
Genome Sequencing of Historical Encephalomyocarditis Viruses from South Africa Links the Historical 1993/4 Savanna Elephant (Loxodonta africana) Outbreak to Cryptic Mastomys Rodents
 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Virus Strains
2.2. RNA Extraction and cDNA Synthesis
2.3. Viral Genome Amplification, Purification, and Sequencing
2.4. Dataset Compilation and Statistics
2.5. Phylogenetic Analysis
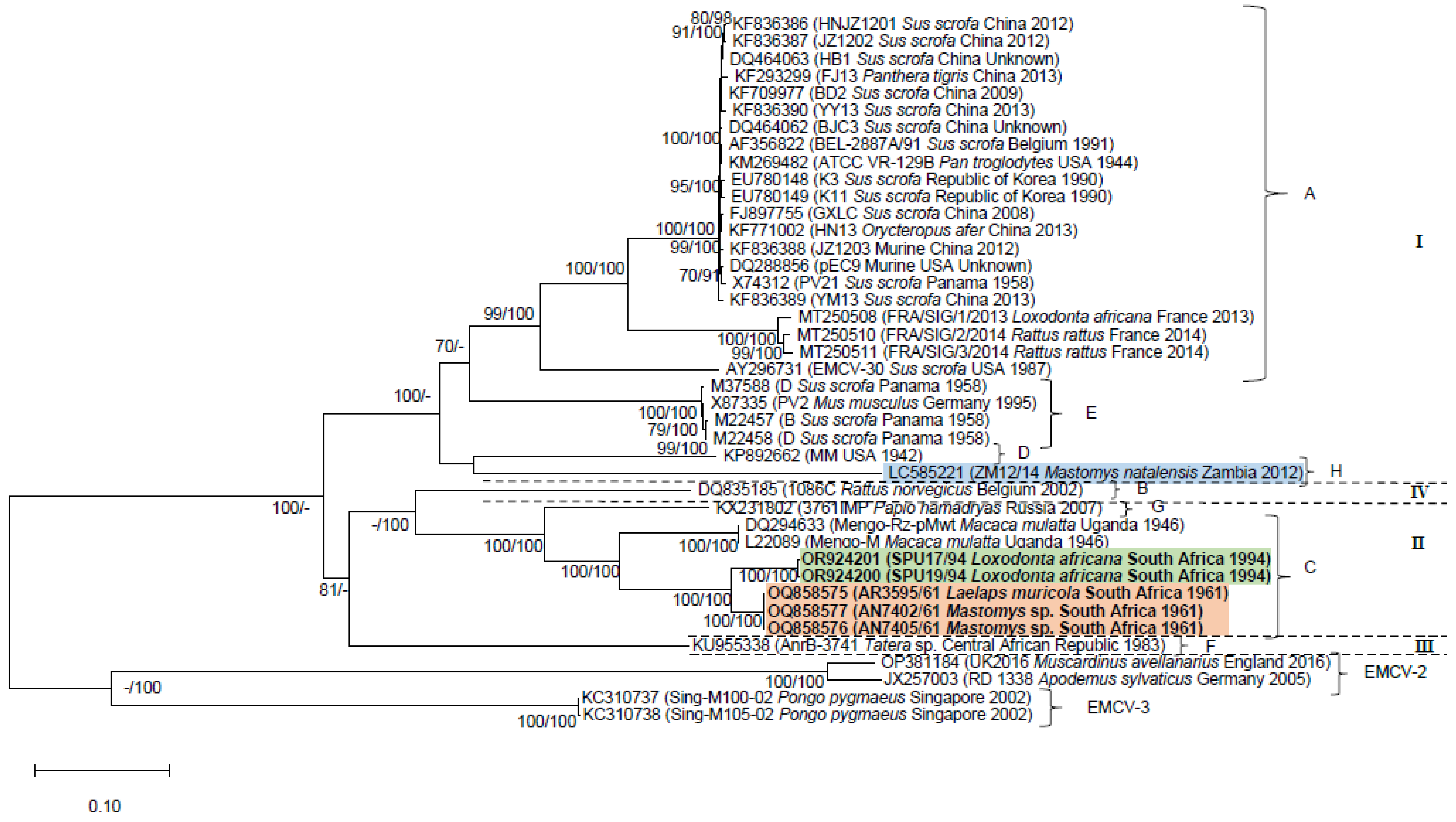
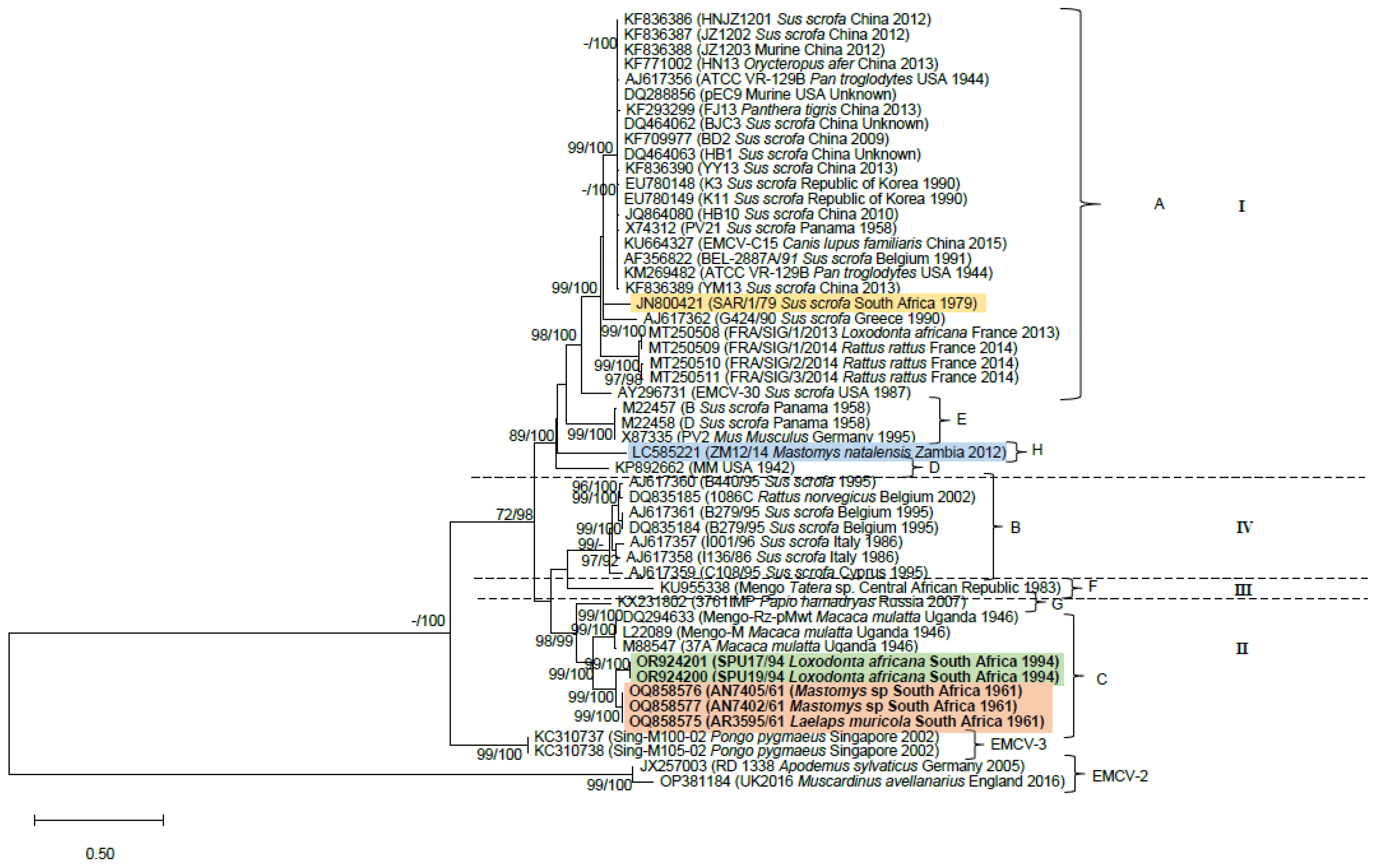
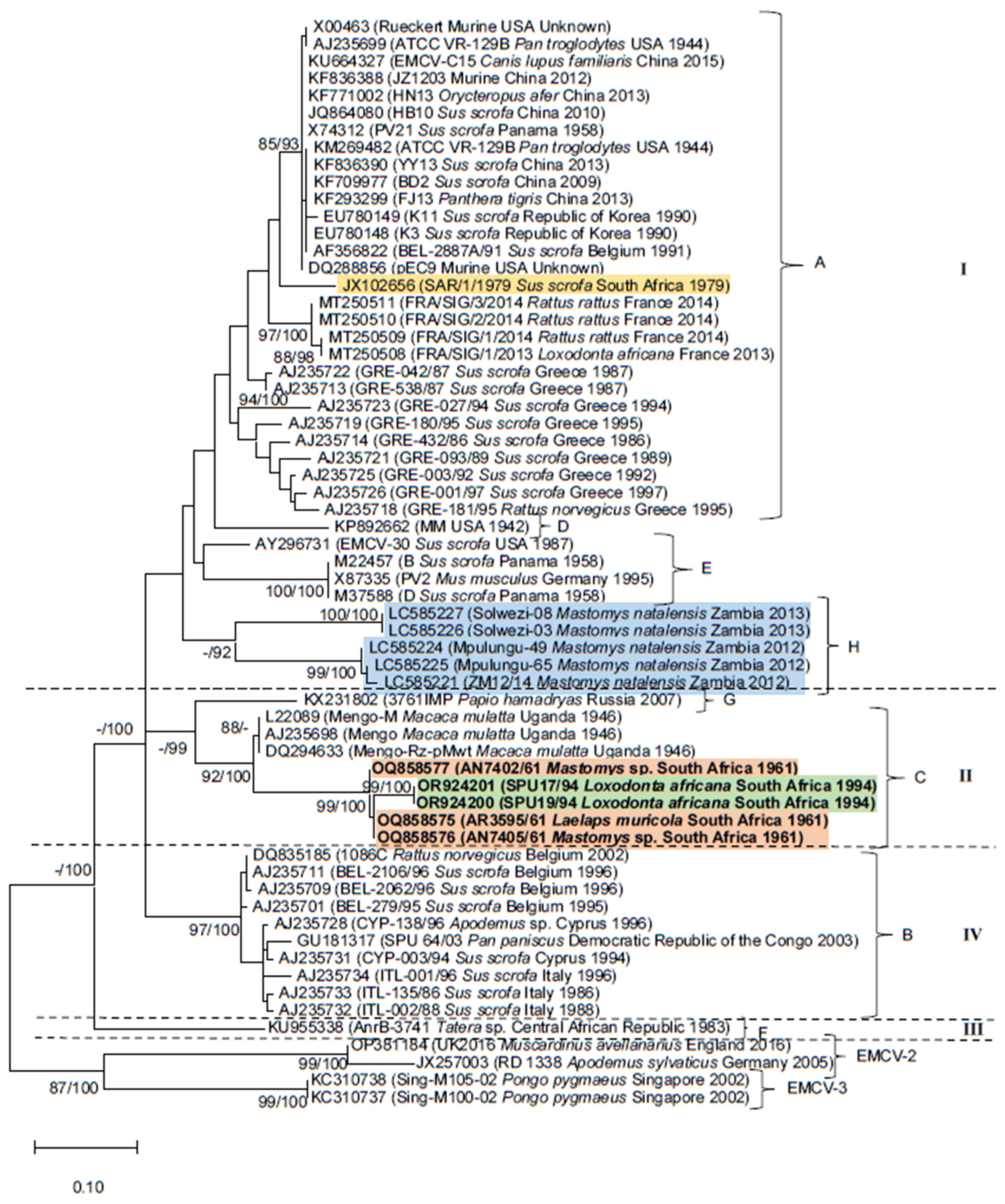
3. Results
3.1. Genome Characterization
3.2. Phylogenetic Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knowles, N.J.; Hovi, T.; Hyypiä, T.; King, A.M.Q.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Simmonds, P.; Skern, T.; Stanway, G. Family Picornaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 855–880. [Google Scholar]
- Yeo, D.S.Y.; Lian, J.E.; Fernandez, C.J.; Lin, Y.N.; Liaw, J.C.W.; Soh, M.L.; Lim, E.A.S.; Chan, K.P.; Ng, M.L.; Tan, H.C.; et al. A highly divergent Encephalomyocarditis virus isolated from nonhuman primates in Singapore. Virol. J. 2013, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV virus taxonomy profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Helwig, F.; Schmidt, C. A filter-passing agent producing interstitial myocarditis in anthropoid apes and small animals. Science 1945, 102, 31–33. [Google Scholar] [CrossRef]
- Gainer, J.H. Encephalomyocarditis virus infections in Florida, 1960-1966. J. Am. Vet. Med. Assoc. 1967, 151, 421–425. [Google Scholar] [PubMed]
- Gainer, J.H.; Sandefur, J.R.; Bigler, W.J. High mortality in a Florida swine herd infected with the encephalomyocarditis virus. An accompanying epizootiologic survey. Cornell Vet. 1968, 58, 31–47. [Google Scholar]
- Williams, M.C. Encephalomyocarditis virus infection. JSAVA 1981, 52, 76. [Google Scholar]
- Maurice, H.; Nielen, M.; Brocchi, E.; Nowotny, N.; Kassimi, L.B.; Billinis, C.; Loukaides, P.; O’Hara, R.S.; Koenen, F. The occurrence of encephalomyocarditis virus (EMCV) in European pigs from 1990 to 2001. Epidemiol. Infect. 2005, 133, 547–557. [Google Scholar] [CrossRef]
