Biogeochemical cycling of chromium and chromium isotopes in the sub-tropical North Atlantic Ocean
 Wenhao Wang1*
Wenhao Wang1*  Heather Goring-Harford1 Korinna Kunde1
Heather Goring-Harford1 Korinna Kunde1  E. Malcolm S. Woodward2
E. Malcolm S. Woodward2  Maeve C. Lohan1
Maeve C. Lohan1  Douglas P. Connelly3
Douglas P. Connelly3  Rachael H. James1
Rachael H. James1- 1School of Ocean and Earth Science, University of Southampton, Southampton, United Kingdom
- 2Plymouth Marine Laboratory, Plymouth, United Kingdom
- 3National Oceanography Centre, Southampton, United Kingdom
Chromium (Cr) is a redox-sensitive element and because Cr isotopes are fractionated by redox and/or biological processes, the Cr isotopic composition of ancient marine sediments may be used to infer changes in past seawater oxygenation or biological productivity. While there appears to be a ‘global correlation’ between the dissolved Cr concentration and Cr isotopic composition of seawater, there is ongoing debate about the relative importance of external sources and internal cycling on shaping the distribution of dissolved Cr that needs to be resolved to validate the efficacy of using Cr isotopes as a paleo proxy. Here, we present full water column depth profiles of total dissolved Cr (Cr(VI)+Cr(III)) and dissolved Cr isotopes (δ53Cr), together with ancillary data, for three stations along a transect (GEOTRACES GApr08) across the sub-tropical North Atlantic. Concentrations of dissolved Cr ranged between 1.84 and 2.63 nmol kg-1, and δ53Cr values varied from 1.06 to 1.42‰. Although atmospheric dust, hydrothermal vents and seabed sediments have the potential to modify the distribution of Cr in the oceans, based on our observations, there is no clear evidence for substantial input of Cr from these sources in our study region although benthic inputs of Cr may be locally important in the vicinity of hydrothermal vents. Subsurface waters (below the surface mixed layer to 700 m water depth) were very slightly depleted in Cr (by up to ~0.4 nmol kg-1), and very slightly enriched in heavy Cr isotopes (by up to ~0.14‰), relative to deeper waters and the lowest Cr concentrations and highest δ53Cr values coincided with lowest concentrations of colloidal (0.02 to 0.2 μm size fraction) Fe. We found no direct evidence for biological uptake of dissolved Cr in the oligotrophic euphotic zone or removal of Cr in modestly oxygen depleted waters (O2 concentrations ~130 μmol kg-1). Rather, we suggest removal of Cr (probably in the form of Cr(III)) in subsurface waters is associated with the formation of colloid aggregates of Fe-(oxyhydr)oxides. This process is likely enhanced by the high lithogenic particle load in this region, and represents a previously unrecognized export flux of Cr. Regeneration of Cr in deeper waters leads to subtly increased levels of Cr alongside decreased δ53Cr values at individual sites, but this trend is more obvious at the global scale, with δ53Cr values decreasing with increasing radiocarbon age of deep waters, from 1.16 ± 0.10‰ (1SD, n=11) in deep Atlantic waters to 0.77 ± 0.10‰ (1SD, n=25) in deep Pacific waters. Removal of relatively isotopically light Cr from subsurface waters onto particulate material and regeneration of this Cr back into the dissolved phase in deep waters partly accounts for the systematic relationship between δ53Cr and Cr concentrations in seawater discussed by other studies.
