
Genetic Diversity of Trypanosomes Infesting Cattle from Savannah District in North of Côte d’Ivoire Using Conserved Genomic Signatures: rRNA, ITS1 and gGAPDH
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection in the Field
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Analysis of Sequences
3. Results
3.1. Sequence Identification
3.2. Mixed Infection and Mixed Identification by Primers
3.3. Sequence Comparison between the Different Trypanosome Species Identified
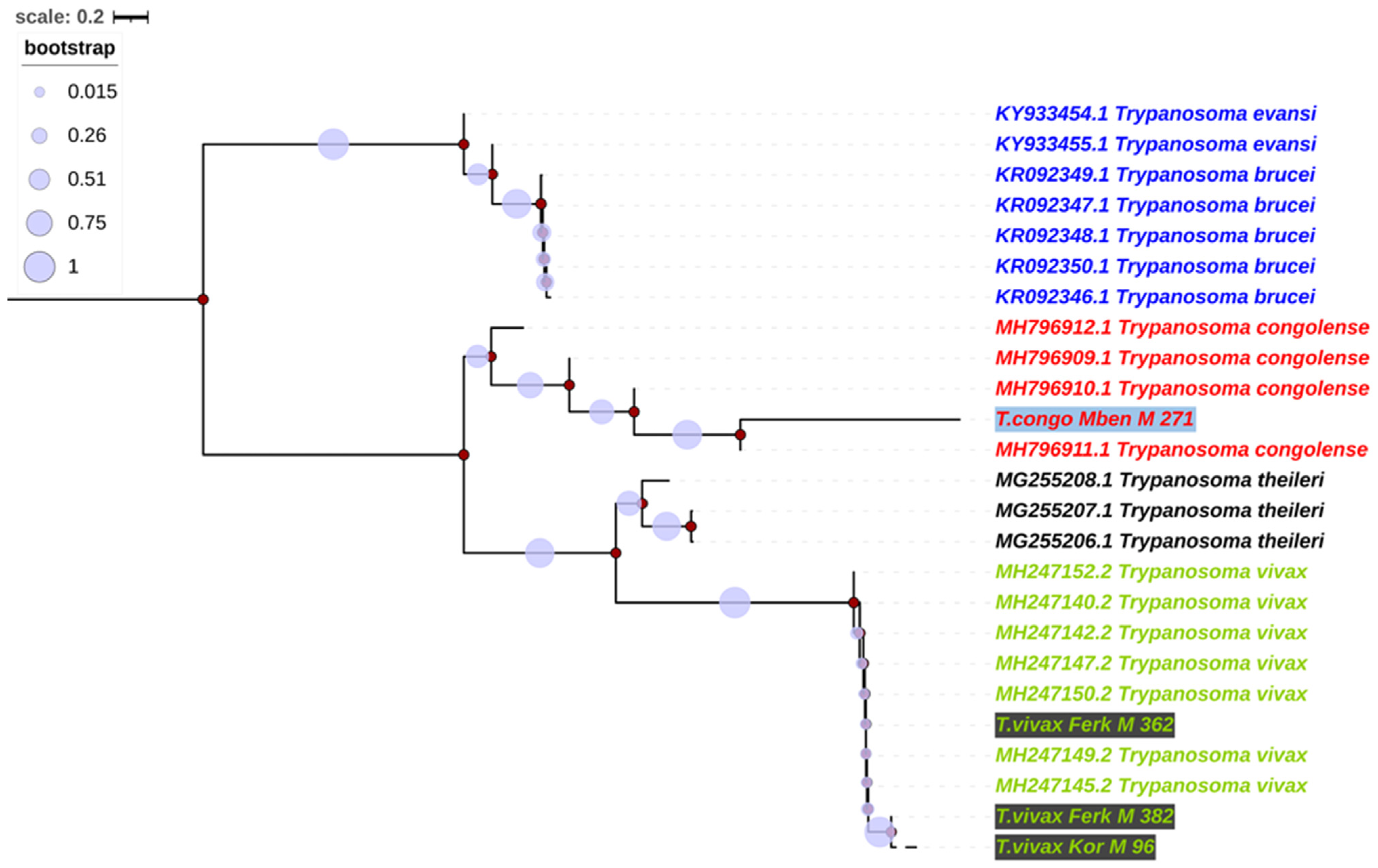
3.4. Alignment and Phylogenetics of Trypanosome Species According to Internal Transcribed Spacer 1 (ITS1)
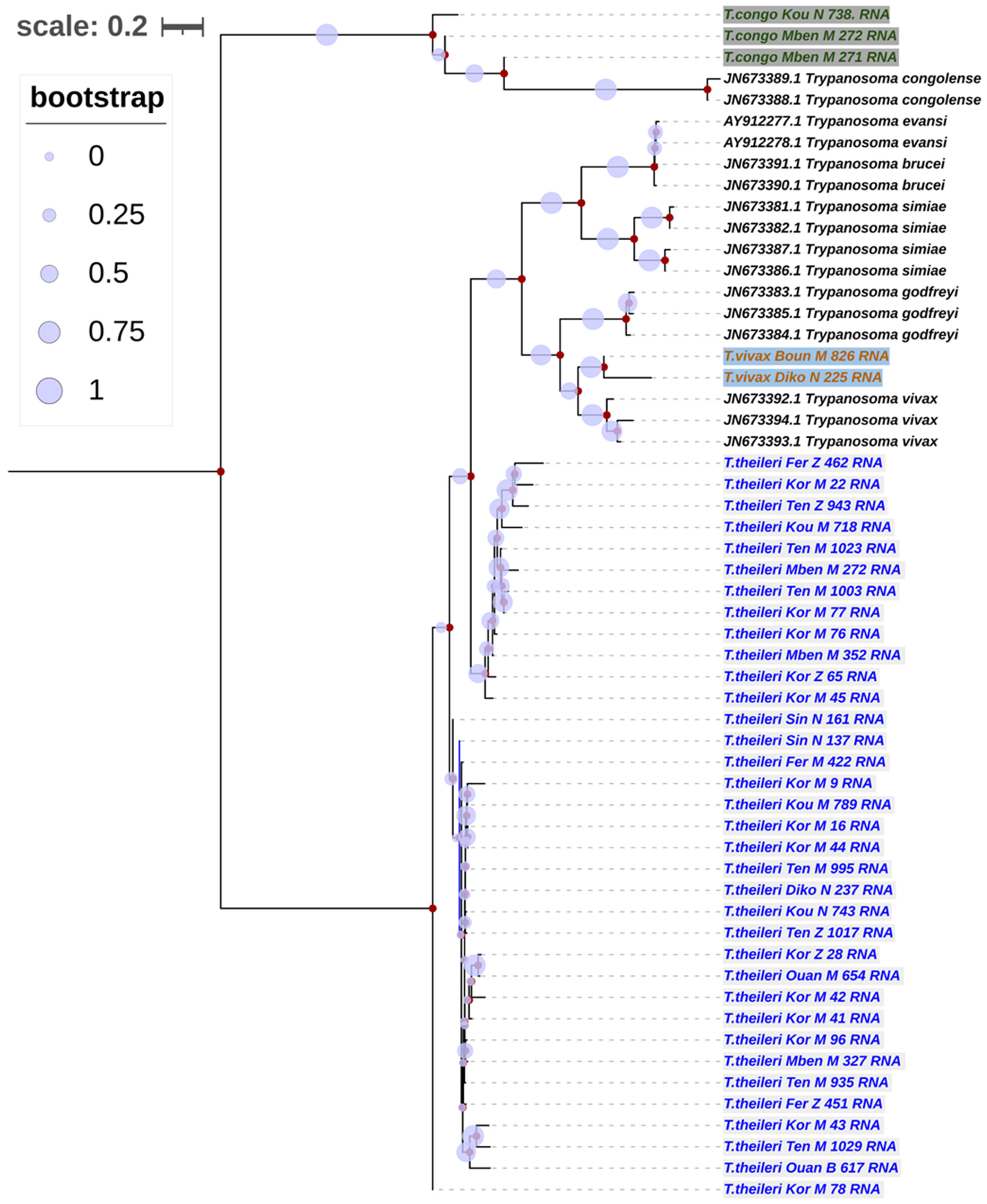
3.5. Phylogenetic Trees of Trypanosomes Species According to Partial 18S, ITS1, 5.8S, ITS2 and Partial 28S rRNA Genes
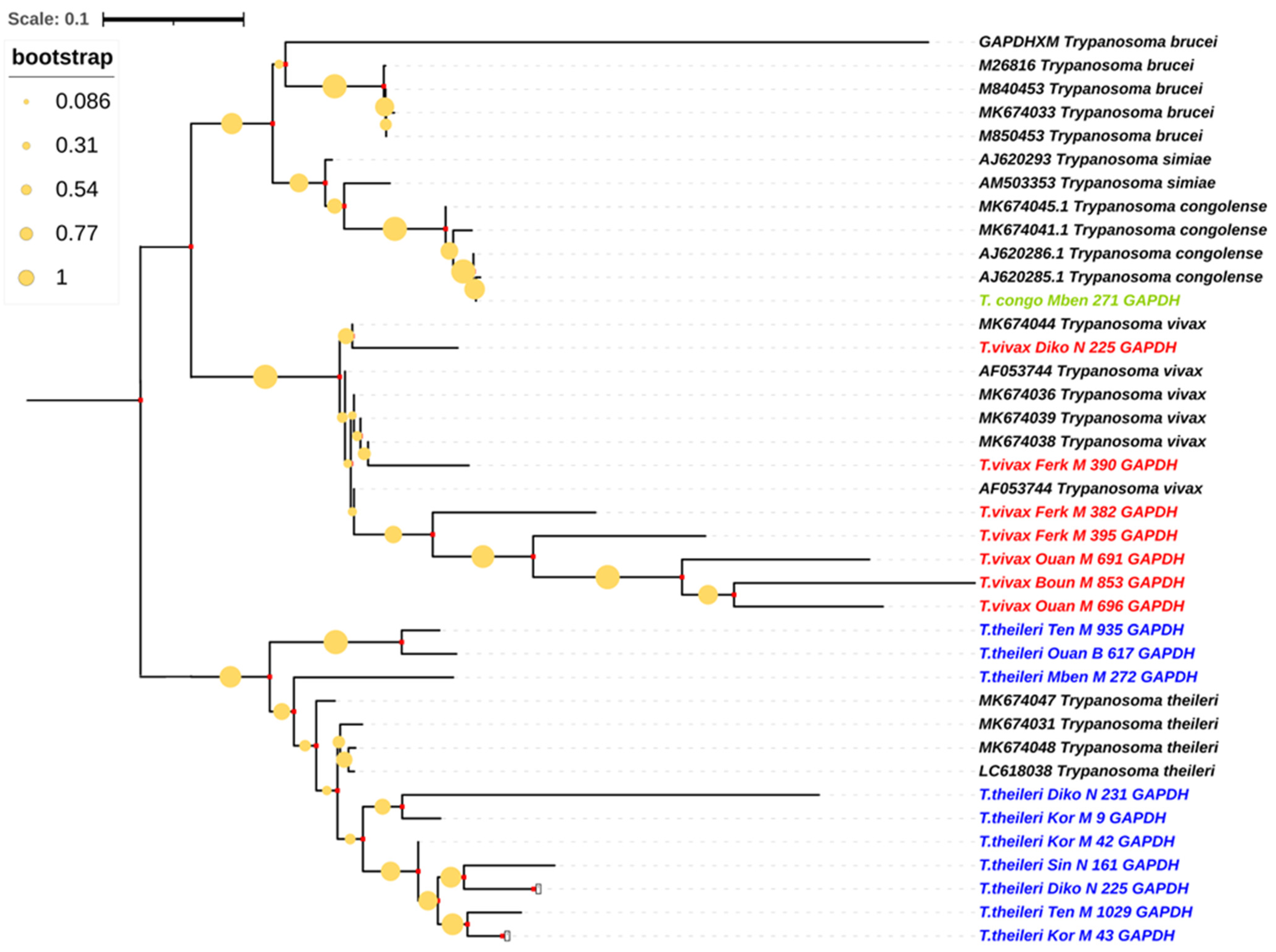
3.6. Phylogenetic Trees of Trypanosome Species According to Glycosomal Glyceraldehyde-3-Phosphate Dehydrogenase Gene (gGAPDH)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semayat, O.; Maireg, H. Review on Prevalence of Bovine Trypanosomosis in Ethiopia. Afr. J. Agric. Res. 2018, 13, 1–6. [Google Scholar] [CrossRef]
- Solano, P.; Ravel, S.; de Meeûs, T. How Can Tsetse Population Genetics Contribute to African Trypanosomiasis Control. Trends Parasitol. 2010, 26, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.J.; Williams, B.G. Tsetse Distribution in Africa: Seeing the Wood and the Trees. In Large Scale Ecology and Conservation Biology: The 35th Symposium of Conservation Biology, University of Southampton, 1993; Wiley–Blackwell: Hoboken, NJ, USA, 1994; pp. 247–271. [Google Scholar]
- Mulandane, F.C.; Snyman, L.P.; Brito, D.R.A.; Bouyer, J.; Fafetine, J.; Van Den Abbeele, J.; Oosthuizen, M.; Delespaux, V.; Neves, L. Evaluation of the Relative Roles of the Tabanidae and Glossinidae in the Transmission of Trypanosomosis in Drug Resistance Hotspots in Mozambique. Parasites Vectors 2020, 13, 219. [Google Scholar] [CrossRef] [PubMed]
- Desquesnes, M.; Bengaly, Z.; Berthier, D.; Bossard, G.; Cene, B.; Ilboudo, H. Compendium of Diagnostic Protocols of the OIE Reference Laboratory for Animal Trypanosomoses of African Origin; OIE: Montpellier, France, 2017. [Google Scholar]