- Foglia, E.A.; Pezzoni, G.; Bonilauri, P.; Torri, D.; Grazioli, S.; Brocchi, E. A recent view about encephalomyocarditis virus circulating in compartmentalised animal population in Northern Italy. Sci. Rep. 2023, 13, 592. [Google Scholar] [CrossRef]
- Amaddeo, D.; Cardeti, G.; Autorino, G.L. Isolation of encephalomyocarditis virus from dormice (Myoxus glis) in Italy. J. Wildl. Dis. 1995, 31, 238–242. [Google Scholar] [CrossRef]
- Grobler, D.G.; Raath, J.P.; Braack, L.E.; Keet, D.F.; Gerdes, G.H.; Barnard, B.J.; Kriek, N.P.; Jardine, J.; Swanepoel, R. An outbreak of encephalomyocarditis-virus infection in free-ranging African elephants in the Kruger National Park. Onderstepoort J. Vet. Res. 1995, 62, 97–108. [Google Scholar]
- Kishimoto, M.; Hang’ombe, B.M.; Hall, W.W.; Orba, Y.; Sawa, H.; Sasaki, M. Mastomys natalensis is a possible natural rodent reservoir for encephalomyocarditis virus. J. Gen. Virol. 2021, 102, 001564. [Google Scholar] [CrossRef]
- Romey, A.; Lamglait, B.; Blanchard, Y.; Touzain, F.; Quenault, H.; Relmy, A.; Zientara, S.; Blaise-Boisseau, S.; Bakkali-Kassimi, L. Molecular characterization of encephalomyocarditis virus strains isolated from an African elephant and rats in a French zoo. J. Vet. Diagn. Investig. 2021, 33, 313–321. [Google Scholar] [CrossRef]
- Joo, H.S.; Kim, S.; Leman, A.D. Detection of antibody to encephalomyocarditis virus in mummified or stillborn pigs. Arch. Virol. 1988, 100, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Koenen, F.; De Clercq, K.; Lefebvre, J.; Strobbe, R. Reproductive failure in sows following experimental infection with a Belgian EMCV isolate. Vet. Microbiol. 1994, 39, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Lamglait, B.; Joris, A.; Romey, A.; Bakkali-Kassimi, L.; Lemberger, K. Fatal encephalomyocarditis virus infection in an African savanna elephant (Loxodonta africana) in a French zoo. J. Zoo Wildl. Med. 2015, 46, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Kilham, L.; Davies, J.N.P.; Mason, P. Host-Virus Relations in Encephalomyocarditis (EMC) Virus Infections. I. Infections of Wild Rats. Am. J. Trop. Med. Hyg. 1956, 5, 647–654. [Google Scholar] [CrossRef]
