
Abstract
Silver is probably the closest isotopic proxy to track monoisotopic gold and has been shown to have great potential to yield new information on the origin and enrichment processes of gold. This study describes the development of a tailored analytical protocol for accurate analysis of Ag isotopes and provides the first Ag isotope data for the Paleoproterozoic Rajapalot Au-Co deposit, Finnish Lapland. Six native Au samples yield ε109Ag values (relative to NIST SRM 978a) from −6.8 to +2.1 and are within the range of Ag isotopic compositions reported for native Au samples. The mean of the analyzed Au samples is ε109Ag = −3.8 ± 1.7 (2SD) with most of the samples with negative ε109Ag values (−6.7 to −2.0); one sample has a positive ε109Ag value of +2.1 ± 0.5. Silver isotope fractionation in the Rajapalot Au deposit was likely associated with physicochemical processes related to deposition and/or re-mobilization of the ore rather than with source region inheritance. It is suggested that redox reactions involving Ag+ ↔ Ag0 phase change primarily account for the isotopic differences within the deposit. Our results also suggest that the Rajapalot Au-Co deposit was formed via multistage ore-forming processes and/or that the primary ore was re-mobilized, which caused isotope fractionation along fluid pathways. Silver isotope variation within a deposit may mark a fractional crystallization trend with the lightest isotopic composition representing the earliest precipitate. Hence, Ag isotopes show potential as an isotopic vectoring tool in search of Au-enriched domains.
Similar content being viewed by others

Introduction
A wide range of Au deposit types and settings has been recognized in Finland. These include porphyry Au-copper, Au-rich volcanogenic massive sulfide (VMS), metamorphosed high-sulfidation epithermal, intrusion-related, IOCG, orogenic Au, and placer and paleoplacer deposits (Eilu et al. 2003; Eilu 2015). The specific Au deposit types have several common characteristics, tectonic setting included, and are related to diverse episodes of Archean and Paleoproterozoic continental evolution (Hagemann and Brown 2000; Robert et al. 2007; Eilu 2015). Several Au deposits, such as the Rompas-type Au-(U) deposit and Rajapalot Au-(Co) deposit in the Peräpohja schist belt (Fig. 1), do not fit the conventional genetic framework, however (Vanhanen et al. 2015; Molnár et al. 2016; Ranta et al. 2018). These deposits possess orogenic Au-like features with geochemical, mineralogical, and petrological characteristics that do not fully match known Au deposit models. Multiple deformation and hydrothermal events, pervasive hydrothermal alteration, and amphibolite-facies metamorphism overprint primary features. In modeling ore-forming processes, a variety of isotope-geochemical tools have been applied, and various origins of fluids and/or different trap conditions have been suggested (Vanhanen et al. 2015; Molnár et al. 2016, 2017; Ranta et al. 2018; Cook et al. 2019; Raič et al. 2022). The geological development of these deposits and their metal sources are still poorly known.

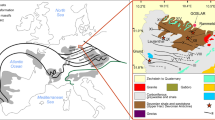
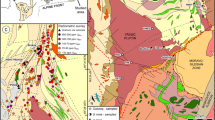
a Simplified geological map of the Peräpohja belt modified from the Bedrock of Finland – DigiKP (Geological Survey of Finland) showing the main lithostratigraphic units of the belt. Location of the Peräpohja belt highlighted in red in the inset map of Finland. The Rompas-Rajapalot project area is marked with diagonal stripes in the northern part of the Peräpohja belt. Legend from the panel b applies. b Close-up showing the location of Raja prospect on a simplified geological map of the Rompas-Rajapalot area. Rompas and Rajapalot prospect areas marked with diagonal stripes. c Simplified drill-core profiles showing the sampling locations of the Au samples. Numbers with red arrows indicate the position of the samples examined in this study (keyed to the last digits of samples JHTE-20190514-(2, 3, 5, 8, 12) and JHTE-20190515-30)
Traditional stable isotopes (e.g., those of O, C, S, B), cannot, however, be used to provide direct information about sources and depositional conditions of Au itself as these elements are not incorporated into the structure of native Au or Au alloys. Gold is a monoisotopic element with only one naturally occurring isotope, 197Au, and therefore Au itself cannot be used as an isotopic tracer to track chemical reactions or source regions. However, transition metals and metalloids (e.g., Cu, Ag, Sb) are frequently present in native Au and Au alloys in significant amounts. Their isotope composition provides a powerful yet rather undeveloped tool in search of a more profound handle on the metal source and conditions under which the deposits were formed. In general, fractionation of transition metal isotopes is highly sensitive to ambient redox conditions, and source regions of metals also leave their footprint to the isotope ratios (Schauble 2004). Stable isotopes of Ag have substantial potential to yield novel information on the origin and enrichment processes of Au as Ag is often closely associated with Au in natural environments. These elements have similar chemical properties and thus behave similarly in geochemical processes (Seward et al. 2014).
Silver geochemistry and isotope systematics
In nature, Ag is a ubiquitous component in natural Au minerals (Boyle 1979). Because of the similar outer shell electron configuration of Au and Ag, their geochemical behavior is quite similar, and Au and Ag are often co-transported in hydrothermal solutions (Seward et al. 2014). Both metals are carried predominantly as monovalent species in aqueous bisulfide (HS)-2 in low-temperature (< 500°C) reduced, near-neutral-alkaline conditions and are deposited through reduction of the monovalent metal ion:
The solubility of Ag in low-temperature environments has been actively studied (Seward 1976; Stefánsson and Seward 2003; Liu et al. 2012). In addition to dominant hydrosulfide complexes, the formation of other Ag and Au complexes with, e.g., chloride Cl-, ammine HN3, and iodide I- ligands, has also been observed, but their contribution to ore formation is still obscure (Perera and Senanayake 2004; Seward et al. 2014). The chalcophile tendency of Ag and Au implies that they interact similarly throughout the ore genesis process (Gammons and Williams-Jones 1995; Seward et al. 2014). Silver can be used as an isotopic proxy and has been suggested to be the closest proxy to Au (Argapadmi et al. 2018; Brügmann et al. 2019).
Silver has two naturally occurring stable isotopes, 109Ag and 107Ag, with relative abundances of 48.161% and 51.839%, respectively. Variation in Ag isotopic composition was first recognized in the 1960s, when Murthy (1962) reported the 107Ag/109Ag ratio in iron meteorites to be 2–3% higher relative to terrestrial Ag. The excess of 107Ag was accounted as radiogenic, resulting from the decay of short-lived radioactive nuclides such as the extinct 107Pd. Recent development of more precise analytical techniques in multi-collector inductively coupled plasma mass spectrometry (MC-ICP/MS) has enabled the determination of more subtle variations of Ag isotope values in terrestrial material. The new MC-ICP-MS technology has reduced the analytical error by an order of magnitude compared to traditional thermal ionization mass spectrometry (Carlson and Hauri 2001; Woodland et al. 2005).
Silver isotopes have been utilized in a variety of fields from cosmochemistry (Carlson and Hauri 2001; Woodland et al. 2005; Schönbächler et al. 2010) to environmental sciences (Luo et al. 2010). Variation in the 109Ag/107Ag ratio has also been used to study past monetary systems by tracking the source and circulation of silver coinage (Desaulty et al. 2011; Desaulty and Albarède 2013; Albarède et al. 2016; Milot et al. 2021, 2022). Limited existing data for terrestrial materials show an Ag isotopic range of ε109Ag from −8.6 to 21.4 (Woodland et al. 2005; Schönbächler et al. 2007; Desaulty et al. 2011; Desaulty and Albarède 2013; Albarède et al. 2016; Argapadmi et al. 2018; Mathur et al. 2018; Brügmann et al. 2019). Only a few recent studies have, however, used Ag isotopes to study ore-forming processes (Chugaev and Chernyshev 2012; Voisey et al. 2017; Argapadmi et al. 2018; Mathur et al. 2018; Yin and Zajacz 2018; Brügmann et al. 2019; Arribas et al. 2020; Wang et al. 2022). Available Ag isotopic data, analyzed from native Ag or Ag-bearing minerals, show a wide range of ε109Ag values covering the whole range of values reported for terrestrial materials (Schönbächler et al. 2010; Chugaev and Chernyshev 2012; Mathur et al. 2018; Arribas et al. 2020). ε109Ag values for native Au in various mineralization types show a more limited range, varying from −8.3 to +8.3 (Chugaev and Chernyshev 2012; Argapadmi et al. 2018; Brügmann et al. 2019; Voisey et al. 2019). The largest variation has been reported in samples from relatively low-temperature mineralization, whereas Ag isotopic values in high-temperature deposits cluster around zero (Mathur et al. 2018).