- Paris, J.; Murray, M.; McOdimba, F. A Comparative Evaluation of the Parasitological Techniques Currently Available for the Diagnosis of African Trypanosomiasis in Cattle. Acta Trop. 1982, 39, 307–316. [Google Scholar] [PubMed]
- Desquesnes, M.; Gonzatti, M.; Sazmand, A.; Thévenon, S.; Bossard, G.; Boulangé, A.; Gimonneau, G.; Truc, P.; Herder, S.; Ravel, S.; et al. A Review on the Diagnosis of Animal Trypanosomoses. Parasites Vectors 2022, 15, 64. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan-a Web and Mobile App for Systematic Reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations. A Field Giude for Diadnosis, Treatment and Preventio of Affrican Animal Trypanosomisis; Food and Agriculture Organization of The United Nations: Rome, Italy, 1998; pp. 99–100. [Google Scholar]
- Solano, P.; Michel, J.F.; Lefrançois, T.; De La Rocque, S.; Sidibé, I.; Zoungrana, A.; Cuisance, D. Polymerase Chain Reaction as a Diagnosis Tool for Detecting Trypanosomes in Naturally Infected Cattle in Burkina Faso. Vet. Parasitol. 1999, 86, 95–103. [Google Scholar] [CrossRef]
- Njiru, Z.K.; Constantine, C.C.; Guya, S.; Crowther, J.; Kiragu, J.M.; Thompson, R.C.A.; Dávila, A.M.R. The Use of ITS1 rDNA PCR in Detecting Pathogenic African Trypanosomes. Parasitol. Res. 2005, 95, 186–192. [Google Scholar] [CrossRef]
- Njiru, Z.K.; Mikosza, A.S.J.; Matovu, E.; Enyaru, J.C.K.; Ouma, J.O.; Kibona, S.N.; Thompson, R.C.A.; Ndung’u, J.M. African Trypanosomiasis: Sensitive and Rapid Detection of the Sub-Genus Trypanozoon by Loop-Mediated Isothermal Am-plification (LAMP) of Parasite DNA. Int. J. Parasitol. 2008, 38, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Malele, I.; Craske, L.; Knight, C.; Ferris, V.; Njiru, Z.; Hamilton, P.; Lehane, S.; Lehane, M.; Gibson, W. The Use of Specific and Generic Primers to Identify Trypanosome Infections of Wild Tsetse Flies in Tanzania by PCR. Infect. Genet. Evol. J. Mol. Epi-demiol. Evol. Genet. Infect. Dis. 2003, 3, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Majiwa, P.A.; Maina, M.; Waitumbi, J.N.; Mihok, S.; Zweygarth, E. Trypanosoma (Nannomonas) Congolense: Molecular Characterization of a New Genotype from Tsavo, Kenya. Parasitology 1993, 106, 151–162. [Google Scholar] [CrossRef]