- Luo, Y.K.; Liang, L.; Tang, Q.H.; Zhou, L.; Shi, L.J.; Cong, Y.Y.; Lin, W.C.; Cui, S.J. Isolation and Characterization of Encephalomyocarditis Virus from Dogs in China. Sci. Rep. 2017, 7, 438. [Google Scholar] [CrossRef]
- Dick, G.W.A.; Haddow, A.J.; Best, A.M.; Smithburn, K.C. Mengo encephalomyelitis; a hitherto unknown virus affecting man. Lancet 1948, 252, 286–289. [Google Scholar] [CrossRef]
- Oberste, M.S.; Gotuzzo, E.; Blair, P.; Nix, W.A.; Ksiazek, T.G.; Comer, J.A.; Rollin, P.; Goldsmith, C.S.; Olson, J.; Kochel, T.J. Human Febrile Illness Caused by Encephalomyocarditis Virus Infection, Peru. Emerg. Infect. Dis. 2009, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Czechowicz, J.; Huaman, J.L.; Forshey, B.M.; Morrison, A.C.; Castillo, R.; Huaman, A.; Caceda, R.; Eza, D.; Rocha, C.; Blair, P.J.; et al. Prevalence and risk factors for encephalomyocarditis virus infection in Peru. Vector Borne Zoonotic Dis. 2011, 11, 367–374. [Google Scholar] [CrossRef]
- Seaman, J.T.; Finnie, E.P. Acute myocarditis in a captive African elephant (Loxodonta africana). J. Wildl. Dis. 1987, 23, 170–171. [Google Scholar] [CrossRef]
- Reddacliff, L.A.; Kirkland, P.D.; Hartley, W.J.; Reece, R.L. Encephalomyocarditis virus infections in an Australian zoo. J. Zoo Wildl. Med. 1997, 28, 153–157. [Google Scholar]
- Krylova, R.I.; Dzhikidze, E.K. Encephalomyocarditis in Monkeys. Bull. Exp. Biol. Med. 2005, 139, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Canelli, E.; Luppi, A.; Lavazza, A.; Lelli, D.; Sozzi, E.; Martin, A.M.; Gelmetti, D.; Pascotto, E.; Sandri, C.; Magnone, W.; et al. Encephalomyocarditis virus infection in an Italian zoo. Virol. J. 2010, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Cordonnier, N.; Mahamba, C.; Burt, F.J.; Rakotovao, F.; Swanepoel, R.; André, C.; Dauger, S.; Bakkali Kassimi, L. Encephalomyocarditis virus mortality in semi-wild bonobos (Pan panicus). J. Med. Primatol. 