Regional geology
The Rompas and Rajapalot Au-U-(Co) prospects are located in the Paleoproterozoic Peräpohja belt in northern Finland (Fig. 1). The area comprises diverse supracrustal sequences deposited on the Archean gneissic basement of the Pudasjärvi complex (Perttunen 1985, 1989; Perttunen et al. 1995). The lowermost part of the Peräpohja belt, the Kivalo group (Perttunen et al. 1995), consists of quartz sandstones with lesser carbonate rocks and conglomerates and was deposited on a low gradient platform or in shallow water in a basin formed by rifting of the Archean continent (Vanhanen et al. 2015). These sediments are accompanied by subaerial mafic lava flows and tuffites. Mafic-ultramafic layered intrusions of ca. 2.44 Ga age predate the supracrustal sequences, yielding a maximum age for the Peräpohja belt (Huhma et al. 1990; Perttunen and Vaasjoki 2001). The supracrustal sequence is cut by 2.22 Ga and 2.14 Ga mafic sills and dykes. Gradual deepening of the basin is seen in the upper part of the Peräpohja belt, where the Paakkola group comprising pillow lavas, mafic and felsic tuffs, turbiditic conglomerates, pelitic schists, and black shales is inferred to have been deposited in a deep-water basin. The Paakkola group sequence is cut by plutonic rocks of the ca. 1.88 Ga Haaparanta suite which sets the upper age for the Peräpohja belt (Perttunen and Vaasjoki 2001). Five structural metamorphic events related to the 1.92–1.79 Ga Svecofennian orogeny have affected the bedrock of the Peräpohja belt (Lahtinen et al. 2015). Metamorphic grade varies along with deformation, increasing from lower greenschist facies in the south to amphibolite facies in the north towards the contact of the Central Lapland Granitoid Complex (Lahtinen et al. 2015; Hölttä and Heilimo 2017).
The Raja prospect, the research target of this study, is part of the larger Rompas-Rajapalot Au-bearing hydrothermal system located in the highly deformed and altered northern part of the Peräpohja belt (Fig. 1). The study area is broadly divided into two sections: Rompas in the northwest and Rajapalot about 6 km east. The Raja prospect lies 1 km south of the Palokas prospect in the Rajapalot area (Fig. 1b; Ranta et al. 2015, 2017, 2018). U-Pb dating of uraninite and monazite and Re-Os dating of molybdenite from the Rompas and Rajapalot prospects indicate that the mineralization was formed by multiple hydrothermal events with several stages of U and Co deposition. Re-mobilization during pre-metamorphic and metamorphic fluid flow events and deposition of Au was genetically linked to fluid circulation generated by post-orogenic granitic magmatism at 1.78–1.75 Ga (Molnár et al. 2017; Ranta et al. 2017; Raič et al. 2022). Boron isotope evidence on tourmaline also indicates a common origin of Au mineralizing fluids in the prospects and in the granitoids (Ranta et al. 2017).
The gold mineralization style in the Raja prospect is different from that in the Rompas deposit despite their proximity and is more like the one in the Palokas prospect. The Rompas Au mineralization is associated with uranium and hosted in calcsilicate veins within mafic volcanic rocks, whereas the Raja mineralization shows no association with U but is closely associated with Co (Vanhanen et al. 2015; Molnár et al. 2017; Raič et al. 2022). Moreover, the Raja Au-Co mineralization is found as sulfide-rich dissemination or in sulfide-bearing fractures with no obvious association to calcsilicate veins that are typical of the Rompas prospect. The adjacent Palokas deposit is hosted by metamorphosed Mg-rich rocks and calcic amphibolites interlayed with pervasively albitized calc-silicate rocks (Ranta et al. 2017).
The Au mineralization in the Raja prospect is mainly hosted by calcsilicate-albite rocks and K-rich metasedimentary rocks or other mica-bearing lithologic units (Taipale 2018; Farajewicz and Cook 2021). Pyrrhotite is the most abundant sulfide mineral and is found with minor pyrite, chalcopyrite, pentlandite, cobaltite, linnaeite, and native Au. The altered host rock mineralogy is characterized by biotite, muscovite, chlorite, quartz, albite, Mg-Fe amphiboles, tourmaline, and scheelite. Gradual change from Ca-amphiboles to Fe-Mg-Mn amphiboles is recognized towards the mineralization, with the highest fraction of Fe-Mg-Mn amphiboles within the mineralized zone. Similarly, the Al-content of micas shows a clear increase towards the mineralization (Taipale 2018). Native Au is found either as disseminated grains at grain boundaries or as inclusions in various minerals in a foliated matrix or in fractures crosscutting the foliation (Farajewicz and Cook 2021; Raič et al. 2022). The mineralization is structurally controlled (Farajewicz and Cook 2021) with an older Co-mineralizing event followed by a subsequent Au-mineralization event (Raič et al. 2022).
Materials and methods
Gold samples
Samples for Ag isotope analyses were collected from six different drill cores (PAL0088, PAL0092, PAL0093, PAL0097, PAL0100, PAL0116) crosscutting the zone of Au-mineralization in the Raja Au-Co prospect (Fig. 1). The selection was targeted on areas with visible Au or high-grade sections based on a pre-existing whole-rock geochemical dataset. In total, 10 samples were selected, and thin sections from them were studied by polarized reflected and transmitted light microscopy.
Au-Co-mineralized samples from the Raja deposit consist of calcsilicate-albite rocks and biotite-rich metasedimentary rocks (Fig. 2). The bulk matrix is characterized by small- to medium-grained granoblastic albite and quartz with elongated biotite and chlorite grains. Accessory minerals include tourmaline and scheelite. The most abundant sulfide is pyrrhotite, which is found as anhedral small, disseminated grains or large networks following the rock fabric (Fig. 2). Chalcopyrite is found intergrown with pyrrhotite. Pyrite is present as large subhedral to euhedral grains associated with pyrrhotite but also as anhedral late vein fillings. The large euhedral crystals show brittle fracturing, whereas the anhedral pyrite is more ductile, filling cracks and voids. Various pyrite forms and associations suggest multiple pyrite populations. Cobaltite is found as subhedral skeletal disseminated grains, often together with pyrrhotite.

Representative images of drill core sections from which Au was acquired for the Ag isotope analyses (see also Fig. 1c). a Sample JHTE-20190514-2 from drill core PAL0092. Fine-grained pervasively albitized metasedimentary rock with Py as inclusions in Po, Ccp intergrown with Po. b Sample JHTE-20190514-3 from drill core PAL0092. Folded and albitized metasedimentary rock, abundant Po following foliation. c Sample JHTE-20190514-5 from drill core PAL0093. Pervasively albitized metasedimentary rock with abundant Po intergrown with Ccp. d Sample JHTE-20190514-8 from drill core from PAL0093. Foliated and altered metasedimentary rock with Po and Ccp following the foliation. e Sample JHTE-20190514-12 from drill core PAL0097. Fine-grained albitized rock with Po dissemination and anhedral Py. f Sample JHTE-20190515-30 from drill core PAL0016 representing foliated metasedimentary rock. Disseminated Po is following the fabric with minor intergrown Ccp. g Sample JHTE-20190514-3 from drill core PAL0092 with Au inclusions in Sch/Ferb in reflected light. Sch and Ferb replacing older Po. h Sample JHTE-20190514-5 from drill core PAL0093 with Au along the grain boundaries of Po/Ccp and silicates, in reflected light. Abbreviations: Po, pyrrhotite; Py, pyrite; Ccp, chalcopyrite; Ferb, ferberite; Sch, scheelite
Visible Au was observed in two of the sections (Fig. 2). In sample JHTE-20190514-3 (Fig. 2g), Au is associated with scheelite and ferberite, which appears to be replacing earlier pyrrhotite. In contrast, in sample JHTE-20190514-5 (Fig. 2h), Au is found at the grain boundary between pyrrhotite, chalcopyrite, and silicate minerals, mainly albite. The gold grains from the two samples were analyzed with SEM to check for the potential for Ag isotope analysis, i.e., to confirm the presence of Ag.
Reagents and materials
Concentrated, high-purity acids were produced in-house from Baker Analyzed™ nitric acid and Merck Emsure® hydrochloric acid grade stocks using a double sub-boiling distillation. Acids of lower molarity were diluted from these by volume with ultrapure water. Any water used during chemistry was high-purity (18.2 MΩ . cm) de-ionized water obtained from a Milli-Q® water purification system.