- Hamilton, P.B.; Adams, E.R.; Malele, I.I.; Gibson, W.C. A Novel, High-Throughput Technique for Species Identification Reveals a New Species of Tsetse-Transmitted Trypanosome Related to the Trypanosoma Brucei Subgenus, Trypanozoon. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2008, 8, 26–33. [Google Scholar] [CrossRef]
- Ekra, J.Y.; N’goran, E.K.; Mboera, L.E.G.; Gragnon, B.G.; Assovié, K.R.N.; Mafie, E.M. Molecular Epidemiological Survey of Pathogenic Trypanosomes in Naturally Infected Cattle in Northern Côte d’ivoire. Parasites Hosts Dis. 2023, 61, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.S.; Gragnon, B.G.; Dri, K.L.N.; Coulibaly, B.; Komono, D. Prévalence Trypanosomienne Dans Le Bassin Cotonnier En Zone Soudanaise de Côte d’Ivoire. IAV Hassan II 2020, 8, 417–424. [Google Scholar]
- N’Djetchi, M.K.; Ilboudo, H.; Koffi, M.; Kaboré, J.; Kaboré, J.W.; Kaba, D.; Courtin, F.; Coulibaly, B.; Fauret, P.; Kouakou, L.; et al. The Study of Trypanosome Species Cir-culating in Domestic Animals in Two Human African Trypanosomiasis Foci of Côte d’Ivoire Identifies Pigs and Cattle as Potential Reservoirs of Trypanosoma Brucei Gambiense. PLoS Negl. Trop. Dis. 2017, 11, e0005993. [Google Scholar] [CrossRef]
- Cox, A.; Tilley, A.; Mcodimba, F.; Fyfe, J.; Eisler, M.; Hide, G.; Welburn, S. A PCR Based Assay for Detection and Differentiation of African Trypanosome Species in Blood. Exp. Parasitol. 2005, 111, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, P.B.; Stevens, J.R.; Gaunt, M.W.; Gidley, J.; Gibson, W.C. Trypanosomes Are Monophyletic: Evidence from Genes for Glyceraldehyde Phosphate Dehydrogenase and Small Subunit Ribosomal RNA. Int. J. Parasitol. 2004, 34, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the Number of Nucleotide Substitutions When There Are Strong Transition-Transversion and G+C-Content Biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nu-cleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for Inferring Very Large Phylogenies by Using the Neighbor-Joining Method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed]
