Diversity and Molecular Evolution of Antimicrobial Peptides in Caecilian Amphibians

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
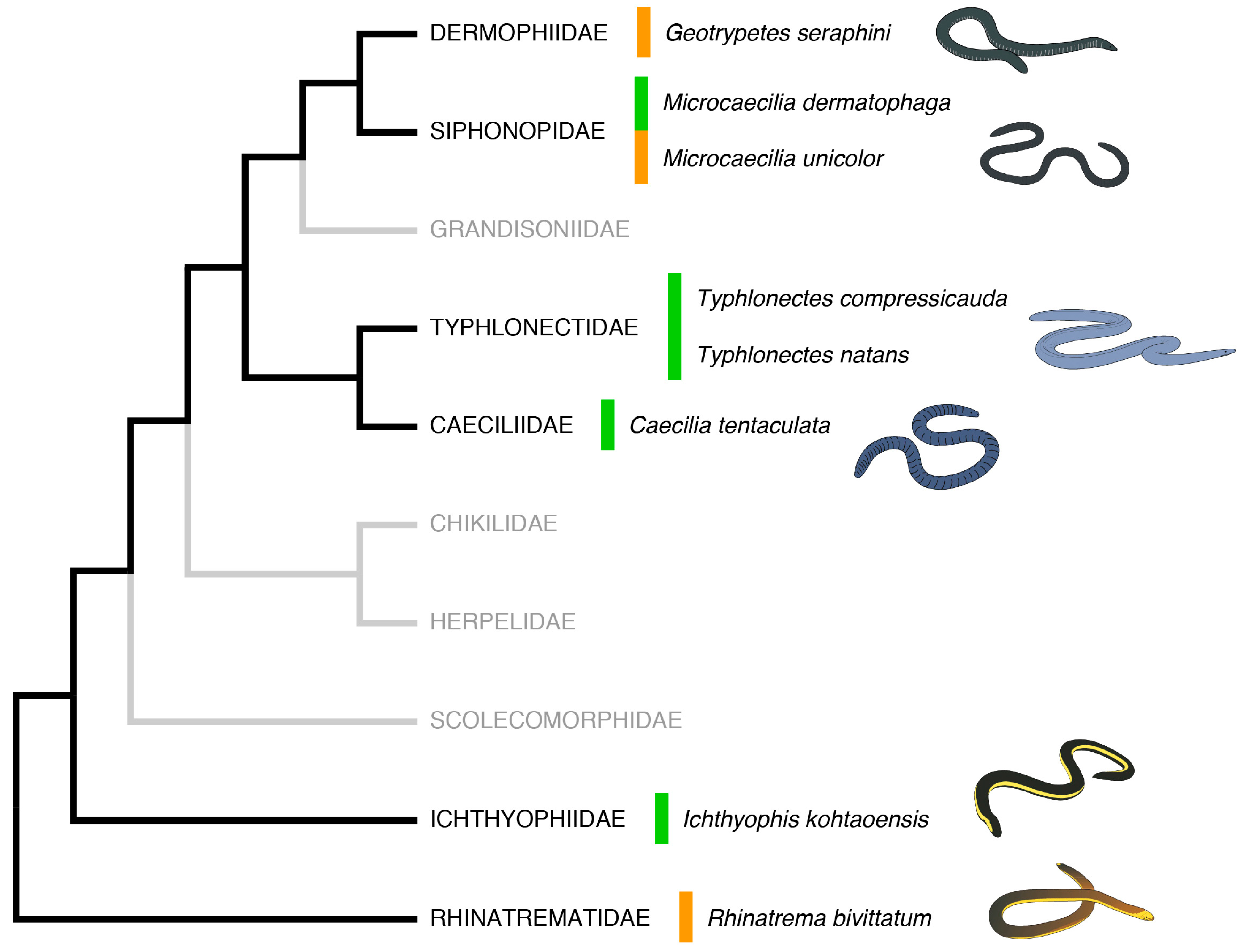
2.1. Sequence-Similarity Searches
2.2. Cataloguing of Potential AMP Sequences
2.3. Phylogenetic and Directional Selection Analyses
2.3.1. Calcitonin and Amylin Tree
2.3.2. Lysozyme G Tree
2.3.3. Cofilin Tree
2.4. Prediction of Antimicrobial Activity of Candidate Caecilian AMPs
2.5. Three-Dimensional Structure of Caecilian GADPH-Related and Histone H2A-Related Peptides
3. Discussion
3.1. Diversity of Candidate AMPs in Caecilians
3.2. Molecular Evolution of Candidate AMPs
3.3. In Silico Activity Prediction of Antimicrobial Properties
3.4. Structural Modelling of Candidate AMPs
4. Conclusions
5. Material and Methods
5.1. Data Retrieval
5.2. Identification of Antimicrobial Peptides
5.3. Alignment, Debugging, and Duplicates Removal
5.4. Phylogenetic and Directional Selection Analyses
5.5. In Silico Prediction of Antimicrobial Properties
5.6. In Silico Prediction of the 3D Structure of Candidate AMPs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Govender, T.; Dawood, A.; Esterhuyse, A.J.; Katerere, D.R. Antimicrobial Properties of the Skin Secretions of Frogs. S. Afr. J. Sci. 2012, 108, 25–30. [Google Scholar] [CrossRef]
- Otero, J.R. Resistencia a Los Antibióticos: La Evolución En Acción. Dendra Médica. Revista de Humanidades 2011, 10, 56–64. [Google Scholar]
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2850. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, J. Frog Skin Hope for HIV Prevention. Drug Discov. Today 2005, 10, 1489–1490. [Google Scholar] [CrossRef] [PubMed]
- Nasseri, S.; Sharifi, M. Therapeutic Potential of Antimicrobial Peptides for Wound Healing. Int. J. Pept. Res. Ther. 2022, 28, 38. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Yan, Z.-B.; Meng, Y.-M.; Hong, X.-Y.; Shao, G.; Ma, J.-J.; Cheng, X.-R.; Liu, J.; Kang, J.; Fu, C.-Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial Peptides of Innate Immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyżewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of Antimicrobial Peptides. A Review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Gallo, R.L. Antimicrobial Peptides: Old Molecules with New Ideas. J. Investig. Dermatol. 2012, 132, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Starr, C.G.; Maderdrut, J.L.; He, J.; Coy, D.H.; Wimley, W.C. Pituitary Adenylate Cyclase-Activating Polypeptide Is a Potent Broad-Spectrum Antimicrobial Peptide: Structure-Activity Relationships. Peptides 2018, 104, 35–40. [Google Scholar] [CrossRef]