1 Introduction
Chromium (Cr) is a transition metal, present in typical concentrations of 0.9 to 6.5 nM in seawater (e.g., Campbell and Yeats, 1981; Cranston, 1983; Jeandel and Minster, 1987; Achterberg and van den Berg, 1997; Sirinawin et al., 2000; Connelly et al., 2006; Scheiderich et al., 2015) and has a relatively long seawater residence time of ~3000 to 9500 years (Reinhard et al., 2013; McClain and Maher, 2016; Pöppelmeier et al., 2021). In some parts of the oceans, concentrations of dissolved Cr are modestly depleted in the surface layer, and Cr has been classified as intermediate between a ‘conservative’ and ‘recycled’ element (Jeandel and Minster, 1987; Sirinawin et al., 2000). In oxic seawater, Cr(VI) is predicted to be the thermodynamically stable form of Cr (Elderfield, 1970), although relatively high concentrations of dissolved Cr(III) have been reported in some studies (e.g., Achterberg and van den Berg, 1997; Connelly et al., 2006). High concentrations of Cr(III) in oxic waters are most likely due to the presence of Cr(III)-organic complexes in the colloidal fraction (Li et al., 2022) that may not be captured by some analytical techniques (Rue et al., 1997; Wang et al., 2019; Huang et al., 2021). Under oxygen deficient conditions, Cr(III) can be the dominant Cr species, accounting for up to ~64% of the total dissolved Cr (Murray et al., 1983; Rue et al., 1997; Huang et al., 2021). However, oxygen is not the sole control on Cr speciation (Goring-Harford et al., 2018; Janssen et al., 2020); field (Connelly et al., 2006; Janssen et al., 2020) and experimental (Døssing et al., 2011; Kitchen et al., 2012) studies have suggested that high levels of biological productivity, as well as the presence of Fe(II) and organic matter in surface waters, can also facilitate Cr reduction even in O2-replete waters (Pettine et al., 1998). At the sea surface, Cr(VI) reduction most likely proceeds via photoreduction (Kieber and Helz, 1992; Kaczynski and Kieber, 1993). Cr(VI) is highly soluble as the chromate (CrO42–) ion in oxic seawater, whilst Cr(III) is particle-reactive and readily adsorbed onto solid surfaces (Rai et al., 1987; Ellis et al., 2002).
Rivers are thought to be the main source of dissolved Cr to the ocean (Bonnand et al., 2013; Reinhard et al., 2013; McClain and Maher, 2016; Pöppelmeier et al., 2021). Chromium is mainly present in the form of Cr(III) in rocks; weathering reactions driven by Mn-oxides oxidise Cr(III) as it is released from silicate rocks (Eary and Rai, 1987; Frei et al., 2014; D’Arcy et al., 2016), so Cr can be in the form of both Cr(III) and Cr(VI) in river waters that are delivered to the oceans (e.g., Cranston and Murray, 1980; Goring-Harford et al., 2020). Atmospheric deposition is a major source of many trace metals to the ocean and can result in elevated concentrations of Cr in surface seawater at some locations (Achterberg and van den Berg, 1997), but not others (Goring-Harford et al., 2018; Janssen et al., 2023). Benthic Cr fluxes can be locally important (e.g., Shaw et al., 1990; Rigaud et al., 2013; Janssen et al., 2021), and may also be important for the overall oceanic Cr inventory (Pöppelmeier et al., 2021). High temperature hydrothermal fluids from the Kermadec Arc have Cr concentrations on the order of 10 to 30 nmol kg-1, distinctly higher than surrounding seawater (Janssen et al., 2023). However, analyses of hydrothermal plume particles indicate that they have higher Cr/Fe ratios than predicted by mixing of vent fluids and seawater, suggesting that hydrothermal activity may be a net sink of Cr as it is scavenged from seawater onto Fe-(oxyhydr)oxides (Trocine and Trefry, 1988; German et al., 1991; Rudnicki and Elderfield, 1993; Feely et al., 1996; Bauer et al., 2019). Cr is removed from the oceanic inventory through reduction of Cr(VI), scavenging onto settling particles and burial as Cr(III) in reducing and/or anoxic sediments (Reinhard et al., 2014; Gueguen et al., 2016). Therefore, marine sediments are considered as the major sink for seawater Cr. The estimated fluxes of Cr inputs to, and outputs from, the ocean reported in the literature are summarized in Table 1.

Table 1 Range of concentrations and fluxes of Cr inputs and outputs to/from the ocean.