- Auty, H.; Anderson, N.E.; Picozzi, K.; Lembo, T.; Mubanga, J.; Hoare, R.; Fyumagwa, R.D.; Mable, B.; Hamill, L.; Cleaveland, S.; et al. Trypanosome Diversity in Wildlife Species from the Serengeti and Luangwa Valley Ecosystems. PLoS Negl. Trop. Dis. 2012, 6, e1828. [Google Scholar] [CrossRef] [PubMed]
- Djohan, V.; Kaba, D.; Rayaissé, J.B.; Dayo, G.K.; Coulibaly, B.; Salou, E.; Dofini, F.; Kouadio, A.D.M.K.; Menan, H.; Solano, P. Detection and Identification of Pathogenic Trypanosome Species in Tsetse Flies along the Comoé River in Côte d’Ivoire. Parasite 2015, 22, 18. [Google Scholar] [CrossRef] [PubMed]
- Acapovi-Yao, G.; Mavoungou, J.; Kohagne, L.; Ta, D. Dynamique et Dispersion Spatiale Des Tabanidae Dans Différents Faciès Écologiques de Korhogo En Côte d’Ivoire. Entomol. Faun.—Faun. Entomol. 2017, 70, 13–22. [Google Scholar]
- Desquesnes, M.; Dia, M.L. Mechanical Transmission of Trypanosoma Congolense in Cattle by the African Tabanid Atylotus Agrestis. Exp. Parasitol. 2003, 105, 226–231. [Google Scholar] [CrossRef]
- Abdi, R.D. Epidemiology of Tsetse Flies and Trypanosomes with a Case Study in Ethiopia. Afr. Focus 2016, 29, 109–116. [Google Scholar] [CrossRef]
- Kostygov, A.Y.; Frolov, A.O.; Malysheva, M.N.; Ganyukova, A.I.; Drachko, D.; Yurchenko, V.; Agasoi, V.V. Development of Two Species of the Trypanosoma Theileri Complex in Tabanids. Parasites Vectors 2022, 15, 95. [Google Scholar] [CrossRef] [PubMed]
- Keita, M.L.; Medkour, H.; Sambou, M.; Dahmana, H.; Mediannikov, O. Tabanids as Possible Pathogen Vectors in Senegal (West Africa). Parasites Vectors 2020, 13, 500. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Linder, C.R.; Warnow, T. Multiple Sequence Alignment: A Major Challenge to Large-Scale Phylogenetics. PLoS Curr. 2010, 2, RRN1198. [Google Scholar] [CrossRef] [PubMed]
- Ekra, J.Y.; N’goran, E.K.; Mboera, L.E.G.; Mafie, E.M. Prevalence of Bovine Trypanosomiasis in Côte d’Ivoire: Systematic Review and Meta-Analysis. Onderstepoort J. Vet. Res. 2023, 90, e1–e8. [Google Scholar] [CrossRef]
- Majiwa, P.A.O.; Hamers, R.; Van Meirvenne, N.; Matthyssens, G. Evidence for Genetic Diversity in Trypanosoma (Nan-nomonas) Congolense. Parasitology 1986, 93, 291–304. [Google Scholar] [CrossRef]
- Desquesnes, M.; Dia, M.L. Trypanosoma Vivax: Mechanical Transmission in Cattle by One of the Most Common African Tabanids, Atylotus Agrestis. Exp. Parasitol. 2003, 103, 35–43. [Google Scholar] [CrossRef]