- Velázquez, J.; Rodríguez-Cornejo, T.; Rodríguez-Ramos, T.; Pérez-Rodríguez, G.; Rivera, L.; Campbell, J.H.; Al-Hussinee, L.; Carpio, Y.; Estrada, M.P.; Dixon, B. New Evidence for the Role of Pituitary Adenylate Cyclase-Activating Polypeptide as an Antimicrobial Peptide in Teleost Fish. Antibiotics 2023, 12, 1484. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Q.; Chen, J.-C.; Cui, Y.-X.; Zhou, B.; Chen, Y.-X.; Zhao, Y.-F.; Li, Y.-M. Antimicrobial Activity of Human Islet Amyloid Polypeptides: An Insight into Amyloid Peptides’ Connection with Antimicrobial Peptides. Biol. Chem. 2012, 393, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Athira, P.P.; Anju, M.V.; Anooja, V.V.; Archana, K.; Neelima, S.; Rosamma, P. A Histone H2A-Derived Antimicrobial Peptide, Hipposin from Mangrove Whip Ray, Himantura Walga: Molecular and Functional Characterisation. 3 Biotech 2020, 10, 467. [Google Scholar] [CrossRef] [PubMed]
- Liepke, C.; Baxmann, S.; Heine, C.; Breithaupt, N.; Ständker, L.; Forssmann, W.-G. Human Hemoglobin-Derived Peptides Exhibit Antimicrobial Activity: A Class of Host Defense Peptides. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2003, 791, 345–356. [Google Scholar] [CrossRef]
- Wagener, J.; Schneider, J.J.; Baxmann, S.; Kalbacher, H.; Borelli, C.; Nuding, S.; Küchler, R.; Wehkamp, J.; Kaeser, M.D.; Mailänder-Sanchez, D.; et al. A Peptide Derived from the Highly Conserved Protein GAPDH Is Involved in Tissue Protection by Different Antifungal Strategies and Epithelial Immunomodulation. J. Investig. Dermatol. 2013, 133, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Sarkar, T.; Chetia, M.; Chatterjee, S. Antimicrobial Peptides and Proteins: From Nature’s Reservoir to the Laboratory and Beyond. Front. Chem. 2021, 9, 691532. [Google Scholar] [CrossRef]
- Chapman, J.R.; Hill, T.; Unckless, R.L. Balancing Selection Drives the Maintenance of Genetic Variation in Drosophila Antimicrobial Peptides. Genome Biol. Evol. 2019, 11, 2691–2701. [Google Scholar] [CrossRef]