Ion exchange resin Bio-Rad AG®1-X8 (100-200 mesh, chloride form) and Triskem Prefilter resin (100–150 μm) were obtained from Bio-Rad Laboratories (Hercules, CA, USA) and Triskem International (Bruz, France), respectively. Ion-exchange columns for Ag separation were made from pre-cleaned disposable polyethylene transfer pipettes with narrow 25 mm × 20 mm resin bed and 1 mL reservoir. Porous polyethylene frits were cut and fitted on the bottom of the resin bed volume to support and retain fine resin particles.
SRM 978a, Ag isotopic standard in AgNO3 form, and SRM 3138, Pd standard in 8% HCl, were provided by the National Institute of Standards and Technology (NIST, Gaithersburg, MD, USA). Silver nitrate, AgNO3, dissolves readily in water, and a 100 ppm 978a stock solution was prepared by quantitatively dissolving the AgNO3 in 2% HNO3. SRM 3138 standard, used for instrumental mass discrimination correction, was provided in glass ampoules containing 10.012 mg/g of Pd in 8% HCl. The acidified solution could not be directly diluted with 2% HNO3 as traces of chloride could induce precipitation of Ag chloride. The content of one ampoule was weighed into a Savillex beaker, and the amount of Pd was calculated. The solution was then evaporated on a 70 °C plate to the last drop. A small amount of 2% HNO3 was added and evaporated again. The procedure was repeated three times to eliminate any HCl remnants. After the last evaporation, the residue was re-dissolved in 1 mL of concentrated HNO3 and left to dissolve. Ultrasonic baths and a 70 °C hot plate were used to enhance the dissolution. Once fully dissolved, the solution was quantitatively diluted with MQ water and 2% HNO3 to produce a 1000 ppm SRM 3138 stock solution in 2% HNO3, which was protected from light exposure using aluminum foil. Working solutions were diluted daily from the stock solutions.
Gold extraction and dissolution
Selected drill core samples, quartered, 5–10 cm in length, were first sampled for thin sections, which were made in the Thin Section Lab in Nancy, France. Gold was extracted from quartered drill cores using an electric pulse fragmentation device (Selfrag; Wang et al. 2012). The use of high-voltage electric pulses allows the sample to break along the phase boundaries minimizing any sample loss that might take place during traditional crushing due to the malleable nature of gold. Fine clay and silt matrix was washed out, and the samples were dried at 90 °C overnight. Heavy minerals were separated with methylene iodide (ρ ≈ 3.32 g/cm3). Because of a high sulfide content, the heavy mineral separates were treated with an excess of 1:1 HNO3 (65%) and water to dissolve any sulfides. The residue was filtered and washed thoroughly with MQ water. Gold grains were then handpicked from the dried samples under a binocular lens. Gold was found either as fully liberated free grains or intergrown with quartz, muscovite, or scheelite/ferberite. In total, Au grains were recovered from only six samples (Fig. 2), most likely because of the limited sample amount and the nugget nature of the Au.
The picked Au grains (<<1 mg) were weighed in Savillex teflon beakers and digested in 1 mL of aqua regia. Closed beakers were left on a hot plate (80 °C) to achieve complete digestion. The samples were evaporated and re-dissolved in 0.5 mL of concentrated 12M HCl. If a clear solution was not achieved, more 12M HCl was added. Once fully digested, a small (2 μL) split was taken and diluted to achieve 0.5M solution to determine Ag concentration. The rest of the sample solution was then diluted with MQ water to achieve a 6M HCl solution.
Ion exchange chromatography
Ag separation from the matrix was carried out in two consecutive stages utilizing ion exchange columns using a procedure adapted from Brügmann et al. (2019). Both ion-exchange separations were carried out in columns made from disposable polyethylene transfer pipettes. Both AG1-X8 and TBP resins were pre-washed before packing them into the columns. Pre-washing of the AG1-X8 resin was done in a small batch measuring approximately 5 mL of resin into a beaker with 6M HCl in excess. The mixture was stirred with a magnetic mixer for 4–6 h to keep the resin in suspension, settled, and the supernatant decanted to remove fine or broken particles. The procedure was repeated three times. After a thorough wash with 6M HCl, the sequence was repeated with ultra-pure water. TBP resin was soaked in dilute 0.5M HCl overnight until it stopped floating and settled on the bottom of the beaker.
The first column was packed with 0.2 mL of pre-washed AG1-X8 anion resin, 100-200 mesh, in chloride form, with a bed length of about 1 cm. The resin was further cleaned and preconditioned with 2 mL of 6M HCl. The sample or a split of a sample ≤1 mL was then introduced in 6M HCl and Ag eluted with 3 mL of 6M HCl. Au and Pd should stay bound to the resin and form a bright orange ring at the top of the resin bed.
The second step was utilized to convert Ag from chloride form to Ag nitrate. The column was packed with 0.4 mL of Triskem TBP resin, 50–100 μm, loaded on top of 0.075 mL of Triskem prefilter medium. The prefilter resin was soaked in batch before use and is employed to remove any organic impurities from the solutions or from the resins. The columns were cleaned with five column volumes of 4M HNO3 and conditioned with 2 mL of MQ-water followed by 2 mL of 0.5M HCl. Samples for the second column were prepared by evaporating the first column eluate, if needed, to contain <1 μg in a 20 μL split and diluted to 0.5M by adding 280 μL of MQ-water. The sample was carefully loaded on top of the resin in three 0.1 mL splits. After loading the sample, 0.1 mL H2O was added to rinse the inner surface of the column and to wash the sample deeper into the resin. To get rid of any chloride ions bound to the inner walls of the column, the resin bed was plugged with a cut pipette head, and the reservoir was rinsed with three volumes of H2O. The end of the columns was rinsed with excess H2O, dipping the end of the column in an H2O beaker. After the reservoir rinse, the sample was washed deeper into the resin with two 0.1 mL H2O volumes. The column rinses, 0.3 mL in total, were collected in a beaker filled with 0.7 mL of 0.5M HCl and analyzed for Ag loss. Ag was collected with 5 mL of 0.38M HNO3. Depending on the Ag concentration, some samples were evaporated to achieve a more concentrated solution and analyzed the next day.
Generally, the Ag loss was found to be <0.5%. Previous studies have reported resolvable Ag isotope fractionation with yields lower than ∼99% during ion exchange chromatography (Schönbächler et al. 2007; Brügmann et al. 2019), and for that reason, high yields are deemed to be mandatory for accurate isotopic measurements. Fractionation related to <5% Ag loss (during the 2nd stage chromatography), however, was found not to induce resolvable isotope fractionation in this study, and hence samples with yield > 95% were accepted. Fractionation behavior of Ag was studied by tracking the fractionation and yield of SRM 978a standard solution after the 2nd stage chromatography.
Mass spectrometry and data processing
Silver isotope measurements were performed on a Nu Instruments TM multi-collector ICPMS at the Geological Survey of Finland. Instrument detector configuration and operating conditions are detailed in Tables 1 and 2. External standardization was used for analysis as Ag has only two isotopes (107Ag and 109Ag) and thus does not allow internal correction of instrumental mass fractionation. Instrumental mass bias was monitored by external standard by doping the sample solution with Pd, NIST SRM 3183, to achieve a 108Pd/107Ag ratio close to 1. The Pd spike was added shortly before the analysis as the isotopic composition of the spiked standard solution was noticed to change after storing overnight. Sample concentrations were matched with the standard concentrations.
Analyses were run in wet mode using a self-aspirating nebulizer (Table 2). On average, 20 ppb solution produced a 109Ag signal of 0.18 V. In dry plasma condition, the water content can be reduced by two orders of magnitude resulting in increased sensitivity (Hattendorf and Günther 2000), and therefore, a desolvating nebulizer (The Teledyne CETAC Aridus3) was tested but found to induce unresolvable variations in fractionation factors between Pd and Ag (Fig. 3). This is likely to be caused by a difference between Ag and Pd interaction between the nebulized sample and the dissolvating membrane during the desolvation process. Similar behavior was also detected by Schönbächler et al. (2007) who reported constant instrumental mass bias for Ag and Pd in wet mode, whereas the relative fractionation of Ag and Pd in dry mode was significantly affected by minor impurities resulting in low analytical precision. Closely matched fractionation behavior between the analyte and the spike is a prerequisite for a viable mass bias correction, and dry plasma conditions were hence ruled out. The slight deviation from the expected slope (Fig. 3b), by exponential mass fractionation law, suggests a subtle difference between the mass bias factors for Pd and Ag in wet mode but yet offers the closest proxy for the mass bias correction. A difference in the fractionation factor has also been recognized in earlier studies (e.g., Woodland et al. 2005).