- Adams, E.R.; Hamilton, P.B.; Rodrigues, A.C.; Malele, I.I.; Delespaux, V.; Teixeira, M.M.G.; Gibson, W. New Trypanosoma (Duttonella) Vivax Genotypes from Tsetse Flies in East Africa. Parasitology 2010, 137, 641–650. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sequence ID | Accession Number | Bovine Race and Location | Length of Sequence (bp) | Similarity (%) | Reference Sequence Accession | Origin Country of Reference |
|---|---|---|---|---|---|---|
| ITS1 | ||||||
| T.vivax_Kor_M_96 a | PP210924 | Méré/Korhogo | 209 | 97.56 | MH247152.2 T. vivax | Kenya |
| T.congo_Mben_M_271 1 | PP210925 | Méré/Mbengue | 666 | 80.57 | MK756202.1 T. congolense | Nigeria |
| T.vivax_Ferk_M_382 | PP210926 | Méré/Ferkéssédougou | 250 | 94.31 | MN213748.1 T. vivax | Sub-Saharan Africa |
| T.vivax_Ferk_M_362 b | PP210927 | Méré/Ferkéssédougou | 250 | 100 | MN213748.1 T. vivax | Sub-Saharan Africa |
| 18S, ITS1, 5.8S, ITS2, 28S | ||||||
| T.theileri_Kor_M_9_RNA 2 | PP188043 | Méré/Korhogo | 932 | 92.60 | LC440407.1 T. theileri | Mongolia |
| T.theileri_Kor_M_16_RNA | PP188044 | Méré/Korhogo | 939 | 99.43 | OQ341207.1 T. theileri | Ecuador |
| T.theileri_Kor_M_22_RNA | OR973744 | Méré/Korhogo | 707 | 84.18 | LC440405.1 T. theileri | Mongolia |
| T.theileri_Kor_Z_28_RNA | OR973745 | Zébu/Korhogo | 872 | 94.13 | OQ341206.1 T. theileri | Ecuador |
| T.theileri_Kor_M_41_RNA | PP188045 | Méré/Korhogo | 930 | 93.87 | LC440408.1 T. theileri | Mongolia |
| T.theileri_Kor_M_42_RNA 5 | OR973746 | Méré/Korhogo | 776 | 92.22 | AB569248.1 T. theileri | Japan |
| T.theileri_Kor_M_43_RNA 6 | OR973747 | Méré/Korhogo | 957 | 90.27 | KY412803.1 T. theileri | Austria |
| T.theileri_Kor_M_44_RNA | OR973748 | Méré/Korhogo | 912 | 99.01 | OQ341206.1 T. theileri | Ecuador |
| T.theileri_Kor_M_45_RNA | PP188046 | Méré/Korhogo | 939 | 98.73 | LC440405.1 T. theileri | Mongolia |
| T.theileri_Kor_Z_52_RNA | OR973749 | Zébu/Korhogo | 930 | 99.45 | OQ341205.1 T. theileri | Ecuador |
| T.theileri_Kor_Z_65_RNA | OR973750 | Zébu/Korhogo | 744 | 90.87 | OQ341215.1 T. theileri | Ecuador |
| T.theileri_Kor_M_76_RNA | OR973751 | Méré/Korhogo | 764 | 94.89 | AB569249.1 T. theileri | Japan |