2011, 40, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yan, Q.; Zhao, B.; Luo, J.; Wang, C.; Du, Y.; Yan, J.; He, H. Isolation, molecular characterization, and phylogenetic analysis of encephalomyocarditis virus from South China tigers in China. Infect. Genet. Evol. 2013, 19, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Recombination and Selection in the Evolution of Picornaviruses and Other Mammalian Positive-Stranded RNA Viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef]
- Vanderhallen, H.; Koenen, F. Identification of encephalomyocarditis virus in clinical samples by reverse transcription-PCR followed by genetic typing using sequence analysis. J. Clin. Microbiol. 1998, 36, 3463–3467. [Google Scholar] [CrossRef] [PubMed]
- Bakkali, L.; Gonzague, M.; Boutrouille, A.; Cruciere, C. Detection of Encephalomyocarditis virus in clinical samples by immunomagnetic separation and one-step RT-PCR. J. Virol. Methods 2002, 101, 197–206. [Google Scholar] [CrossRef]
- Denis, P.; Liebig, H.D.; Nowotny, N.; Billinis, C.; Papadopoulos, O.; O’Hara, R.S.; Knowles, N.J.; Koenen, F. Genetic variability of encephalomyocarditis virus (EMCV) isolates. Vet. Microbiol. 2006, 113, 1–12. [Google Scholar] [CrossRef]
- van Sandwyk, J.H.d.T.; Bennett, N.C.; Swanepoel, R.; Bastos, A.D.S. Retrospective genetic characterisation of Encephalomyocarditis viruses from African elephant and swine recovers two distinct lineages in South Africa. Vet. Microbiol. 2013, 162, 23–31. [Google Scholar] [CrossRef]
- Gordon, D.H.; Watson, C.R.B. Identification of cryptic species of rodents (Mastomys, Aethomys, Saccostomus) in the Kruger National Park. Afr. Zool. 1986, 21, 95–99. [Google Scholar] [CrossRef]
- Venturi, F.P.; Chimimba, C.T.; van Aarde, R.J.; Fairall, N. The distribution of two medically and agriculturally important cryptic rodent species, Mastomys natalensis and M. coucha (Rodentia: Muridae) in South Africa. Afr. Zool. 2004, 39, 235–245. [Google Scholar] [CrossRef]
- Meyer, R.F.; Brown, C.C.; House, C.; House, J.A.; Molitor, T.W. Rapid and sensitive detection of foot-and-mouth disease virus in tissues by enzymatic RNA amplification of the polymerase gene. J. Virol. Methods 1991, 34, 161–172. [Google Scholar] [CrossRef]