- Duda, T.F.; Vanhoye, D.; Nicolas, P. Roles of Diversifying Selection and Coordinated Evolution in the Evolution of Amphibian Antimicrobial Peptides. Mol. Biol. Evol. 2002, 19, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-H.; Li, Y.; Lai, R.; Li, S.; Zhang, Y.; Wang, W. Variety of Antimicrobial Peptides in the Bombina Maxima Toad and Evidence of Their Rapid Diversification. Eur. J. Immunol. 2005, 35, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.A. Molecular Evolution of Animal Antimicrobial Peptides: Widespread Moderate Positive Selection. J. Evol. Biol. 2005, 18, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.A.; Blouin, M.S. Balancing Selection at a Frog Antimicrobial Peptide Locus: Fluctuating Immune Effector Alleles? Mol. Biol. Evol. 2008, 25, 2669–2680. [Google Scholar] [CrossRef] [PubMed]
- Vanhoye, D.; Bruston, F.; Nicolas, P.; Amiche, M. Antimicrobial Peptides from Hylid and Ranin Frogs Originated from a 150-Million-Year-Old Ancestral Precursor with a Conserved Signal Peptide but a Hypermutable Antimicrobial Domain. Eur. J. Biochem. 2003, 270, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xia, R.; Ji, J.J.; Zhu, Q.; Li, X.P.; Ma, Y.; Xu, Y.C. Diversity of Antimicrobial Peptides in Three Partially Sympatric Frog Species in Northeast Asia and Implications for Evolution. Genes 2020, 11, 158. [Google Scholar] [CrossRef]
- Chen, X.; Liu, S.; Fang, J.; Zheng, S.; Wang, Z.; Jiao, Y.; Xia, P.; Wu, H.; Ma, Z.; Hao, L. Peptides Isolated from Amphibian Skin Secretions with Emphasis on Antimicrobial Peptides. Toxins 2022, 14, 722. [Google Scholar] [CrossRef]
- Clarke, B.T. The Natural History of Amphibian Skin Secretions, Their Normal Functioning and Potential Medical Applications. Biol. Rev. Camb. Philos. Soc. 1997, 72, 365–379. [Google Scholar] [CrossRef]
- Xu, X.; Lai, R. The Chemistry and Biological Activities of Peptides from Amphibian Skin Secretions. Chem. Rev. 2015, 115, 1760–1846. [Google Scholar] [CrossRef]
- Duellman, W.E.; Trueb, L. Biology of Amphibians; JHU Press: Baltimore, MD, USA, 1994; ISBN 978-0-8018-4780-6. [Google Scholar]
- Rollins-Smith, L.A. The Importance of Antimicrobial Peptides (AMPs) in Amphibian Skin Defense. Dev. Comp. Immunol. 2023, 142, 104657. [Google Scholar] [CrossRef]
- Habermehl, G. Salamander Alkaloids. In Progress in Organic Chemistry: Volume 7; Cook, J., Carruthers, W., Eds.; Springer US: Boston, MA, USA, 1968; pp. 35–47. ISBN 978-1-4899-7315-3. [Google Scholar]
- Hanifin, C.T. The Chemical and Evolutionary Ecology of Tetrodotoxin (TTX) Toxicity in Terrestrial Vertebrates. Mar. Drugs 2010, 8, 577–593. [Google Scholar] [CrossRef]