Stable chromium isotope ratios are expressed in delta notation relative to the NBS979 standard, as:
The δ53Cr values of silicate rocks show a narrow range, −0.12 ± 0.10‰ (Schoenberg et al., 2008). Groundwaters with low levels of oxygen are relatively enriched in heavy Cr isotopes (δ53Cr = 0.7 to 5.8‰: Ellis et al., 2002; Izbicki et al., 2008), which is thought to reflect isotope fractionation during oxidation of Cr(III) to Cr(VI) followed by partial back reduction of the Cr(VI). Theoretical studies and laboratory experiments have shown that reduction of Cr(VI) leads to large mass-dependent fractionation, with enrichment of light Cr isotopes in the Cr(III) that forms (Ellis et al., 2002; Døssing et al., 2011; Basu and Johnson, 2012; Kitchen et al., 2012). The direction of fractionation remains the same regardless of the reductant (Fe(II), Fe(II)-bearing minerals, organic matter), but the kinetic fractionation factors are variable (δ53Cr(III) – δ53Cr(VI) = −1.5 to −4.2‰). To date, there have only been three reports of the Cr isotopic composition of single Cr species in seawater in the peer-reviewed literature (Wang et al., 2019; Davidson et al., 2020; Huang et al., 2021). These studies all show that Cr(III) is isotopically lighter than co-existing Cr(VI), by between −0.07 to −3.1‰ (Davidson et al., 2020; Huang et al., 2021), consistent with the theoretical and laboratory studies.
There is a rapidly growing dataset on the distribution of dissolved Cr isotopes in the open ocean with δ53Cr values ranging from 0.60 to 1.71‰ (Scheiderich et al., 2015; Goring-Harford et al., 2018; Moos and Boyle, 2019; Rickli et al., 2019; Janssen et al., 2020; Moos et al., 2020; Nasemann et al., 2020; Huang et al., 2021; Janssen et al., 2021). The average seawater δ53Cr value yielded from the emerging dataset is 1.04 ± 0.19‰ (1SD, n=347; see Section 4.5 for more details), slightly higher than that of the global average river input (0.49 ± 0.23‰, 1SD, n=49: Frei et al., 2014; Paulukat et al., 2015; D’Arcy et al., 2016; Wu et al., 2017; Andronikov et al., 2019). Although some studies show evidence for fractionation of Cr isotopes during estuarine mixing (Roseburrough and Wang, 2021) and others do not (Goring-Harford et al., 2020), the difference between the mean river water and seawater values nevertheless suggests that the Cr isotopic signature of seawater is likely influenced by input of Cr from other sources (such as seabed sediments) and/or is modified by fractionation of Cr as it is removed from seawater (e.g., by reduction of Cr(VI) on the surface of particles that scavenge the Cr(III) that forms). The measured δ53Cr values in the authigenic fractions of reducing and anoxic marine sediments are reported to be between 0.45 and 0.61‰ in Peru margin sediments (Gueguen et al., 2016; Bruggmann et al., 2019) and between 0.38 and 0.53‰ in Cariaco Basin sediments (Reinhard et al., 2014; Gueguen et al., 2016). These values are within the range of the δ53Cr values of the riverine input, but are lower than most seawater values, supporting the proposition that Cr isotope fractionation occurs during reduction and scavenging of Cr (Janssen et al., 2022).
Scheiderich et al. (2015) showed that the δ53Cr composition of waters in the Arctic and Pacific Ocean was highly heterogeneous (0.61 to 1.55‰) and hypothesized that this can be attributed to Cr reduction and scavenging in surface waters and oxygen minimum zones (OMZs), with subsequent release of Cr from sinking particles in deeper waters. These processes were used to explain the inverse correlation between δ53Cr value and logarithmic Cr concentration in the global ocean, from which they derived an isotope fractionation factor (ϵ, the difference between the δ53Cr value of Cr(III) and the δ53Cr value of Cr(VI)) of ~ –0.8‰ (Scheiderich et al., 2015). More recent studies of the Cr isotopic composition of seawater from the Pacific (including oxygen-deficient zones) and the Southern Ocean (Moos and Boyle, 2019; Rickli et al., 2019; Janssen et al., 2020; Moos et al., 2020; Nasemann et al., 2020; Huang et al., 2021; Janssen et al., 2021) also follow this ‘global correlation’. Whilst evidence for the roles of redox transformations (Nasemann et al., 2020; Huang et al., 2021; Wang, 2021), water mass mixing (Rickli et al., 2019) and biological uptake and regeneration (Janssen et al., 2020; Janssen et al., 2021) on seawater δ53Cr is emerging, the underlying mechanisms that produce the apparent correlation between δ53Cr and Cr concentration are still under debate.