| T.theileri_Kor_M_77_RNA | OR973752 | Méré/Korhogo | 939 | 97.23 | AB569249.1 T. theileri | Japan |
| T.theileri_Kor_M_78_RNA | OR973753 | Méré/Korhogo | 687 | 88.35 | AB569248.1 T. theileri | Japan |
| T.theileri_Kor_M_96_RNA a | OR973754 | Méré/Korhogo | 914 | 98.57 | KY412803.1 T. theileri | Austria |
| T.theileri_Sin_N_119_RNA | OR973755 | N’Dama/Sinématiali | 617 | 88.77 | OQ341215.1 T. theileri | Ecuador |
| T.theileri_Sin_N_137_RNA | OR973756 | N’Dama/Sinématiali | 716 | 99.16 | LC440408.1 T. theileri | Mongolia |
| T.theileri_Sin_N_161_RNA | PP188047 | N’Dama/Sinématiali | 931 | 98.83 | OQ341205.1 T. theileri | Ecuador |
| T.vivax_Diko_N_225_RNA 3 | PP188048 | N’Dama/Dikodougou | 575 | 84.88 | KM391836.1 T. vivax | Nigeria |
| T.theileri_Diko_N_237_RNA | OR973757 | N’Dama/Dikodougou | 925 | 98.26 | OQ341205.1 T. theileri | Ecuador |
| T.congo_Mben_M_271_RNA 1 | PP188049 | Méré/M’Bengué | 815 | 85.40 | U22319.1 T. congolense | Ecuador |
| T.theileri_Mben_M_272_RNA c | PP188050 | Méré/M’Bengué | 918 | 93.69 | OQ341215.1 T. theileri | Ecuador |
| T.congo_Mben_M_272_RNA c 9 | PP188052 | Méré/M’Bengué | 710 | 85.76 | U22319.1 T. congolense | Kenya |
| T.theileri_Mben_M_327_RNA | OR973758 | Méré/M’Bengué | 819 | 97.68 | LC440408.1 T. theileri | Ecuador |
| T.theileri_Mben_M_352_RNA | OR973759 | Méré/M’Bengué | 750 | 96.55 | AB569249.1 T. theileri | Japan |
| T.theileri_Fer_M_362_RNA b | OR973760 | Méré/Ferkéssédougou | 708 | 95.28 | AB569249.1 T. theileri | Japan |
| T.theileri_Fer_M_422_RNA | OR973761 | Méré/Ferkéssédougou | 757 | 97.74 | OQ341209.1 T. theileri | Ecuador |
| T.theileri_Fer_Z_451_RNA | OR973762 | Zébu/Ferkéssédougou | 904 | 96.50 | KY412803.1 T. theileri | Austria |
| T.theileri_Fer_Z_462_RNA | OR973763 | Zébu/Ferkéssédougou | 763 | 85.87 | LC440405.1 T. theileri | Mongolia |
| T.theileri_Ouan_B_617_RNA 4 | OR973764 | Baoulé/Ouangolodougou | 700 | 87.43 | JX178185.1 T. theileri | USA |
| T.theileri_Ouan_M_654_RNA | OR973765 | Méré/Ouangolodougou | 880 | 95.72 | OQ341206.1 T. theileri | Ecuador |
| T.theileri_Kou_M_718_RNA | OR973766 | Méré/Kouto | 931 | 89.04 | AB569249.1 T. theileri | Japan |
| T.congo_Kou_N_738_RNA | PP188051 | N’Dama/Kouto | 1457 | 85.64 | U22319.1 T. congolense | Ecuador |