- Knepper, J.; Lüddecke, T.; Preißler, K.; Vences, M.; Schulz, S. Isolation and Identification of Alkaloids from Poisons of Fire Salamanders (Salamandra salamandra). J. Nat. Prod. 2019, 82, 1319–1324. [Google Scholar] [CrossRef]
- Jiang, W.-B.; Hakim, M.; Luo, L.; Li, B.-W.; Yang, S.-L.; Song, Y.-Z.; Lai, R.; Lu, Q.-M. Purification and Characterization of Cholecystokinin from the Skin of Salamander Tylototriton verrucosus. Zool. Res. 2015, 36, 174–177. [Google Scholar]
- Plácido, A.; Bueno, J.; Barbosa, E.A.; Moreira, D.C.; Dias, J.d.N.; Cabral, W.F.; Albuquerque, P.; Bessa, L.J.; Freitas, J.; Kuckelhaus, S.A.S.; et al. The Antioxidant Peptide Salamandrin-I: First Bioactive Peptide Identified from Skin Secretion of Salamandra Genus (Salamandra salamandra). Biomolecules 2020, 10, 512. [Google Scholar] [CrossRef]
- Vasconcelos, I.A.d.; Souza, J.O.d.; de Castro, J.S.; Santana, C.J.C.d.; Magalhães, A.C.M.; Castro, M.d.S.; Pires Júnior, O.R. Salamanders and Caecilians, Neglected from the Chemical Point of View. Toxin Rev. 2022, 41, 1304–1332. [Google Scholar] [CrossRef]
- König, E.; Bininda-Emonds, O.R.P.; Shaw, C. The Diversity and Evolution of Anuran Skin Peptides. Peptides 2015, 63, 96–117. [Google Scholar] [CrossRef]
- Raaymakers, C.; Verbrugghe, E.; Hernot, S.; Hellebuyck, T.; Betti, C.; Peleman, C.; Claeys, M.; Bert, W.; Caveliers, V.; Ballet, S.; et al. Antimicrobial Peptides in Frog Poisons Constitute a Molecular Toxin Delivery System against Predators. Nat. Commun. 2017, 8, 1495. [Google Scholar] [CrossRef]
- Wilkinson, M. Caecilians. Curr. Biol. 2012, 22, R668–R669. [Google Scholar] [CrossRef] [PubMed]
- Wake, M. The Osteology of Caecilians. Amphib. Biol. 2003, 5, 1809–1876. [Google Scholar]
- Arun, D.; Sandhya, S.; Akbarsha, M.A.; Oommen, O.V.; Divya, L. An Insight into the Skin Glands, Dermal Scales and Secretions of the Caecilian Amphibian Ichthyophis beddomei. Saudi J. Biol. Sci. 2020, 27, 2683–2690. [Google Scholar] [CrossRef]
- Torres-Sánchez, M.; Wilkinson, M.; Gower, D.J.; Creevey, C.J.; San Mauro, D. Insights into the Skin of Caecilian Amphibians from Gene Expression Profiles. BMC Genom. 2020, 21, 515. [Google Scholar] [CrossRef] [PubMed]
- A Handbook of Tropical Soil Biology: Sampling and Characterization of Below-Ground Biodiversity. Available online: https://www.routledge.com/A-Handbook-of-Tropical-Soil-Biology-Sampling-and-Characterization-of-Below-ground/Moreira-Huising-Bignell/p/book/9781844075935 (accessed on 9 September 2023).