Correlation trends between Ag and Pd fractionation factors for dry and wet plasma. a Poor correlation between natural logarithms of 108Pd/105Pd and 109Ag/107Ag indicating different fractionation behavior between Pd and Ag in dry plasma. b Good correlation between natural logarithms of 108Pd/105Pd and 109Ag/107Ag in wet plasma
Data were collected in static mode monitoring masses 105–111 amu with a mass separation of 0.5. A single measurement cycle included three consecutive washes with 0.5M HNO3, a 60s background measurement on mass and 60 isotope ratio measurements in one block with 5s integration time. The signals were corrected for background using the background data measured prior to every analysis. Cadmium interference on mass 106, 108, and 110 was corrected by monitoring 111Cd. Instrumental mass bias correction was done using internal normalization to a known 108Pd/105Pd ratio of 1.18899 (Kelly and Wasserburg 1978), following the exponential mass fractionation law (Maréchal et al. 1999) and assuming alike mass bias for Ag and Pd even though the respective ionization energy of Ag and Pd are 7.5765 and 8.3369 eV, respectively. In addition, standard-sample-standard bracketing was applied to improve the accuracy. A new batch of Pd (SRM3138) spiked SRM 978a, used as the bracketing standard, was prepared daily. The performance of the instrument was assessed with several runs of an in-house standard solution at the beginning and at the end of every session and between every 10 analyses of unknowns.
The long-term average of Pd bias corrected 109Ag/107Ag for SRM 978a was 1.07982 ± 0.0005 (n = 165). The data were collected from repeated measurements of the SRM 3138 spiked SRM 978a standard in 15 sessions over 6 months (Fig. 4). The obtained value is indistinguishable from the values reported in previous studies (Woodland et al. 2005; Schönbächler et al. 2007; Brügmann et al. 2019; Voisey et al. 2019). On average, the reproducibility of the measurements within a single run for ε109Ag was 0.2 × 10−4 (2SD). Systematic long-term bias for the in-house standard was ε109Ag −0.013 ± 0.26 (n=66).

Average value of NIST SRM 978a Ag isotope standard measured in this study, in comparison with reported values. Error bars are at 2SD
Variations in Ag isotopic composition are here reported using the epsilon notation ε109Ag = [(109Ag/107Ag)sample/(109Ag/107Ag)standard−1] × 104, where the Ag isotopic composition is reported as the relative difference between the Ag isotopic composition in the sample and in the NIST SRM 978a international Ag standard in parts per 10,000 (Desaulty and Albarède 2013; Albarède et al. 2016; Argapadmi et al. 2018; Mathur et al. 2018; Brügmann et al. 2019). This is in line with the stable isotope convention of using the heavy isotope as the numerator in the equation, i.e., reporting the heavy/light ratio (Kendall and Caldwell 1998). The notation is different from cosmochemistry, where ε107Ag notation is used to account for the radiogenic decay of 107Pd to 107Ag (Hauri et al. 2000; Schönbächler et al. 2010). ε107Ag notation has been used in some earlier studies (Woodland et al. 2005; Chugaev and Chernyshev 2012; Voisey et al. 2019) to report the Ag isotopic composition in terrestrial samples.
Results
Silver isotopic composition was measured from six native Au samples purified in the two-step chromatographic separation. The 109Ag/107Ag isotopic compositions relative to the SRM 978a standard and Ag concentrations of the samples are summarized in Table 3. Silver content in the analyzed Au samples was calculated from sample weights and analyzed Ag concentrations and should only be regarded as indicative because of the presence of other minerals within the hand-picked Au samples (poor liberation) and the weighing balance error for such a small volume. Nevertheless, Ag concentrations show large variation with a range from 0.4 to 11.2 wt%. Variation was verified with SEM analysis on two samples with visible Au (Table 3). SEM analysis of Au in sample JHTE-20190514-3 shows a relatively low Ag concentration of 0.94 wt%, whereas Au in sample JHTE-20190514-5 is much more abundant (8.9 wt%). The difference between the calculated and measured (by SEM) Ag concentration in sample JHTE-20190514-3 is likely related to the fact that only two small Au grains were analyzed with SEM, whereas the dissolved sample (and calculated wt%) comprised several Au grains and represents a much larger sample volume, i.e., intergranular variation in Ag concentration. Moreover, heterogenous distribution of Ag within the Au grains may also contribute to the difference. Silver content for sample JHTE-20190514-8 is not reported as the volume/mass of the hand-picked sample was too low to be weighed (< 10 μg).
The measured ε109Ag values show a fairly limited range from −6.7 to +2.1 with a total variation of 8.8 ε-units (Table 3). This is in line with and well within the range of ε109Ag values reported for native Ag (from −8.6 to +21.4) and Au samples (from −8.3 to +8.3) (Argapadmi et al. 2018; Mathur et al. 2018; Brügmann et al. 2019; Voisey et al. 2019; Arribas et al. 2020). Most of the analyzed samples have negative ε109Ag values between −6.7 and −2.0, falling below the Bulk Silicate Earth (BSE) of +2.2 ± 0.7 ε109Ag (Schönbächler et al. 2010). One of the samples has a positive ε109Ag value of +2.1 ± 0.5, close to BSE (Fig. 5). The mean value of the analyzed samples is ε109Ag = −3.8 ± 1.7 (2SD).

a Frequency distribution histogram showing Ag isotopic composition of the Au samples analyzed in this study relative to published data from native Au and Ag by (Chugaev and Chernyshev 2012; Argapadmi et al. 2018; Mathur et al. 2018; Brügmann et al. 2019; Voisey et al. 2019; Arribas et al. 2020). Bulk Silicate Earth (BSE) composition after Schönbächler et al. (2010). b Histogram showing Ag isotopic composition of the Au samples analyzed in this study relative to published data from Au deposits (Chugaev and Chernyshev 2012; Argapadmi et al. 2018; Brügmann et al. 2019; Voisey et al. 2019)
Discussion
The origin, depositional environment, and ore-forming processes in the Rajapalot Au deposit are controversial as the deposit carries features that do not fit the conventional classification of the deposit as an orogenic Au deposit with atypical metal association (Molnár et al. 2016; Ranta et al. 2017, 2018). In this work, silver isotope analyses were designed to develop a general working protocol for Ag isotope analysis in Co-enriched Au deposits to give implications on the Au enrichment processes in the Rajapalot deposit. This is based on the similar geochemical properties of Ag and Au in hydrothermal solutions and their presence in electrum in the Rajapalot deposit (Seward et al. 2014).
Isotopic inheritance
Early research on Ag isotopes suggests that varying ε109Ag values could be inherited from the source rock (Chugaev and Chernyshec 2009; Hauri et al. 2000; Chugaev and Chernyshev 2012), and hence the isotopic composition of Ag could potentially be used for source region footprinting. Resolvable variation in Ag isotopic composition has indeed been described for common terrestrial bulk rocks such as basalts and sedimentary rocks, with a total variation of 11 ε-units (Hauri et al. 2000; Woodland et al. 2005; Schönbächler et al. 2007, 2010). This strong incentive inspired researchers to use the variation in the 109Ag/107Ag ratio in provenance tracing to study Ag circulation in past monetary systems by analysis of ε109Ag values from old coinage (Desaulty et al. 2011; Desaulty and Albarède 2013; Albarède et al. 2016, 2021; Milot et al. 2021, 2022). The use of Ag isotopes as a regional footprint might not be that unambiguous, however, at least not in the context of Au deposits (tracer of Au), as large isotopic variation has been reported within deposits. Overall variation of 7.9 (± 0.4 2SD) ε109Ag has been analyzed from hydrothermal Au deposits in the Barberton greenstone belt (Argapadmi et al. 2018) where Ag and Au are believed to have originated from a single, common source. An even wider range of ε109Ag values has been reported from orogenic Au from the Bendigo zone in the Victorian Goldfields, showing a variation of 14.9 ε-units for samples in Au deposits sharing similar settings (Voisey et al. 2019). As it seems that the Ag isotopic composition can vary significantly between deposits within a single mining camp or between deposits that share common age, tectonic history, structural setting, and host rock, the inheritance is unlikely to be the major control on Ag isotope systematics in hydrothermal gold. The variation of 8.8 (± 0.6 2SD) ε-units in the studied samples is well within the range of variation reported earlier giving no direct implications on two differing metal sources. The origin of the positive ε109Ag signal in one of the samples while other samples have consistently negative ε109Ag values is, however, intriguing. If the isotopic signature of Ag isotopes was inherited from the source rock and the possible partitioning during transportation and deposition was considered negligible, the bimodal distribution of the ε109Ag values would indicate two distinct metal sources—one with Ag isotopic composition of Au close to BSE and one with clearly negative Ag isotopic composition. Inheritance from two sources with vastly different ε109Ag compositions for gold seems unlikely in the Rajapalot deposit. Therefore, it is suggested that the large intradeposit variation is related to other factors such as physicochemical processes during the ore formation.