| T.theileri_Kou_N_743_RNA | OR973767 | N’Dama/Kouto | 934 | 98.93 | OQ341206.1 T. theileri | Ecuador |
| T.theileri_Kou_M_789_RNA | OR973768 | Méré/Kouto | 924 | 99.14 | KY412803.1 T. theileri | Austria |
| T.vivax_Boun_M_826_RNA | PP188053 | Méré/Boundiali | 549 | 96.72 | KC196660.1 T. vivax | Gambia |
| T.vivax_Boun_M_854_RNA | OR973769 | Méré/Boundiali | 740 | 83.02 | AB569249.1 T. theileri | Japan |
| T.theileri_Ten_M_935_RNA | OR973770 | Méré/Tengréla | 922 | 99.23 | KY412803.1 T. theileri | Austria |
| T.theileri_Ten_Z_943_RNA | OR973771 | Zébu/Tengréla | 888 | 89.19 | OQ341215.1 T. theileri | Ecuador |
| T.theileri_Ten_M_995_RNA | OR973772 | Méré/Tengréla | 937 | 98.79 | OQ341205.1 T. theileri | Ecuador |
| T.theileri_Ten_M_1003_RNA | OR973773 | Méré/Tengréla | 925 | 96.95 | AB569249.1 T. theileri | Japan |
| T.theileri_Ten_Z_1017_RNA | OR973774 | Zébu/Tengréla | 1048 | 97.97 | OQ341206.1 T. theileri | Ecuador |
| T.theileri_Ten_M_1023_RNA | OR973775 | Méré/Tengréla | 940 | 97.12 | AB569249.1 T. theileri | Japan |
| T.theileri_Ten_M_1029_RNA 8 | OR973776 | Méré/Tengréla | 1168 | 91.27 | KY412803.1 T. theileri | Austria |
| gGAPDH | ||||||
| T.theileri_Kor_M_9_GAPDH 2 | OR966684 | Méré/Korhogo | 865 | 97.38 | XM_029023637.1 T. theileri | United Kingdom |
| T.vivax_Diko_N_225_GAPDH 3 | OR966685 | N’Dama/Dikodougou | 752 | 90.83 | MK674038.1 T. vivax | Cameroon |
| T.vivax_Ferk_M_390_GAPDH | OR966686 | Méré/Ferkéssédougou | 424 | 91.23 | MK674038.1 T. vivax | Cameroon |
| T.vivax_Ferk_M_395_GAPDH | OR966687 | Méré/Ferkéssédougou | 391 | 89.88 | MK674038.1 T. vivax | Cameroon |
| T.theileri_Ouan_B_617_GAPDH 4 | OR966688 | Baoulé/Ouangolodougou | 342 | 93.08 | XM_029023637.1 T. theileri | United Kingdom |
| T.vivax_Ouan_M_691_GAPDH | OR966689 | Méré/Ouangolodougou | 331 | 84.72 | MK674007.1 T. vivax | Cameroon |
| T.vivax_Ouan_M_696_GAPDH | OR966690 | Méré/Ouangolodougou | 339 | 84.55 | MK674007.1 T. vivax | Cameroon |
| T.vivax_Boun_M_853_GAPDH | OR966691 | Méré/Boundiali | 317 | 85.96 | MK674007.1 T. vivax | Cameroon |
| T.theileri_Ten_M_935_GAPDH 7 | OR966692 | Méré/Tengréla | 349 | 95.13 | XM_029023637.1 T. theileri | United Kingdom |