- Bastos, A.D. Detection and characterization of foot-and-mouth disease virus in sub-Saharan Africa. Onderstepoort J. Vet. Res. 1998, 65, 37–47. [Google Scholar] [PubMed]
- de Bruyn, P.J.N.; Bastos, A.D.S.; Eadie, C.; Tosh, C.A.; Bester, M.N. Mass Mortality of Adult Male Subantarctic Fur Seals: Are Alien Mice the Culprits? PLoS ONE 2008, 3, e3757. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. 2018. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh.
- Koenen, F.; Vanderhallen, H.; Dickinson, N.D.; Knowles, N.J. Phylogenetic analysis of European encephalomyocarditis viruses: Comparison of two genomic regions. Arch. Virol. 1999, 144, 893–903. [Google Scholar] [CrossRef]
- Vyshemirskii, O.I.; Agumava, A.A.; Kalaydzyan, A.A.; Leontyuk, A.V.; Kuhn, J.H.; Shchetinin, A.M.; Vishnevskaya, T.V.; Eremyan, A.A.; Alkhovsky, S.V. Isolation and genetic characterization of encephalomyocarditis virus 1 from a deceased captive hamadryas baboon. Virus Res. 2018, 244, 164–172. [Google Scholar] [CrossRef]
- Chang, H.; Chen, L.; Zhao, J.; Wang, X.; Yang, X.; Yao, H.; Wang, C. Complete genome sequence of porcine encephalomyocarditis virus from an aardvark in China. Genome Announc. 2014, 2, e00017-14. [Google Scholar] [CrossRef]
- LaRue, R.; Myers, S.; Brewer, L.; Shaw, D.P.; Brown, C.; Seal, B.S.; Njenga, M.K. A wild-type porcine encephalomyocarditis virus containing a short poly(C) tract is pathogenic to mice, pigs, and cynomolgus macaques. J. Virol. 2003, 77, 9136–9146. [Google Scholar] [CrossRef]
- Fata-Hartley, C.L.; Palmenberg, A.C. Dipyridamole reversibly inhibits mengovirus RNA replication. J. Virol. 2005, 79, 11062–11070. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.H.; Naviaux, R.K.; vanden Brink, K.M.; Jordan, G.W. Comparison of the nucleotide sequences of diabetogenic and nondiabetogenic encephalomyocarditis virus. Virology 1988, 166, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Eun, H.M.; Yoon, J.W. Genomic differences between the diabetogenic and nondiabetogenic variants of encephalomyocarditis virus. Virology 1989, 170, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Nelsen-Salz, B.; Zimmermann, A.; Wickert, S.; Arnold, G.; Botta, A.; Eggers, H.J.; Kruppenbacher, J.P. Analysis of sequence and pathogenic properties of two variants of encephalomyocarditis virus differing in a single amino acid in VP1. Virus Res. 1996, 41, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Berthet, N.; Descorps-Declère, S.; Nkili-Meyong, A.A.; Nakouné, E.; Gessain, A.; Manuguerra, J.-C.; Kazanji, M. Improved assembly procedure of viral RNA genomes amplified with Phi29 polymerase from new generation sequencing data. Biol. Res. 2016, 49, 39. [Google Scholar] [CrossRef] [PubMed]
- Heath, L.; Van Der Walt, E.; Varsani, A.; Martin, D.P. Recombination Patterns in Aphthoviruses Mirror Those Found in Other Picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Welch, J. Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Hicks, A.L.; Duffy, S. Genus-specific substitution rate variability among picornaviruses. J. Virol. 2011, 85, 7942–7947. [Google Scholar] [CrossRef]
- Eigen, M. Viral Quasispecies. Sci. Am. 1993, 269, 42–49. [Google Scholar] [CrossRef]
- Diaz Arenas, C.; Lehman, N. Quasispecies-like behavior observed in catalytic RNA populations evolving in a test tube. BMC Evol. Biol. 2010, 10, 80. [Google Scholar] [CrossRef]
- Aplin, K.P.; Suzuki, H.; Chinen, A.A.; Chesser, R.T.; Have, J.T.; Donnellan, S.C.; Austin, J.; Frost, A.; Gonzalez, J.P.; Herbreteau, V.; et al. multiple geographic origins of commensalism and complex dispersal history of black rats. PLoS ONE 2011, 6, e26357. [Google Scholar] [CrossRef]
- Hulme, P.E. Invasion pathways at a crossroad: Policy and research challenges for managing alien species introductions. J. Appl. Ecol. 2015, 52, 1418–1424. [Google Scholar] [CrossRef]
- Bastos, A.; Chimimba, C.; Von Maltitz, E.; Kirsten, F.; Belmain, S. Identification of rodent species that play a role in disease transmission to humans in South Africa. In Proceedings of the South African Society for Veterinary Epidemiology and Preventive Medicine, Leriba Lodger, Pretoria, South Africa, 11–12 August 2005. [Google Scholar] [CrossRef]