- Steffan, J.J.; Derby, J.A.; Brevik, E.C. Soil Pathogens That May Potentially Cause Pandemics, Including Severe Acute Respiratory Syndrome (SARS) Coronaviruses. Curr. Opin. Environ. Sci. Health 2020, 17, 35–40. [Google Scholar] [CrossRef] [PubMed]
- San Mauro, D.; Gower, D.J.; Müller, H.; Loader, S.P.; Zardoya, R.; Nussbaum, R.A.; Wilkinson, M. Life-History Evolution and Mitogenomic Phylogeny of Caecilian Amphibians. Mol. Phylogenet. Evol. 2014, 73, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, G.; Engsontia, P. Diversity of the Antimicrobial Peptide Genes in Collembola. Insects 2023, 14, 215. [Google Scholar] [CrossRef]
- Yi, Y.; You, X.; Bian, C.; Chen, S.; Lv, Z.; Qiu, L.; Shi, Q. High-Throughput Identification of Antimicrobial Peptides from Amphibious Mudskippers. Mar. Drugs 2017, 15, 364. [Google Scholar] [CrossRef]
- Irisarri, I.; Baurain, D.; Brinkmann, H.; Delsuc, F.; Sire, J.-Y.; Kupfer, A.; Petersen, J.; Jarek, M.; Meyer, A.; Vences, M.; et al. Phylotranscriptomic Consolidation of the Jawed Vertebrate Timetree. Nat. Ecol. Evol. 2017, 1, 1370–1378. [Google Scholar] [CrossRef]
- San Mauro, D. A Multilocus Timescale for the Origin of Extant Amphibians. Mol. Phylogenet. Evol. 2010, 56, 554–561. [Google Scholar] [CrossRef]
- Shamova, O.V.; Orlov, D.S.; Balandin, S.V.; Shramova, E.I.; Tsvetkova, E.V.; Panteleev, P.V.; Leonova, Y.F.; Tagaev, A.A.; Kokryakov, V.N.; Ovchinnikova, T.V. Acipensins—Novel Antimicrobial Peptides from Leukocytes of the Russian Sturgeon Acipenser Gueldenstaedtii. Acta Naturae 2014, 6, 99–109. [Google Scholar] [CrossRef]
- Klüter, T.; Fitschen-Oestern, S.; Lippross, S.; Weuster, M.; Mentlein, R.; Steubesand, N.; Neunaber, C.; Hildebrand, F.; Pufe, T.; Tohidnezhad, M.; et al. The Antimicrobial Peptide Lysozyme Is Induced after Multiple Trauma. Mediat. Inflamm. 2014, 2014, 303106. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gong, Y.; Chen, Y.; Qu, B.; Zhang, S. Identification and Functional Characterization of Cofilin-1 as a New Member of Antimicrobial Protein. Dev. Comp. Immunol. 2022, 127, 104281. [Google Scholar] [CrossRef]
- Ibrahim, H.R.; Imazato, K.; Ono, H. Human Lysozyme Possesses Novel Antimicrobial Peptides within Its N-Terminal Domain That Target Bacterial Respiration. J. Agric. Food Chem. 2011, 59, 10336–10345. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Sillard, R.; Kleemeier, B.; Klüver, E.; Maronde, E.; Conejo-García, J.R.; Forssmann, W.G.; Schulz-Knappe, P.; Nehls, M.C.; Wattler, F.; et al. Isolation and Biochemical Characterization of LEAP-2, a Novel Blood Peptide Expressed in the Liver. Protein Sci. 2003, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- El Karim, I.A.; Linden, G.J.; Orr, D.F.; Lundy, F.T. Antimicrobial Activity of Neuropeptides against a Range of Micro-Organisms from Skin, Oral, Respiratory and Gastrointestinal Tract Sites. J. Neuroimmunol. 2008, 200, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Pavia, K.E.; Spinella, S.A.; Elmore, D.E. Novel Histone-Derived Antimicrobial Peptides Use Different Antimicrobial Mechanisms. Biochim. Biophys. Acta 2012, 1818, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, J.; Zhu, C.; Yang, L.; Ren, Y.; Ruan, J.; Fan, G.; Hu, J.; Xu, W.; Bi, X.; et al. African Lungfish Genome Sheds Light on the Vertebrate Water-to-Land Transition. Cell 2021, 184, 1362–1376.e18. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R. Animal Genome Size Database 2024. Available online: http://www.genomesize.com (accessed on 1 February 2024).