Here, we report total dissolved Cr (Cr(VI) + Cr(III)) concentrations and total dissolved Cr isotope compositions (δ53Cr) for full-depth water column profiles for three stations from a GEOTRACES transect along the 22°N sub-tropical Atlantic Ocean (Figure 1A). This fills an important knowledge gap, because few full-depth water column profiles of Cr isotopes, together with ancillary data (biogeochemical parameters, concentrations of other trace metals, etc.) that aid the interpretation of Cr behavior, are available for the Atlantic ocean. The potential sources of Cr to the North Atlantic Ocean as well as the effects of internal cycling are explicitly investigated, and by considering these new data together with other data from the literature, we are able to provide new insights as to the removal and regeneration processes that shape the global distributions of Cr and Cr isotopes in seawater.
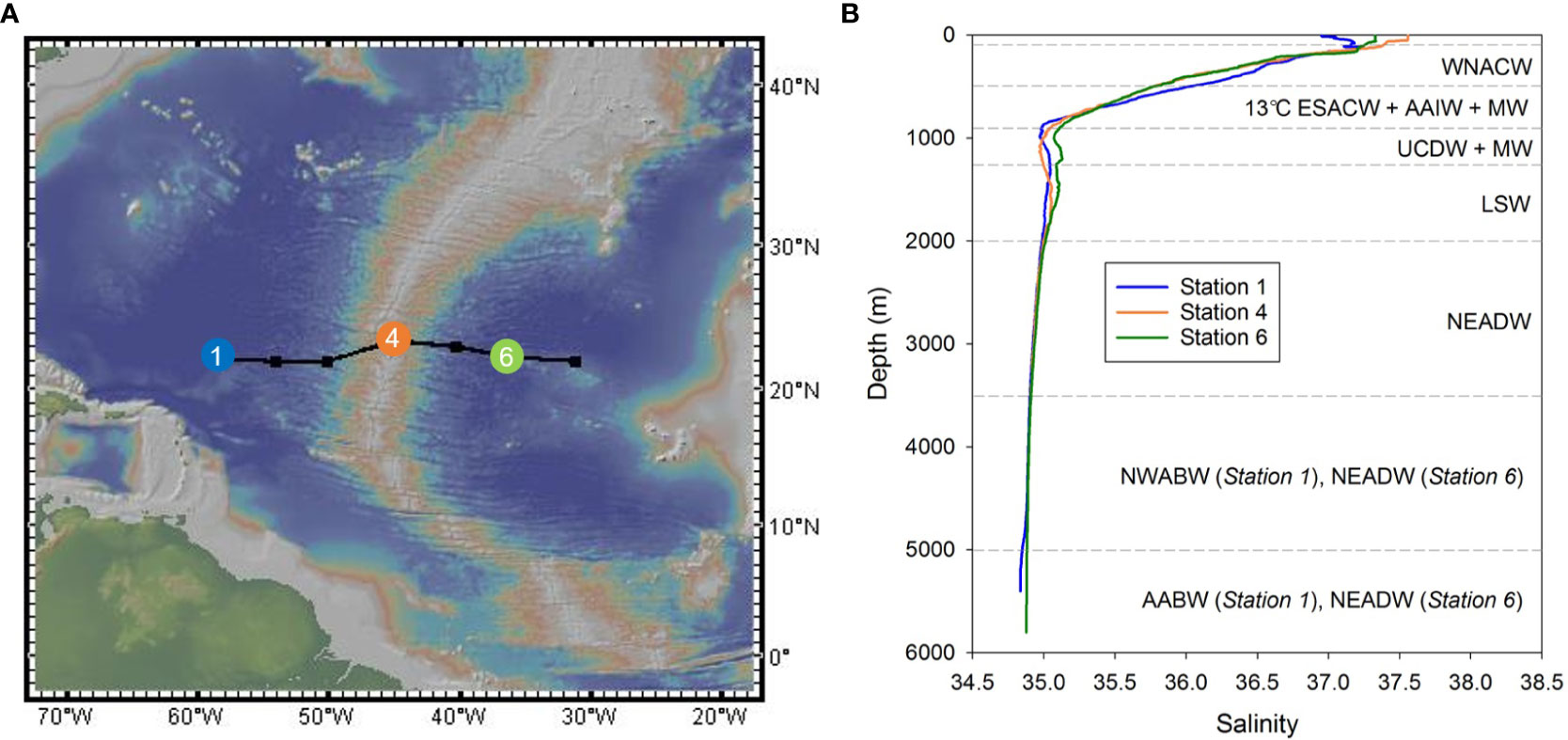
Figure 1 (A) Locations of sampling stations (1, 4 and 6) in the sub-tropical North Atlantic Ocean. Map courtesy of http://www.geomapapp.org (B) Salinity profiles for the three sampling stations. Water masses are delimited by horizontal dashed lines. WNACW, West North Atlantic Central Water; ESACW, East South Atlantic Central Water; AAIW, Antarctic Intermediate Water; MW, Mediterranean Water; UCDW, Upper Circumpolar Deep Water; LSW, Labrador Sea Water; NEADW, North East Atlantic Deep Water; NWABW, North West Atlantic Bottom Water; AABW, Antarctic Bottom Water. Water mass analysis after Artigue et al. (2020).
2 Materials and methods
2.1 Sample collection and oceanographic setting
Samples for this study were collected from Stations 1, 4, and 6 in the sub-tropical North Atlantic (Figure 1A) during RRS James Cook cruise JC150 (UK GEOTRACES GApr08 process cruise) between 26th of June and 12th of August 2017. The water depths for the three stations were 5408 m, 3505 m and 5810 m respectively. The cruise was specifically designed to investigate macro- and micro-nutrient co-limitation on nitrogen fixation in the oligotrophic gyre; thus, nutrient and trace metal distributions (e.g., Kunde et al., 2019; Artigue et al., 2021) were carefully quantified.