- Matthee, C.A.; Engelbrecht, A.; Matthee, S. Comparative phylogeography of parasitic Laelaps mites contribute new insights into the specialist-generalist variation hypothesis (SGVH). BMC Evol. Biol. 2018, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.C.; Lee, P.L.; Wang, H.C. Molecular detection of Rickettsia species and host associations of Laelaps mites (Acari: Laelapidae) in Taiwan. Exp. Appl. Acarol. 2020, 81, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.A.; Smithburn, K.C.; Haddow, A.J. Mengo encephalomyelitis virus isolation and immunological properties. Br. J. Exp. Pathol. 1948, 29, 547–558. [Google Scholar]
- Dick, G.W.A. Epidemiological notes on some viruses isolated in Uganda (Yellow fever, Rift Valley fever, Bwamba fever, West Nile, Mengo, Semliki forest, Bunyamwera, Ntaya, Uganda S and Zika viruses). Trans. R. Soc. Trop. Med. Hyg. 1953, 47, 13–48. [Google Scholar] [CrossRef]
- Causey, O.; Shope, R.; Laemmert, H. Report of an epizootic of encephalomyocarditis virus in Pará, Brazil. Rev. Serv. Espec. Saude Publica 1962, 12, 47–50. [Google Scholar]
- Paul, S.D.; Pavri, K.M.; D’Lima, L.V.; Rajagopalan, P.K.; Goverdhan, M.K.; Singh, K.R. Isolation of encephalomyocarditis (EMC) virus strains in India. Indian J. Med. Res. 1968, 56, 264–274. [Google Scholar]
- Vanella, J.M.; Kissling, R.E.; Chamberlain, R.W. Transmission Studies with Encephalomyocarditis Virus. J. Infect. Dis. 1956, 98, 98–102. [Google Scholar] [CrossRef]
- Wells, S.K.; Gutter, A.E.; Soike, K.F.; Baskin, G.B. Encephalomyocarditis Virus: Epizootic in a Zoological Collection. J. Zoo Wildl. Med. 1989, 20, 291–296. [Google Scholar]
- Masek-Hammerman, K.; Miller, A.D.; Lin, K.C.; MacKey, J.; Weissenbock, H.; Gierbolini, L.; Burgos, A.; Perez, H.; Mansfield, K.G. Epizootic myocarditis associated with encephalomyocarditis virus in a group of rhesus macaques (Macaca mulatta). Vet. Pathol. 2012, 49, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Azeem, S.; Bengis, R.; Van Aarde, R.; Bastos, A.D.S. Mass Die-Off of African Elephants in Botswana: Pathogen, Poison or a Perfect Storm? Afr. J. Wildl. Res. 2020, 50, 149–156. [Google Scholar] [CrossRef]