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of Duplicate Genes by Complementary, Degenerative Mutations. Genetics 1999, 151, 1531–1545. [Google Scholar] [CrossRef]
- Lynch, M.; Force, A. The Probability of Duplicate Gene Preservation by Subfunctionalization. Genetics 2000, 154, 459–473. [Google Scholar] [CrossRef]
- Calhoun, D.M.; Woodhams, D.; Howard, C.; LaFonte, B.E.; Gregory, J.R.; Johnson, P.T.J. Role of Antimicrobial Peptides in Amphibian Defense Against Trematode Infection. EcoHealth 2016, 13, 383–391. [Google Scholar] [CrossRef]
- Kobel, H.R.; Du Pasquier, L. Genetics of Polyploid Xenopus. Trends Genet. 1986, 2, 310–315. [Google Scholar] [CrossRef]
- Mechkarska, M.; Eman, A.; Coquet, L.; Jérôme, L.; Jouenne, T.; Vaudry, H.; King, J.D.; Takada, K.; Conlon, J.M. Genome Duplications within the Xenopodinae Do Not Increase the Multiplicity of Antimicrobial Peptides in Silurana Paratropicalis and Xenopus Andrei Skin Secretions. Comp. Biochem. Physiol. Part D Genom. Proteom. 2011, 6, 206–212. [Google Scholar] [CrossRef]
- Lu, B. Evolutionary Insights into the Relationship of Frogs, Salamanders, and Caecilians and Their Adaptive Traits, with an Emphasis on Salamander Regeneration and Longevity. Animals 2023, 13, 3449. [Google Scholar] [CrossRef]
- Wilkinson, M.; San Mauro, D.; Sherratt, E.; Gower, D.J. A Nine-Family Classification of Caecilians (Amphibia: Gymnophiona). Zootaxa 2011, 2874, 41–64. [Google Scholar] [CrossRef]
- Koenig, D.; Hagmann, J.; Li, R.; Bemm, F.; Slotte, T.; Neuffer, B.; Wright, S.I.; Weigel, D. Long-Term Balancing Selection Drives Evolution of Immunity Genes in Capsella. eLife 2019, 8, e43606. [Google Scholar] [CrossRef]
- Rieseberg, L.H.; Widmer, A.; Arntz, A.M.; Burke, J.M. Directional Selection Is the Primary Cause of Phenotypic Diversification. Proc. Natl. Acad. Sci. USA 2002, 99, 12242–12245. [Google Scholar] [CrossRef] [PubMed]
- Des Marais, D.L.; Rausher, M.D. Escape from Adaptive Conflict after Duplication in an Anthocyanin Pathway Gene. Nature 2008, 454, 762–765. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vaisman, I.I.; van Hoek, M.L. Machine Learning Prediction of Antimicrobial Peptides. In Methods Molecular Biology; Springer: Clifton, NJ, USA, 2022; Volume 2405, pp. 1–37. [Google Scholar] [CrossRef]
- Richter, A.; Sutherland, D.; Ebrahimikondori, H.; Babcock, A.; Louie, N.; Li, C.; Coombe, L.; Lin, D.; Warren, R.L.; Yanai, A.; et al. Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects. Antibiotics 2022, 11, 1710. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.L.; Akhtar, S.; Bajpai, P. In Silico Protein Modeling: Possibilities and Limitations. EXCLI J. 2014, 13, 513–515. [Google Scholar] [PubMed]
- Wang, G. Discovery, Classification and Functional Diversity of Antimicrobial Peptides. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; Wang, G., Ed.; CABI: Wallingford, UK, 2017; pp. 1–19. ISBN 978-1-78639-039-4. [Google Scholar]