Physicochemical fractionation
The bimodal distribution of the ε109Ag values in Au in the Rajapalot deposit may reflect physicochemical fractionation of Ag during transportation and/or deposition processes with similar behavior reported for, e.g., Cu and Fe isotopes (Zhao et al. 2017, 2019; Zhu et al. 2018; Ding et al. 2019). Several processes could induce mass-dependent Ag isotope fractionation during the ore-forming processes. These include temperature changes, fractional crystallization, boiling, phase separation, secondary remobilization, fluctuating redox conditions, or combinations thereof (Argapadmi et al. 2018; Fujii and Albarède 2018; Mathur et al. 2018; Voisey et al. 2019). Major temperature-dependent fractionation is commonly related to low-temperature environments where equilibrium fractionation is more extensive (Schauble 2004), and only subtle Ag isotope fractionation has been related to hypogene Au deposits compared to significant fractionation found in supergene deposits (Mathur et al. 2018; Arribas et al. 2020). Therefore, temperature alone cannot be considered the primary variable that controls the Ag fractionation in the Rajapalot Au deposit. Equilibrium fractionation can vary considerably depending on temperature, and thus temperature may have an accompanying effect on the Ag fractionation.
Equilibrium and Rayleigh-type fractionation
Processes that involve equilibrium fractionation (such as boiling or other liquid-vapor phase separation processes) of isotopes are well recognized for other elements/isotope systems. For example, Fe isotope fractionation between liquid and vapor phase can reach 0.15 ± 0.05‰ δ56Fe (Syverson et al. 2014), whereas Cu fractionation may reach values up to 0.69‰ ± 0.05‰ δ65Cu (Rempel et al. 2012). Both systems show gradual enrichment of heavy 56Fe or 65Cu in the residual liquid as the vapor phase favors the light isotope, offering a basis for the gradual precipitation of metals with systematically varying isotopic composition. As Ag and Au are similar transition metals, the same principles may be applicable to Ag isotope systematics in Au deposits as well.
Silver isotope fractionation during phase separation was confirmed and quantified by Wang et al. (2022) who demonstrated that Ag isotope fractionation resulting from vapor-liquid phase separation varies from −3.0 to +4.0 ε109Ag. This implies that the total variation of ε109Ag of 8.8 in the samples analyzed in this study could largely be explained by phase separation, which can account for up to 7.0 ε-unit changes in the Ag isotopic composition. If vapor-liquid phase separation took place in the Rajapalot deposit, the vapor phase would have been enriched in the light isotope 107Ag because of its higher diffusion rate relative to the liquid, which in turn becomes relatively enriched in the heavy isotope 109Ag—as observed for the Cu and Fe isotope systems (Rempel et al. 2012; Syverson et al. 2014). This would presumably result in the concentration of light isotopes in the shallow parts of the systems as the vapor rises.
Rayleigh-type fractional crystallization can, however, result in identical gradual changes and has been shown to be the major cause of Ag isotope variation from −4.2 to +3.6 ε109Ag (total variation of 7.9 ε-units) in hydrothermal Au in the Barberton greenstone belt (Argapadmi et al. 2018). The compositional change during fractional crystallization is likely driven by changes in Ag redox state. In a low-temperature reduced hydrothermal solution, Ag is carried predominantly as monovalent species in aqueous (HS)-2 complexes with lesser chloride or hydroxide complexes, with main oxidation state of +1 (Seward 1976; Migdisov and Williams-Jones 2013; Seward et al. 2014), whereas in native Ag, or Ag alloys, Ag is found in its reduced form Ag0 (Seward 1976; Migdisov and Williams-Jones 2013; Seward et al. 2014). Similarly, Au is carried predominantly as monovalent species in aqueous (HS)-2 complexes and is deposited through reduction of the Au+ ion to Au0. Coprecipitation of Ag with Au in the form of electrum in hydrothermal systems involves the reduction of both elements, irrespective of the cause of the precipitation. For native Ag precipitating from hydrothermal solution, the reduction process should favor the light isotope 107Ag as heavy isotopes prefer high valence and low coordination to achieve lower zero-point energy and the greatest possible bond stiffness (Bigeleisen and Mayer 1947; Young et al. 2015). The bond stiffness is greater for shorter and stronger bonds, and these properties correlate to a high oxidation state (Schauble 2004). If this general stable isotope fractionation theory is applied, the fractionation should leave the residue enriched in 109Ag, and the enrichment would gradually increase towards the periphery of the deposit. This is in line with laboratory experiments involving the precipitation of Ag onto MnO2 that showed significant fractionation of Ag isotopes up to 6.8 ε-units with Ag0 favoring the light isotope (Mathur et al. 2018). Moreover, native Ag has been reported to have a lighter isotopic composition compared to trace Ag found in the lattice of pyrite as Ag+ (Woodland et al. 2005).
Building on the fact that Ag is found as a ubiquitous element in Au in the Rajapalot deposit, and the assumption that Ag and Au track together during hydrothermal ore formation because of their similar geochemical properties, the observed variation in the ε109Ag in the Rajapalot Au could be explained by phase separation and fractional crystallization of the coprecipitating Au and Ag. Both the abovementioned physicochemical mechanisms have similar effects on the Ag isotope fractionation, leading to enrichment of the heavy isotope in the residue, and could both explain the isotope values. Even though we cannot come to a simple conclusion regarding the exact mechanism because of the rather limited number of samples, physicochemical changes during the ore precipitation are considered to be the main reason for Ag isotope fractionation in the Rajapalot deposit.
Nuclear volume effect
The nuclear volume effect (NVE), also known as nuclear field shift, has also been described to contribute on isotope fractionation in natural systems. The effect has raised particular interest among researchers studying heavy isotopes that are known to show limited equilibrium mass-dependent fractionation because of very small differences in zero-point vibrational energies between the studied isotopes (Bigeleisen and Mayer 1947). The nuclear volume effect is caused by a shift in the nuclear field leading to preferential enrichment of the heavy isotope for the chemical species with the smallest number of s-electrons in the outer orbital (Bigeleisen 1996). For heavy elements such as Hg and Tl, NVE has been reported to be the dominant cause of equilibrium fractionation resulting in up to 3‰ fractionation at room temperature (Schauble 2007). Lighter elements have also been suggested to show isotope fractionation related to NVE (Fujii et al. 2006; Moynier et al. 2013) indicating that the s-electron occupational change between the Ag+ ([Kr]4d105s0) and Ag0 ([Kr]4d105s1) structures would theoretically lead to concentration of the heavy Ag109 into the oxidized Ag+ species. Analytical observations on NVE are difficult with only two isotopes, but the effect, associated with redox processes, has been predicted to induce 1 ε-unit fractionation between Ag0 and dissolved Ag-sulfides at low temperature (Fujii and Albarède 2018). Moreover, NVE has been suggested to be one of the controlling factors on Ag fractionation in orogenic Au systems (Voisey et al. 2019) and Ag ore-forming hydrothermal systems (Wang et al. 2022). For these reasons, NVE associated with oxidation state changes may have contributed to Ag fractionation in the Rajapalot deposit. It is not, however, the only or primary factor influencing isotope fractionation because of the limited fractionation predicted in the previous studies. Further evaluation of the intensity of NVE and its contribution to Ag isotope fractionation in the Rajapalot reposit is beyond this study, however.
