122, 8594–8757 (2022).
Giannakakis, G., Mitchell, S. & Pérez-Ramírez, J. Single-atom heterogeneous catalysts for sustainable organic synthesis. Trends Chem. 4, 264–276 (2022).
Clarke, C. J., Tu, W.-C., Levers, O., Bröhl, A. & Hallett, J. P. Green and sustainable solvents in chemical processes. Chem. Rev. 118, 747–800 (2018).
Ni, S. et al. Mechanochemical solvent-free catalytic C−H methylation. Angew. Chem. Int. Ed. 60, 6660–6666 (2021).
Mi, R. et al. Solvent-free heterogeneous catalytic hydrogenation of polyesters to diols. Angew. Chem. Int. Ed. 62, e202304219 (2023).
Miao, Y. et al. Photothermal recycling of waste polyolefin plastics into liquid fuels with high selectivity under solvent-free conditions. Nat. Commun. 14, 4242 (2023).
Shi, X. et al. High performance and active sites of a ceria-supported palladium catalyst for solvent-free chemoselective hydrogenation of nitroarenes. Chem. Cat. Chem. 9, 3743–3751 (2017).
Sun, Z. et al. The solvent-free selective hydrogenation of nitrobenzene to aniline: an unexpected catalytic activity of ultrafine Pt nanoparticles deposited on carbon nanotubes. Green Chem. 12, 1007–1011 (2010).
Di Liberto, G., Tosoni, S., Cipriano, L. A. & Pacchioni, G. A few questions about single-atom catalysts: When modeling helps. Acc. Mater. Res. 3, 986–995 (2022).
Liang, X., Fu, N., Yao, S., Li, Z. & Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 144, 18155–18174 (2022).
Li, H., Li, R., Liu, G., Zhai, M. & Yu, J. Noble-metal-free single- and dual-atom catalysts for artificial photosynthesis. Adv. Mater. 13, 2301307 (2023).
Liu, L. & Corma, A. Isolated metal atoms and clusters for alkane activation: translating knowledge from enzymatic and homogeneous to heterogeneous systems. Chem 7, 2347–2384 (2021).
Li, Z., Wang, D., Wu, Y. & Li, Y. Recent advances in the precise control of isolated single-site catalysts by chemical methods. Natl. Sci. Rev. 5, 673–689 (2018).
Swain, S., Altaee, A., Saxena, M. & Samal, A. K. A comprehensive study on heterogeneous single atom catalysis: current progress, and challenges. Coord. Chem. Rev. 470, 214710 (2022).
Ji, S. et al. Construction of a single-atom palladium catalyst by electronic metal-support interaction and interface confinement effect with remarkable performance in Suzuki coupling reaction. Chem. Eng. J. 452, 139205 (2023).
Li, Z. et al. Engineering the electronic structure of single-atom iron sites with boosted oxygen bifunctional activity for zinc-air batteries. Adv. Mater. 35, 2209644 (2023).
Chen, C. et al. Zero-valent palladium single-atoms catalysts confined in black phosphorus for efficient semi-hydrogenation. Adv. Mater. 33, 2008471 (2021).
Yang, H. et al. Catalytically active atomically thin cuprate with periodic Cu single sites. Natl. Sci. Rev. 10, https://doi.org/10.1093/nsr/nwac100 (2023).
Li, Z. et al. A heterogeneous single atom cobalt catalyst for highly efficient acceptorless dehydrogenative coupling reactions. Small 19, 2207941 (2023).
Dong, X. et al. Molten salt-induction of geometrically deformed ruthenium single atom catalysts with high performance for aerobic oxidation of alcohols. Chem. Eng. J. 451, 138660 (2023).
Guo, Y., Jing, Y., Xia, Q. & Wang, Y. NbOx-based catalysts for the activation of C-O and C-C bonds in the valorization of waste carbon resources. Acc. Chem. Res. 55, 1301–1312 (2022).