- Hunter, P.; Swanepoel, S.P.; Esterhuysen, J.J.; Raath, J.P.; Bengis, R.G.; Van der Lugt, J.J. The efficacy of an experimental oil-adjuvanted encephalomyocarditis vaccine in elephants, mice and pigs. Vaccine 1998, 16, 55–61. [Google Scholar] [CrossRef] [PubMed]
- McLelland, D.J.; Kirkland, P.D.; Rose, K.A.; Dixon, R.J.; Smith, N. Serologic responses of Barbary sheep (Ammotragus lervia), Indian antelope (Antilope cervicapra), wallaroos (Macropus robustus), and chimpanzees (Pan troglodytes) to an inactivated encephalomyocarditis virus vaccine. J. Zoo Wildl. Med. 2005, 36, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Kilburn, J.J.; Murphy, D.P.; Titus, M.; Payton, M.E.; Backues, K.A. Vaccination of llamas, Llama glama, with an experimental killed encephalomyocarditis virus vaccine. J. Zoo Wildl. Med. 2011, 42, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Scollo, A.; Mazzoni, C.; Luppi, A. Management of encephalomyocarditis virus infection in Italian pig farms: A case report. BMC Vet. Res. 2023, 19, 54. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Meer, V.; Pawęska, J.T.; Swanepoel, R.; Grobbelaar, A.; Bastos, A.D. Genome Sequencing of Historical Encephalomyocarditis Viruses from South Africa Links the Historical 1993/4 Savanna Elephant (Loxodonta africana) Outbreak to Cryptic Mastomys Rodents. Pathogens 2024, 13, 261. https://doi.org/10.3390/pathogens13030261
van Meer V, Pawęska JT, Swanepoel R, Grobbelaar A, Bastos AD. Genome Sequencing of Historical Encephalomyocarditis Viruses from South Africa Links the Historical 1993/4 Savanna Elephant (Loxodonta africana) Outbreak to Cryptic Mastomys Rodents. Pathogens. 2024; 13(3):261. https://doi.org/10.3390/pathogens13030261
Chicago/Turabian Stylevan Meer, Vanessa, Janusz T. Pawęska, Robert Swanepoel, Antoinette Grobbelaar, and Armanda D. Bastos. 2024. "Genome Sequencing of Historical Encephalomyocarditis Viruses from South Africa Links the Historical 1993/4 Savanna Elephant (Loxodonta africana) Outbreak to Cryptic Mastomys Rodents" Pathogens 13, no. 3: 261. https://doi.org/10.3390/pathogens13030261