- Benfield, A.H.; Henriques, S.T. Mode-of-Action of Antimicrobial Peptides: Membrane Disruption vs. Intracellular Mechanisms. Front. Med. Technol. 2020, 2, 610997. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, α-Helical Antimicrobial Peptides. Pept. Sci. 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Li, J.; Koh, J.-J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef]
- Koehbach, J.; Craik, D.J. The Vast Structural Diversity of Antimicrobial Peptides. Trends Pharmacol. Sci. 2019, 40, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards Complete and Error-Free Genome Assemblies of All Vertebrate Species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Ovchinnikov, V.; Uliano-Silva, M.; Wilkinson, M.; Wood, J.; Smith, M.; Oliver, K.; Sims, Y.; Torrance, J.; Suh, A.; McCarthy, S.A.; et al. Caecilian Genomes Reveal the Molecular Basis of Adaptation and Convergent Evolution of Limblessness in Snakes and Caecilians. Mol. Biol. Evol. 2023, 40, msad102. [Google Scholar] [CrossRef] [PubMed]
- Torres-Sánchez, M.; Creevey, C.J.; Kornobis, E.; Gower, D.J.; Wilkinson, M.; San Mauro, D. Multi-Tissue Transcriptomes of Caecilian Amphibians Highlight Incomplete Knowledge of Vertebrate Gene Families. DNA Res. 2019, 26, 13–20. [Google Scholar] [CrossRef]
- Kamei, R.G.; San Mauro, D.; Gower, D.J.; Van Bocxlaer, I.; Sherratt, E.; Thomas, A.; Babu, S.; Bossuyt, F.; Wilkinson, M.; Biju, S.D. Discovery of a New Family of Amphibians from Northeast India with Ancient Links to Africa. Proc. R. Soc. B Biol. Sci. 2012, 279, 2396–2401. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Lorente-Martínez, H.; Agorreta, A.; San Mauro, D. Genomic Fishing and Data Processing for Molecular Evolution Research. Methods Protoc. 2022, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Martínez, H.; Agorreta, A.; Irisarri, I.; Zardoya, R.; Edwards, S.V.; San Mauro, D. Multiple Instances of Adaptive Evolution in Aquaporins of Amphibious Fishes. Biology 2023, 12, 846. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Cases in Which Parsimony or Compatibility Methods Will Be Positively Misleading. Syst. Zool. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A Useful Resource for Research on Antimicrobial Peptides. Nucleic Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.; Kamath, U.; Shehu, A. Deep Learning Improves Antimicrobial Peptide Recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of Antimicrobial/Cytotoxic Activity and Structure of Peptides as a Resource for Development of New Therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Grigolava, M.; Managadze, G.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Pirtskhalava, M. Comparative Analysis of Machine Learning Algorithms on the Microbial Strain-Specific AMP Prediction. Brief. Bioinform. 2022, 23, bbac233. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for Structure Building and Analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R. bivittatum G+T | I. kohtaoensis T | C. tentaculata T | T. natans T | T. compressicauda T | M. unicolor G+T | M. dermatophaga T | G. seraphini G+T | |
|---|---|---|---|---|---|---|---|---|
| AMP families (primary antimicrobial function) | ||||||||
| LEAP2 | X | X | X | X | X | |||