Jing, Y., Xin, Y., Guo, Y., Liu, X. & Wang, Y. Highly efficient Nb2O5 catalyst for aldol condensation of biomass-derived carbonyl molecules to fuel precursors. Chin. J. Catal. 40, 1168–1177 (2019).
Jing, Y. et al. Towards the circular economy: converting aromatic plastic waste back to arenes over a Ru/Nb2O5 catalyst. Angew. Chem. Int. Ed. 60, 5527–5535 (2021).
Shao, Y. et al. Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat. Commun. 8, 16104 (2017).
Xia, J. et al. Identifying the activity origin of a single-atom Au1/Nb2O5 catalyst for hydrodeoxygenation of methylcatechol: A stable substitutional Au+ site. ACS Catal. 13, 6093–6103 (2023).
Lim, E. et al. Facile synthesis of Nb2O5@carbon core–shell nanocrystals with controlled crystalline structure for high-power anodes in hybrid supercapacitors. ACS Nano 9, 7497–7505 (2015).
Zheng, Y. et al. Defect-concentration-mediated T-Nb2O5 anodes for durable and fast-charging Li-ion batteries. Adv. Funct. Mater. 32, 2107060 (2022).
Chen, D. et al. Unraveling the nature of anomalously fast energy storage in T-Nb2O5. J. Am. Chem. Soc. 139, 7071–7081 (2017).
Zheng, Y. et al. Triple conductive wiring by electron doping, chelation coating and electrochemical conversion in fluffy Nb2O5 anodes for fast-charging Li-ion batteries. Adv. Sci. 9, 2202201 (2022).
Zhang, Y., Song, T., Zhou, X. & Yang, Y. Oxygen-vacancy-boosted visible light driven photocatalytic oxidative dehydrogenation of saturated N-heterocycles over Nb2O5 nanorods. Appl. Catal. B Environ. 316, 121622 (2022).
Liu, R. et al. Design of aligned porous carbon films with single-atom Co-N-C sites for high-current-density hydrogen generation. Adv. Mater. 33, 2103533 (2021).
Liu, Y. et al. Improving CO2 photoconversion with ionic liquid and Co single atoms. Nat. Commun. 14, 1457 (2023).
Li, L., Dong, L., Liu, X., Guo, Y. & Wang, Y. Selective production of ethylbenzene from lignin oil over FeOx modified Ru/Nb2O5 catalyst. Appl. Catal. B Environ. 260, 118143 (2020).
Su, K. et al. Tuning the Pt species on Nb2O5 by support-induced modification in the photocatalytic transfer hydrogenation of phenylacetylene. Appl. Catal. B Environ. 298, 120554 (2021).
Li, Y. et al. A single site ruthenium catalyst for robust soot oxidation without platinum or palladium. Nat. Commun. 14, 7149 (2023).
Shi, X. et al. Protruding Pt single-sites on hexagonal ZnIn2S4 to accelerate photocatalytic hydrogen evolution. Nat. Commun. 13, 1287 (2022).
Singh, J. A. et al. Understanding the active sites of CO hydrogenation on Pt–Co catalysts prepared using atomic layer deposition. J. Phys. Chem. C 122, 2184–2194 (2018).
Kumar, N., Jothimurugesan, K., Stanley, G. G., Schwartz, V. & Spivey, J. J. In situ FT-IR study on the effect of cobalt precursors on CO adsorption behavior. J. Phys. Chem. C 115, 990–998 (2011).
Song, D., Li, J. & Cai, Q. In situ diffuse reflectance FTIR study of CO adsorbed on a cobalt catalyst supported by silica with different pore sizes. J. Phys. Chem. C 111, 18970–18979 (2007).
Kumar, P. et al. High-density cobalt single-atom catalysts for enhanced oxygen evolution reaction. J. Am. Chem. Soc. 145, 8052–8063 (2023).
Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829–7836 (2023).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the Ir/Nb2O5-x catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022).