Variation in Ag isotopic composition in the Rajapalot deposit and implications for deposition of Au
Ag isotopic composition within the Rajapalot deposit shows systematic, clearly trackable changes, irrespective of the driving forces of the fractionation. Fractionation processes in the hydrothermal system of the Rajapalot deposit are considered to be related to physicochemical changes during deposition and/or re-mobilization of the ore rather than isotopic inheritance from the source rock. Any of the abovementioned physicochemical factors could explain the Ag isotopic composition and the rather limited fractionation of 8.8 ε-units seen in the analyzed Au samples. When the theoretical fractionation principles, i.e., the relative partitioning of the heavier isotope to the oxidized species (described above), are applied to the Rajapalot deposit, we should be able to track systematic changes in Ag isotopic composition within the deposit. In its simplest form, the first particles of silver precipitating with gold would have a low ε109Ag value compared to the initial bulk ε109Ag composition, whereas the remaining Ag+(aq) in the hydrothermal solution would be isotopically heavy. Silver and Au precipitating from the remaining solution would be gradually heavier. The limited available data show a possible fractionation trend between ε109Ag and Au concentration, and a positive correlation between ε109Ag and Ag concentration analyzed from bulk drillcore samples (Fig. 6) suggesting that Ag isotope fractionation is following a gradual trend. If the bulk of the Au-Ag alloy was precipitated at the first stage according to reaction (2), the lightest isotopic composition would represent the first precipitates, which is in line with the trend recorded by the samples. Hence, the samples with the heavier isotopic composition and lower Au concentration are suggested to represent later precipitates and have formed later in the process. Similar behavior of Ag isotopes in gold has been reported by Argapadmi et al. (2018). If this is the case, Ag isotopes could potentially provide a vectoring tool that could be used as a guide towards the mineralization. More extensive studies with a greater number of samples across the full mineralization would be needed to test this hypothesis.

Correlation trends between analyzed ε109Ag values and Au or Ag concentrations of the bulk rock. The size of the symbols is relative to Au concentration in the samples. a Correlation between analyzed ε109Ag values and Au concentration of the bulk rock in the sampling interval. Sample name is shown for a single outlier. Error bars are at 2SD. b Correlation between analyzed ε109Ag values and Ag concentration of the bulk rock. Sample JHTE-20190514-5 with anomalously high e109Ag is labeled
Interestingly, in the Rajapalot deposit, the sample with the heaviest, and only positive, ε109Ag value falls away of the indicated “fractionation” trendline (Fig. 6). This sample could represent a different fractionation line compared to the rest of the samples and/or be part of an alternative process resulting in Au precipitation. As part of a secondary dissolvation-precipitation cycle, the Ag-bearing Au would be dissolved to produce Ag1Cl or Ag1(HS)2- complex enriched in the heavy 109Ag according to theoretical predictions. Precipitation of the dissolved secondary Ag together with Au would result in the formation of isotopically heavy particles compared to the primary Au. The relatively heavy Ag isotope composition in the deviant sample and the fact that the sample is sitting outside of the trendline fit this interpretation. A fully new mineralization pulse cannot, however, be ruled out based on the existing data and could similarly explain the differing isotopic composition of the deviant sample. Nonetheless, the bimodal distribution of the analyzed ε109Ag values and different fractionation trends together with varying Ag concentration suggest a multistage ore paragenesis with either remobilization of pre-existing silver mineralization or a new mineralizing pulse or both. Distinct differences between sulfide mineralogy and Au texture in the two samples with visible gold further support the concept of (at least) two stages of mineralization in the Rajapalot deposit. Gold is found at the grain boundary of pyrrhotite, chalcopyrite, and albite in the deviant sample, whereas the other sample with visible gold shows an association with scheelite and ferberite. The association of Au in the remaining samples is unknown. This concurs with previous work on the Rompas-Rajapalot area that indicates the existence of multiple pyrite generations (Raič et al. 2022) with at least two Au-bearing sulfurous fluids (Vanhanen et al. 2015). The sulfide mineralogy of the deposit does not fully coincide with the abovementioned fractionation trend and shows that further studies on the Ag isotope systematics are needed. The number of analyzed samples is rather limited, for comprehensive conclusions on the cause of the applicability of the results.
Concluding remarks
Silver isotopic composition was measured from six native Au samples from the Rajapalot Au-Co deposit that is characterized by an atypical metal association. A tailored two-step ion-exchange procedure and a high-precision MC-ICP-MS analysis with Pd-doping and standard bracketing were utilized. The long-term average of 109Ag/107Ag for SRM 978a was 1.07982 ± 0.0005 (n = 165), and the average reproducibility of the measurements within a single run for ε109Ag was 0.2 ε-units (2SD). Silver concentration in the analyzed Au samples varies from 0.4 to 11.2 wt%. The measured ε109Ag values yield a fairly limited range from −6.8 to +2.1, well in line with Ag isotopic compositions reported for native Au ores. Moreover, the results show that Ag isotope heterogeneity exists on a depositional scale. Despite the small number of samples analyzed, utilization of ε109Ag values as a source region footprinting tool is considered to be limited. Variations in the source region ε109Ag signal are easily masked by Ag isotope fractionation related to transport, deposition, and re-mobilization processes. It is suggested that the ε109Ag variation in the Rajapalot deposit is related to redox reactions involving Ag+ ↔ Ag0 phase change facilitated by various physicochemical changes during deposition and/or re-mobilization. Bimodal distribution of the ε109Ag values and Ag concentrations suggests either a multistage ore formation with at least two distinct fluid compositions or remobilization of the primary precipitates or both. This study indicates that the fractionation is systematic and gradual, but the limited number of samples examined does not allow a comprehensive evaluation of isotopic partitioning within the Rajapalot deposit. Further studies using the sample preparation and analytical protocol presented here are needed to assess the full significance of our results and their bearing on the understanding of the potential of Ag isotopes as a mineralization vectoring tool.
References
Albarède F, Blichert-Toft J, de Callataÿ F, Davis G, Debernardi P, Gentelli L, Gitler H, Kemmers F, Klein S, Malod-Dognin C, Milot J, Télouk P, Vaxevanopoulos M, Westner K (2021) From commodity to money: the rise of silver coinage around the Ancient Mediterranean (sixth–first centuries bce). Archaeometry 63:142–155. https://doi.org/10.1111/arcm.12615
Albarède F, Blichert-Toft J, Rivoal M, Telouk P (2016) A glimpse into the Roman finances of the Second Punic War through silver isotopes. Geochem Perspect Lett 2:127–137. https://doi.org/10.7185/geochemlet.1613
Argapadmi W, Toth ER, Fehr MA, Schönbächler M, Heinrich CA (2018) Silver isotopes as a source and transport tracer for gold: a reconnaissance study at the Sheba and New Consort gold mines in the Barberton greenstone belt, Kaapvaal craton, South Africa. Econ Geol 113:1553–1570. https://doi.org/10.5382/econgeo.2018.4602
Arribas A, Mathur R, Megaw P, Arribas I (2020) The isotopic composition of silver in ore minerals. Geochem Geophys 21:1–17. https://doi.org/10.1029/2020GC009097