Seawater was collected using pre-cleaned 10 L Ocean Test Equipment (OTE) water sampling bottles that were mounted on a titanium rosette system and deployed from a Kevlar wire. On recovery, the OTE bottles were transferred into a trace metal clean container for sub-sampling. Seawater was filtered through a Sartobran 300 (Sartorius) filter capsule (0.2 µm) under gentle pressure and was collected into 2 L acid-cleaned low density polyethylene (LDPE) bottles. Filtered seawater samples were acidified with UpA-grade hydrochloric acid (HCl, Romil) to 0.024 M, and were stored for at least a year before the Cr isotope analysis, allowing complete conversion of Cr species to Cr (III) (Semeniuk et al., 2016).
Analyses of the water masses along the transect based on combined hydrographic and nutrient data are reported in Artigue et al. (2020) and summarized in Figure 1B. Briefly, surface waters were occupied by a shallow form of Eastern South Atlantic Central Water (ESACW), with a small contribution from the Amazon plume in the west. Below the surface, the waters mainly consisted of West North Atlantic Central Water (WNACW, 100-500 m) and 13°C-ESACW (500-900 m), although at approximately 700 m water depth, Antarctic Intermediate Water (AAIW) and Mediterranean Water (MW) were present, respectively, to the west and to the east of the Mid-Atlantic Ridge (MAR). Upper Circumpolar Deep Water (UCDW) was generally observed between 900-1250 m, below which Labrador Sea Water (LSW) was centered at ~1500 m. North East Atlantic Deep Water (NEADW), which includes a contribution from Iceland-Scotland Overflow Water (ISOW), was found below LSW and was centered at ~2500 m. At Station 1, to the west of the MAR, North West Atlantic Bottom Water (NWABW) was present between 3500 and 5000 m water depth, and deeper waters (>5000 m) consisted of Antarctic Bottom Water (AABW). The deep waters at Station 6 were dominated by NEADW. Note that LSW, NEADW and NWABW all contribute to North Atlantic Deep Water (NADW). Hereafter, we define waters from below the surface mixed layer to 700 m depth as subsurface waters (including the thermocline which was located between ~300 and ~700 m; Artigue et al., 2020), waters from between 700 and 2000 m depth as intermediate waters, and waters below ~2000 m water depth as deep waters (Sarmiento et al., 2007).