Chen, X. et al. Structure-dependence and metal-dependence on atomically dispersed Ir catalysts for efficient n-butane dehydrogenation. Nat. Commun. 14, 2588 (2023).
Feng, Z. et al. Asymmetric sites on the ZnZrOx catalyst for promoting formate formation and transformation in CO2 hydrogenation. J. Am. Chem. Soc. 145, 12663–12672 (2023).
Lee, J. D. et al. Facilitating hydrogen dissociation over dilute nanoporous Ti–Cu catalysts. J. Am. Chem. Soc. 144, 16778–16791 (2022).
Zhao, H. et al. The role of Cu1-O3 species in single-atom Cu/ZrO2 catalyst for CO2 hydrogenation. Nat. Catal. 5, 818–831 (2022).
Kyriakou, G. et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209–1212 (2012).
Liu, W. et al. Highly-efficient RuNi single-atom alloy catalysts toward chemoselective hydrogenation of nitroarenes. Nat. Commun. 13, 3188 (2022).
Richner, G., van Bokhoven, J. A., Neuhold, Y.-M., Makosch, M. & Hungerbühler, K. In situ infrared monitoring of the solid/liquid catalyst interface during the three-phase hydrogenation of nitrobenzene over nanosized Au on TiO2. Phys. Chem. Chem. Phys. 13, 12463–12471 (2011).
Liu, L., Concepción, P. & Corma, A. Modulating the catalytic behavior of non-noble metal nanoparticles by inter-particle interaction for chemoselective hydrogenation of nitroarenes into corresponding azoxy or azo compounds. J. Catal. 369, 312–323 (2019).
Zhou, H. et al. Hydrogenolysis cleavage of the Csp2–Csp3 bond over a metal-free NbOPO4 catalyst. ACS Catal. 12, 4806–4812 (2022).
Yu, M. et al. High coverage H2 adsorption and dissociation on fcc Co surfaces from DFT and thermodynamics. Int. J. Hydrog. Energy 43, 5576–5590 (2018).
Acknowledgements
This work was supported by the Outstanding Youth Project of the Natural Science Foundation of Heilongjiang Province (YQ2022B002), the Scientific Research Foundation for Returned Scholars of Heilongjiang Province of China (719900091), and the Heilongjiang Touyan Innovation Team Program. Y.W. acknowledges financial supports from the Natural Science Foundation of China (22203044) and the Jiangsu Specially Appointed Professor Plan. The authors thank beamline 1W1B of Beijing Synchrotron Radiation Facility (BSRF) in Beijing and beamline of MCD-A and MCD-B (Soochow Beamline for Energy Materials) of National Synchrotron Radiation Laboratory in Hefei for XAS measurements. The authors also acknowledge the Catalysis and Surface Science Endstation at beamline BL11U for support in ultraviolet photoemission spectroscopy (UPS) measurements.
Author information
Authors and Affiliations
Contributions
Z.L. conceived the idea, supervised the project, and wrote the paper. Xi.L. synthesized the catalysts, performed the catalytic reactions, and analyzed the data. Co.G. and Y.W. conducted the DFT calculations. S.J., H.L., Ch.G., Xu.L., and S.X. assisted with the material synthesis and characterizations. S.J. and H.L. performed the in-situ FT-IR measurements and analyzed the data. B.L. and W.W. performed H2-TPR, H2-TPD, H2-D2 exchange, and TGA measurements. C.W. and W.Y. helped with the analysis of the XAFS spectra. J.H.H. contributed to the discussion on the experiment. All authors contributed to the overall scientific interpretation and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huizhen Liu, Jiong Lu and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Z., Lu, X., Guo, C. et al. Solvent-free selective hydrogenation of nitroaromatics to azoxy compounds over Co single atoms decorated on Nb2O5 nanomeshes. Nat Commun 15, 3195 (2024). https://doi.org/10.1038/s41467-024-47402-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47402-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