Bigeleisen J (1996) Nuclear size and shape effects in chemical reactions. Isotope chemistry of the heavy elements. J Am Chem Soc 118:3676–3680. https://doi.org/10.1021/ja954076k
Bigeleisen J, Mayer MG (1947) Calculation of equilibrium constants for isotopic exchange reactions. J Chem Phys 15:261–267. https://doi.org/10.1063/1.1746492
Boyle R (1979) The geochemistry of gold and its deposits. Canadian Geological Survey Bulletin 280:584
Brügmann G, Brauns M, Maas R (2019) Silver isotope analysis of gold nuggets: an appraisal of instrumental isotope fractionation effects and potential for high-resolution tracing of placer gold. Chem Geol 516:59–67. https://doi.org/10.1016/j.chemgeo.2019.03.015
Carlson R, Hauri E (2001) Extending the 107 Pd- 107 Ag chronometer to low Pd / Ag meteorites with multicollector plasma-ionization mass spectrometry. Geochim Cosmochim Acta 65:1839–1848
Chugaev AV, Chernyshec IV (2009) Variations of 107Ag/109Ag isotope ratio in ore deposits by high-precision MC-ICP- MS. Geochim Cosmochim Acta 73:A225
Chugaev AV, Chernyshev IV (2012) High-noble measurement of 107Ag/ 109Ag in native silver and gold by multicollector inductively coupled plasma mass spectrometry (MC-ICP-MS). Geochemistry International 50:899–910. https://doi.org/10.1134/S0016702912110055
Cook N, Molnár F, O’Brien H, Lahaye Y, Tapio J (2019) The evolution of the Peräpohja Belt as viewed from within the Rompas-Rajapalot project. In: NEXT - 3rd Progress Meeting 7-10, October, Pohtimolampi, Rovaniemi, Finland
Desaulty AM, Albarède F (2013) Copper, lead, and silver isotopes solve a major economic conundrum of Tudor and early Stuart Europe. Geology 41:135–138. https://doi.org/10.1130/G33555.1
Desaulty AM, Telouk P, Albalat E, Albarède F (2011) Isotopic Ag-Cu-Pb record of silver circulation through 16th-18th century Spain. Proc Natl Acad Sci U S A 108:9002–9007. https://doi.org/10.1073/pnas.1018210108
Ding X, Ripley EM, Wang W, Li C, Huang F (2019) Iron isotope fractionation during sulfide liquid segregation and crystallization at the Lengshuiqing Ni-Cu magmatic sulfide deposit, SW China. Geochim Cosmochim Acta 261:327–341. https://doi.org/10.1016/j.gca.2019.07.015
Eilu P (2015) Overview on gold deposits in Finland. In: Maier et al (eds) Mineral deposits of Finland, 1st edn. Elsevier, pp 377–410. https://doi.org/10.1016/B978-0-12-410438-9.00015-7
Eilu P, Sorjonen-Ward P, Nurmi P, Niiranen T (2003) A review of gold mineralization styles in Finland. Econ Geol 98:1329–1353. https://doi.org/10.2113/gsecongeo.98.7.1329
Farajewicz M, Cook N (2021) Sample selection for geometallurgical characterization in the Rajapalot Deposit BATCircle Project Report 02-WP1 Task 1.2. Geol S Finl, Rep of Inves 9/2021, p 29
Fujii T, Albarède F (2018) 109Ag–107Ag fractionation in fluids with applications to ore deposits, archeometry, and cosmochemistry. Geochim Cosmochim Acta 234:37–49. https://doi.org/10.1016/j.gca.2018.05.013
Fujii T, Moynier F, Telouk P, Albarède F (2006) Mass-independent isotope fractionation of molybdenum and ruthenium and the origin of isotopic anomalies in Murchison. Astrophys J 647:1506–1516
Gammons CH, Williams-Jones AE (1995) Hydrothermal geochemistry of electrum: thermodynamic constraints. Econ Geol 90:420–432. https://doi.org/10.2113/gsecongeo.90.2.420
Hagemann SG, Brown PE (2000) Gold in 2000: An introduction. Rev Econ Geol 13:1–7. https://doi.org/10.5382/Rev.13
Hattendorf B, Günther D (2000) Characteristics and capabilities of an ICP-MS with a dynamic reaction cell for dry aerosols and laser ablation. J Anal At Spectrom 15:1125–1131. https://doi.org/10.1039/B001677M
Hauri EH, Carlson RW, Bauer J (2000) The timing of core formation and volatile depletion in solar system objects from high-precision 107Pd-107Ag isotope systematics. 31st Annual Lunar and Planetary Science Conference, March 13-17, 2000, Houston, Texas, abstract no. 1812
Hölttä P, Heilimo E (2017) Metamorphic map of Finland. Geological Survey of Finland, Special Paper 60:75–126
Huhma H, Cliff RA, Perttunen V, Sakko M (1990) Sm-Nd and Pb isotopic study of mafic rocks associated with early Proterozoic continental rifting: the Peräpohja schist belt in northern Finland. Contrib Mineral Petrol 104:369–379. https://doi.org/10.1007/BF00321491
Kelly W, Wasserburg G (1978) Evidence for the existence of 107Pd in the early solar system. Geophys Res Lett 5:1079–1082
Kendall C, Caldwell EA (1998) Fundamentals of isotope geochemistry. In: Kendall, McDonnell (eds) Isotope tracers in catchment hydrology, 1st edn. Elsevier, pp 51–86. https://doi.org/10.1016/b978-0-444-81546-0.50009-4
Lahtinen R, Huhma H, Lahaye Y, Kousa J, Luukas J (2015) Archean-Proterozoic collision boundary in central Fennoscandia: revisited. Precambrian Res 261:127–165. https://doi.org/10.1016/j.precamres.2015.02.012
Liu X, Lu X, Wang R, Zhou H (2012) Silver speciation in chloride-containing hydrothermal solutions from first principles molecular dynamics simulations. Chem Geol 294–295:103–112. https://doi.org/10.1016/j.chemgeo.2011.11.034
Luo Y, Dabek-Zlotorzynska E, Celo V, Muir DCG, Yang L (2010) Accurate and precise determination of silver isotope fractionation in environmental samples by multicollector-ICPMS. Anal Chem 82:3922–3928. https://doi.org/10.1021/ac100532r
Maréchal CN, Télouk P, Albarède F (1999) Precise analysis of copper and zinc isotopic compositions by plasma-source mass spectrometry. Chem Geol 156:251–273. https://doi.org/10.1016/S0009-2541(98)00191-0
Mathur R, Arribas A, Megaw P, Wilson M, Stroup S, Meyer-Arrivillaga D, Arribas I (2018) Fractionation of silver isotopes in native silver explained by redox reactions. Geochim Cosmochim Acta 224:313–326. https://doi.org/10.1016/j.gca.2018.01.011
Migdisov AA, Williams-Jones AE (2013) A predictive model for metal transport of silver chloride by aqueous vapor in ore-forming magmatic-hydrothermal systems. Geochim Cosmochim Acta 104:123–135. https://doi.org/10.1016/j.gca.2012.11.020
Milot J, Blichert-Toft J, Sanz MA, Malod-Dognin C, Télouk P, Albarède F (2022) Silver isotope and volatile trace element systematics in galena samples from the Iberian Peninsula and the quest for silver sources of Roman coinage. Geology 50:422–426. https://doi.org/10.1130/g49690.1
Milot J, Malod-Dognin C, Blichert-Toft J, Télouk P, Albarède F (2021) Sampling and combined Pb and Ag isotopic analysis of ancient silver coins and ores. Chem Geol 564:1–14. https://doi.org/10.1016/j.chemgeo.2020.120028
Molnár F, O’Brien H, Stein H, Cook NDJ (2017) Geochronology of hydrothermal processes leading to the formation of the Au-U mineralization at the Rompas prospect, Peräpohja Belt, Northern Finland: application of paired U-Pb dating of uraninite and Re-Os dating of molybdenite to the identification of multiple hydrothermal events in a metamorphic terrane. Minerals 7:171. https://doi.org/10.3390/min7090171
Molnár F, Oduro H, Cook NDJ, Pohjolainen E, Takács Á, O’Brien H, Pakkanen L, Johanson B, Wirth R (2016) Association of gold with uraninite and pyrobitumen in the metavolcanic rock hosted hydrothermal Au-U mineralisation at Rompas, Peräpohja Schist Belt, northern Finland. Miner Depos 51:681–702. https://doi.org/10.1007/s00126-015-0636-6
Moynier F, Fujii T, Brennecka GA, Nielsen SG (2013) Nuclear field shift in natural environments. CR Geosci 345:150–159. https://doi.org/10.1016/j.crte.2013.01.004
Murthy VR (1962) The isotopic composition of silver in iron meteorites. Geochim Cosmochim Acta 26:481–488. https://doi.org/10.1016/0016-7037(62)90093-5
Perera WN, Senanayake G (2004) The ammine, thiosulfato, and mixed ammine/thiosulfato complexes of silver(I) and gold(I). Inorg Chem 43:3048–3056. https://doi.org/10.1021/ic035301z
Perttunen V (1985) On the Proterozoic stratigraphy and exogenic evolution of the Perapohja area, Finland. Bull Geol Surv Finland 331:131–141
Perttunen V (1989) Volcanic rocks in the Peräpohja area, Northern Finland. Geol S Finl, Rep of Inves 92, p 40