| T.theileri_Kor_M_42_GAPDH 5 | OR966693 | Méré/Korhogo | 504 | 97.35 | XM_029023637.1 T. theileri | United Kingdom |
| T.theileri_Kor_M_43_GAPDH 6 | OR966694 | Méré/Korhogo | 294 | 97.29 | XM_029023637.1 T. theileri | United Kingdom |
| T.theileri_Sin_N_161_GAPDH | OR966695 | N’Dama/Sinématiali | 323 | 89.30 | LC618038.1 T. theileri | Japan |
| T.theileri_Diko_N_231_GAPDH | OR966696 | N’Dama/Dikodougou | 245 | 88.36 | HQ664784.1 T. theileri | Brazil |
| T.congolense_Mben_M_271_GAPDH | OR966697 | Méré/M’Bengué | 270 | 90.08 | AJ620289.1 T. congolense | Cameroon |
| T.theileri_Mben_M_272_GAPDH | OR966698 | Méré/M’Bengué | 303 | 96.08 | AJ620282.1 T. theileri | Cameroon |
| T.vivax_Ferk_M_382_GAPDH | OR966699 | Méré/Ferkéssédougou | 300 | 91.04 | MK674044.1 T. vivax | Cameroon |
| T.theileri_Ouan_B_617_GAPDH | OR966700 | Baoulé/Ouangolodougou | 337 | 93.31 | XM_029023637.1 T. theileri | United Kingdom |
| T.vivax_Boun_N_816_GAPDH | OR966701 | N’Dama/Boundiali | 481 | 88.12 | MK674044.1 T. vivax | Cameroon |
| T.theileri_Ten_M_935_GAPDH 7 | OR966702 | Méré/Tengréla | 338 | 97.98 | LC618038.1 T. theileri | Japan |
| T.theileri_Ten_M_1029_GAPDH 8 | OR966703 | Méré/Tengréla | 328 | 92.76 | XM_029023637.1 T. theileri | United Kingdom |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ekra, J.-Y.; Mafie, E.M.; N’Goran, E.K.; Kaba, D.; Gragnon, B.G.; Srinivasan, J. Genetic Diversity of Trypanosomes Infesting Cattle from Savannah District in North of Côte d’Ivoire Using Conserved Genomic Signatures: rRNA, ITS1 and gGAPDH. Pathogens 2024, 13, 262. https://doi.org/10.3390/pathogens13030262
Ekra J-Y, Mafie EM, N’Goran EK, Kaba D, Gragnon BG, Srinivasan J. Genetic Diversity of Trypanosomes Infesting Cattle from Savannah District in North of Côte d’Ivoire Using Conserved Genomic Signatures: rRNA, ITS1 and gGAPDH. Pathogens. 2024; 13(3):262. https://doi.org/10.3390/pathogens13030262
Chicago/Turabian StyleEkra, Jean-Yves, Eliakunda Michael Mafie, Edouard K. N’Goran, Dramane Kaba, Biégo Guillaume Gragnon, and Jagan Srinivasan. 2024. "Genetic Diversity of Trypanosomes Infesting Cattle from Savannah District in North of Côte d’Ivoire Using Conserved Genomic Signatures: rRNA, ITS1 and gGAPDH" Pathogens 13, no. 3: 262. https://doi.org/10.3390/pathogens13030262