2.2 Cr isotope analysis
All acids used for chemical processing were thermally distilled. Milli-Q (MQ) water was used for diluting and for cleaning. LDPE bottles and Perfluoroalkoxy (PFA) vials were thoroughly cleaned for trace metal purposes. Samples were handled under laminar flow hoods, set within Class 100 clean laboratories at the National Oceanography Centre Southampton.
Dissolved Cr concentrations were initially determined using a Mg(OH)2 co-precipitation method (Rickli et al., 2019; Davidson et al., 2020). Approximately 50 mL of filtered seawater was transferred into an acid-cleaned centrifuge tube, weighed, and amended with 10 ng of a 53Cr single spike. Ammonia solution (SpA-grade, Romil) was then added to the sample until Mg(OH)2 formed. For 50 mL of acidified seawater (pH ~1.7), ~500 µL concentrated ammonia was required; the size of Mg(OH)2 pellet was kept as small as possible to minimize the potential for matrix effects. After centrifugation and removal of the supernatant, the Cr precipitate was re-dissolved in 5 mL of 0.45 M HNO3. The Cr concentration of the seawater sample was derived by isotope dilution, based on the 52Cr/53Cr ratio of the sample/spike mixture measured by inductively coupled plasma mass spectrometry (ICP-MS; Thermo Scientific Element). These Cr concentration data were used to estimate the volume of water required for pre-concentration and to optimize the amount of double spike added. The accuracy of the Mg(OH)2 co-precipitation method was assessed through the analysis of (1) the NASS-6 certified reference material (National Research Council Canada), which gave Cr = 1.95 ± 0.23 nmol kg-1 (n=1; 2SE internal error), compared to certified value of 2.17 ± 0.15 nmol kg-1 (2SD); and (2) an OSIL Atlantic seawater salinity standard (http://www.osil.co.uk), which gave Cr = 2.96 ± 0.12 nmol kg-1 (n=1; 2SE internal error), consistent with a previously published value for the same OSIL batch (3.06 ± 0.04 nmol kg-1 (2SD, n=6); Goring-Harford et al., 2018).
The Cr isotope compositions of seawater samples were determined using a method adapted from Bonnand et al. (2013). Samples of 1-2 L volume were amended with a 50Cr-54Cr double spike to achieve optimal target isotope ratios (Goring-Harford et al., 2018) and were left to equilibrate for ~24 hrs. Information on our double spike technique is given in Bonnand et al. (2011). The sample pH was then adjusted to pH 8-9 to facilitate precipitation of Cr. A freshly prepared suspended precipitate of Fe(II) hydroxide, made by addition of ammonia to a fresh ammonium Fe(II) sulfate solution, was added to the samples (10 mL L-1 seawater), allowing oxidation of the Fe(II) hydroxide and reduction of any remaining Cr(VI). The Fe(III) hydroxide scavenges the Cr(III), resulting in quantitative precipitation of dissolved inorganic Cr (Connelly et al., 2006).
The precipitate was separated from the solution via vacuum filtration through pre-cleaned PTFE membrane filters (1 µm, Millipore Omnipore), and was subsequently leached from the filters using 6 M HCl before being dried down and taken up in 6 mL of 7 M HCl. The Cr was first separated from the Fe by anion exchange chromatography (~2 mL of Bio Rad AG1-X8 resin loaded in a Bio Rad Poly-Prep column). The resin was extensively cleaned with concentrated HNO3, 0.5 M HCl and concentrated HCl, and was pre-conditioned with 7M HCl. The sample was loaded in 6 mL of 7M HCl onto the resin. The eluent was collected, dried down and then reconstituted in 6 mL of 0.5 M HCl. The column was cleaned with 0.5 M HCl to remove Fe and stored in 0.5 M HCl.