Perttunen V, Hanski E, Väänänen J (1995) Stratigraphical map of the Peräpohja Schist Belt. In: Kohonen T, Lindberg B (eds) The 22nd nordic geological winter meeting, Turku-Åbo, Finland, 8-11 January 1996, Abstracts of oral and poster sessions, p 152
Perttunen V, Vaasjoki M (2001) U-Pb geochronology of the Peräpohja Schist Belt, northwestern Finland. In: Vaasjoki M (ed) Radiometric age determinations from Finnish Lapland and their bearing on the timing of Precambrian volcano-sedimentary sequences. Geol S Finl, Special Paper 33:45–84
Raič S, Molnár F, Cook N, O’Brien H, Lahaye Y (2022) Application of lithogeochemical and pyrite trace element data for the determination of vectors to ore in the Raja Au-Co prospect, northern Finland. Solid Earth 13:271–299. https://doi.org/10.5194/se-13-271-2022
Ranta J, Lauri LS, Hanski E, Huhma H, Lahaye Y, Vanhanen E (2015) U – Pb and Sm – Nd isotopic constraints on the evolution of the. Precambrian Res 266:246–259
Ranta J-P, Hanski E, Cook N, Lahaye Y (2017) Source of boron in the Palokas gold deposit, northern Finland: evidence from boron isotopes and major element composition of tourmaline. Miner Depos 52:733–746. https://doi.org/10.1007/s00126-016-0700-x
Ranta JP, Molnár F, Hanski E, Cook N (2018) Epigenetic gold occurrence in a paleoproterozoic meta-evaporitic sequence in the Rompas-Rajapalot Au system, Peräpohja belt, northern Finland. Bull Geol Soc Finl 90:69–108. https://doi.org/10.17741/bgsf/90.1.004
Rempel KU, Liebscher A, Meixner A, Romer RL, Heinrich W (2012) An experimental study of the elemental and isotopic fractionation of copper between aqueous vapour and liquid to 450°C and 400bar in the CuCl-NaCl-H 2O and CuCl-NaHS-NaCl-H 2O systems. Geochim Cosmochim Acta 94:199–216. https://doi.org/10.1016/j.gca.2012.06.028
Robert F, Brommecker R, Bourne BT, Dobak PJ, McEwan CJ, Rowe RR, Zhou X (2007) Models and exploration methods for major gold deposit types. In: Milkereit B (ed) Proceeding of Exploration 07: Fifth Decennial International Conference on Mineral Exploration, Toronto, Canada, 9-12 September 2007, pp 691–711
Schauble EA (2004) Applying stable isotope fractionation theory to new systems. Rev Mineral Geochem 55:65–111. https://doi.org/10.2138/gsrmg.55.1.65
Schauble EA (2007) Role of nuclear volume in driving equilibrium stable isotope fractionation of mercury, thallium, and other very heavy elements. Geochim Cosmochim Acta 71:2170–2189. https://doi.org/10.1016/j.gca.2007.02.004
Schönbächler M, Carlson RW, Horan MF, Mock TD, Hauri EH (2007) High precision Ag isotope measurements in geologic materials by multiple-collector ICPMS: an evaluation of dry versus wet plasma. Int J Mass Spectrom 261:183–191. https://doi.org/10.1016/j.ijms.2006.09.016
Schönbächler M, Carlson RW, Horan MF, Mock TD, Hauri EH (2010) Heterogenous accretion and the moderately volatile element budget of earth. Science 328:884–887
Seward TM (1976) The stability of chloride complexes of silver in hydrothermal solutions up to 350°C. Geochim Cosmochim Acta 40:1329–1341. https://doi.org/10.1016/0016-7037(76)90122-8
Seward TM, Williams-Jones AE, Migdisov AA (2014) The chemistry of metal transport and deposition by ore-forming hydrothermal fluids. In: Holland, Turekian (eds) Treatise on Geochemistry, 2nd edn. Elsevier, pp 29–57. https://doi.org/10.1016/B978-0-08-095975-7.01102-5
Stefánsson A, Seward TM (2003) Experimental determination of the stability and stoichiometry of sulphide complexes of silver(I) in hydrothermal solutions to 400°C. Geochim Cosmochim Acta 67:1395–1413. https://doi.org/10.1016/S0016-7037(02)01093-1
Syverson DD, Pester NJ, Craddock PR, Seyfried WE (2014) Fe isotope fractionation during phase separation in the NaCl-H2O system: an experimental study with implications for seafloor hydrothermal vents. Earth Planet Sci Lett 406:223–232. https://doi.org/10.1016/j.epsl.2014.09.020
Taipale N (2018) Mineralogy of the Paleoproterozoic Raja Au-Co Prospect, Northern Finland. Master’s thesis,. University of Oulu, Finland, p 61
Vanhanen E, Cook NDJ, Hudson MR, Dahlenborg L, Ranta JP, Havela T, Kinnunen J, Molnár F, Prave AR, Oliver NHS (2015) The Rompas Prospect, Peräpohja Schist Belt, Northern Finland. In: Maier et al (eds) Mineral deposits of Finland, 1st edn. Elsevier, pp 467–484. https://doi.org/10.1016/B978-0-12-410438-9.00018-2
Voisey CR, Maas R, Tomkins AG, Brauns M (2017) The Silver Bullet: Ag isotope systematics in native gold from the central Victorian goldfields, Australia. Proceedings of the 14th Biennial SGA Meeting, Quebéc, Canada SY01:115-118.
Voisey CR, Maas R, Tomkins AG, Brauns M, Brügmann G (2019) Extreme silver isotope variation in orogenic gold systems implies multistaged metal remobilization during ore genesis. Econ Geol 114:233–242. https://doi.org/10.5382/econgeo.2019.4629
Wang E, Shi F, Manlapig E (2012) Mineral liberation by high voltage pulses and conventional comminution with same specific energy levels. Miner Eng 27–28:28–36. https://doi.org/10.1016/j.mineng.2011.12.005
Wang JL, Wei HZ, Williams-Jones AE, Dong G, Zhu YF, Jiang SY, Ma J, Hohl SV, Liu X, Li YC, Lu JJ (2022) Silver isotope fractionation in ore-forming hydrothermal systems. Geochim Cosmochim Acta 322:24–42. https://doi.org/10.1016/j.gca.2022.01.024
Woodland SJ, Rehkämper M, Halliday AN, Lee DC, Hattendorf B, Günther D (2005) Accurate measurement of silver isotopic compositions in geological materials including low Pd/Ag meteorites. Geochim Cosmochim Acta 69:2153–2163. https://doi.org/10.1016/j.gca.2004.10.012
Yin Y, Zajacz Z (2018) The solubility of silver in magmatic fluids: implications for silver transfer to the magmatic-hydrothermal ore-forming environment. Geochim Cosmochim Acta 238:235–251. https://doi.org/10.1016/j.gca.2018.06.041
Young ED, Manning CE, Schauble EA, Shahar A, Macris CA, Lazar C, Jordan M (2015) High-temperature equilibrium isotope fractionation of non-traditional stable isotopes: experiments, theory, and applications. Chem Geol 395:176–195. https://doi.org/10.1016/j.chemgeo.2014.12.013
Zhao Y, Xue C, Liu SA, Mathur R, Zhao X, Yang Y, Dai J, Man R, Liu X (2019) Redox reactions control Cu and Fe isotope fractionation in a magmatic Ni–Cu mineralization system. Geochim Cosmochim Acta 249:42–58. https://doi.org/10.1016/j.gca.2018.12.039
Zhao Y, Xue C, Liu SA, Symons DTA, Zhao X, Yang Y, Ke J (2017) Copper isotope fractionation during sulfide-magma differentiation in the Tulaergen magmatic Ni–Cu deposit, NW China. Lithos 286–287:206–215. https://doi.org/10.1016/j.lithos.2017.06.007
Zhu ZY, Jiang SY, Mathur R, Cook NJ, Yang T, Wang M, Ma L, Ciobanu CL (2018) Iron isotope behavior during fluid/rock interaction in K-feldspar alteration zone – a model for pyrite in gold deposits from the Jiaodong Peninsula, East China. Geochim Cosmochim Acta 222:94–116. https://doi.org/10.1016/j.gca.2017.10.001
Funding
Open Access funding provided by University of Helsinki (including Helsinki University Central Hospital).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Johanna Tepsell with assistance from Yann Lahaye. The first draft of the manuscript was written by Johanna Tepsell, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Acknowledgements
Mawson Gold Ltd. is acknowledged for providing access to their drill core facility, sample material, and geochemical datasets. We are grateful to Leena Järvinen and Minna Myllyperkiö for their invaluable help in the laboratory. Journal referee Ryan Mathur and Editor-in-Chief Bernd Lehmann provided comments that improved the content and layout of the paper.
Funding
This work was supported by the K.H. Renlund Foundation and the Geological Survey of Finland.
Additional information
Editorial handling: B. Lehmann
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tepsell, J., Lahaye, Y., Molnár, F. et al. Silver isotope analysis and systematics of native gold from the Rajapalot Co-enriched gold deposit, Finnish Lapland. Miner Deposita (2023). https://doi.org/10.1007/s00126-023-01239-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00126-023-01239-y