| Lysozyme C | X | X | X | X | X | X | X | |
| Lysozyme G | X | X | X | X | X | X | ||
| Cathelicidin * | X | X | X | X | X | X | ||
| Protein (sub)families with a possible secondary function as AMP | ||||||||
| A1P/Antitrypsin | X | X | X | X | X | X | ||
| Adrenomedulin | X | X | X | X | X | X | ||
| Apelin | X | |||||||
| Beta-amyloid | X | X | X | X | X | X | X | |
| Amylin/Calcitonin | X | X | X | |||||
| Chemokine | X | X | X | X | X | X | ||
| Chrombacin/Secretogranin | X | X | X | X | X | X | X | |
| Cofilin | X | X | X | X | X | X | X | X |
| DBI (Diazepam Binding Inhibitor) | X | X | X | X | X | X | X | |
| Enolase | X | X | X | X | X | X | X | |
| GADPH (Glyceraldehyde phosphate dehydrogenase) | X | X | X | X | X | X | X | X |
| Granulin | X | X | X | X | X | X | X | |
| Histone H2A—Acipensin 6 region | X | X | X | X | X | X | X | X |
| Histone H2A—Hipposin region | X | X | X | X | X | X | X | X |
| Histone H2B | X | X | X | X | X | X | ||
| Histone H3 | X | X | X | X | X | X | X | |
| IBP (Insulin-like growth factor binding protein) | X | X | X | X | X | X | X | X |
| Neuropeptide W | X | X | X | |||||
| Neuropeptide YY | X | X | X | X | X | X | ||
| PACAP (Pituitary adenylate cyclase-activating polypeptide) | X | X | X | X | X | X | ||
| POMC (Pro-opiomelanocortin) | X | X | X | |||||
| Proenkephalin | X | X | X | X | ||||
| Thymosin | X | X | X | |||||
| Vasostatin | X | X | X | X | X | X | X | |
| Cystatin/Kininogen * | X | X | X | X | X | X | ||
| AA | Site | Bias | Bayes Factor | AA Site Composition | Inferred Substitutions History |
|---|---|---|---|---|---|
| Lysozyme C (clade 1) | |||||
| F | 54 | 25.09 | 122.56 | F8, Y15 | Y->F(4) |
| H | 107 | 34.01 | 293.40 | A1, D13, E1, H3, N4, S1 | D->A(1)E(1)H(2), N->D(1)H(1)S(1) |
| Lysozyme C (clade 2) | |||||
| S | 10 | 34.13 | 234.10 | S15 | - |
| Cathelicidin | |||||
| H | 754 | 21.06 | 447.56 | H28, I1, L12, V1 | H->L(1), L->I(1)V(1) |
| I | 360 | 36.83 | 124.09 | I12, R35 | R->I(2) |
| Histone H2A—Hipposin region | |||||
| D | 41 | 41.49 | 165.23 | D3, E43 | E->D(2) |
| Histone H2B | |||||
| G | 39 | 29.83 | 100.12 | A23, G7, S1 | A->G(3)S(1) |
| PACAP | |||||
| N | 28 | 36.74 | 150.08 | N6, T5 | T->N(2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benítez-Prián, M.; Lorente-Martínez, H.; Agorreta, A.; Gower, D.J.; Wilkinson, M.; Roelants, K.; San Mauro, D. Diversity and Molecular Evolution of Antimicrobial Peptides in Caecilian Amphibians. Toxins 2024, 16, 150. https://doi.org/10.3390/toxins16030150
Benítez-Prián M, Lorente-Martínez H, Agorreta A, Gower DJ, Wilkinson M, Roelants K, San Mauro D. Diversity and Molecular Evolution of Antimicrobial Peptides in Caecilian Amphibians. Toxins. 2024; 16(3):150. https://doi.org/10.3390/toxins16030150
Chicago/Turabian StyleBenítez-Prián, Mario, Héctor Lorente-Martínez, Ainhoa Agorreta, David J. Gower, Mark Wilkinson, Kim Roelants, and Diego San Mauro. 2024. "Diversity and Molecular Evolution of Antimicrobial Peptides in Caecilian Amphibians" Toxins 16, no. 3: 150. https://doi.org/10.3390/toxins16030150