Any residual salts were removed by cation exchange chromatography (2.9 mL of BioRad AG 50W-X12 resin loaded in a 30 mL PFA Savillex column; Trinquier et al., 2008). The resin was cleaned with 10 mL of 8 M HNO3, 30 mL of 6 M HCL and 30 mL of MQ water and was pre-conditioned with 12 mL of 0.5 M HCl. The sample was loaded in 6 mL of 0.5 M HCl and the Cr was immediately eluted and collected in a 15 mL Savillex vial. A further 4 mL of 0.5 M HCl was added to the column and collected. The resin was cleaned with 6M HCl to remove the remaining cations and stored in 0.5 M HCl. The Cr fraction was evaporated to dryness and was treated with 50 μL of concentrated H2O2 and HNO3, respectively, to oxidize any remaining organic material, before being dried down once again and re-dissolved in 0.45 M HNO3.
The isotopic composition of Cr was determined by multicollector inductively coupled plasma mass spectrometry (MC-ICP-MS; Thermo Fisher Neptune Plus) at the University of Southampton, using a method similar to that described in Goring-Harford et al. (2018). Purified samples at a concentration of ~50 ppb Cr were introduced using an Aridus 3 desolvator and signals from 50Cr, 52Cr, 53Cr, 54Cr and 49Ti, 51V, 56Fe were quantified. Medium resolution setting was used and a mass resolution of >5000 was achieved. The NBS979 standard was analyzed after every 3 sample measurements in the analytical sequence. Each sample/standard analysis consisted of 100 individual measurements. Polyatomic interferences were avoided by making measurements on peak shoulders. The typical ion beam size was 0.15-0.24 volts per ppb for 52Cr on a 1011 Ω amplifier. The mean signal intensity of a blank solution that was analyzed before and after each sample/standard was subtracted. Despite the large quantities of Fe added to the samples during co-precipitation, efficient removal of Fe by anion exchange chromatography ensured that the 56Fe/54Cr of the samples was always <0.50 and typically around 0.03, which would have no resolvable effect on δ53Cr values (Bonnand et al., 2011). The raw Cr data were corrected for mass bias using an iterative deconvolution procedure (Albarède and Beard, 2004) and for the total procedural blank contribution. The final Cr isotope value is expressed in delta notation relative to the NBS979 isotope standard.
The Cr blank for the Fe(II) co-precipitation method was relatively constant, 18 ± 0.9 ng, the majority of which (~93%) came from the Fe(II) salt (Goring-Harford et al., 2018). The δ53Cr value of the ammonium Fe(II) sulphate solution used in this study was –0.16 ± 0.04 ‰ (n=1; 2SE internal error), within the error of that measured previously (–0.34 ± 0.32‰, 2SD, n=6; Goring-Harford et al., 2018). The effect of the blank on the uncertainties of Cr and δ53Cr is estimated to be ± 0.005 nmol kg-1 and ± 0.02‰ (2SD), respectively (Goring-Harford et al., 2018).
The Cr concentration of each sample was determined simultaneously with the isotope ratios using isotope dilution equations, based on the known sample volume and the quantity of added spike. The results were generally within 10% of the concentrations derived from the Mg(OH)2 co-precipitation method; the reported Cr concentration data are from the MC-ICP-MS measurements because of the better precision.
Analyses of the NBS979 chromium isotope standard gave δ53Cr = 0.00 ± 0.05‰ (2SD, n=43). The precision of the methods was further assessed through multiple analyses of an OSIL Atlantic seawater salinity standard that yielded Cr = 3.07 ± 0.058 nmol kg-1 (2SD, n=4) and δ53Cr = 0.99 ± 0.04‰ (2SD, n=4) (Supplementary Info. 1; Table S1). These data are consistent with previously reported values (Cr = 3.10 ± 0.04 nmol kg-1, δ53Cr = 0.97 ± 0.10‰, 2SD, n=6; Goring-Harford et al., 2018) for the same batch of OSIL. By compiling the repeat analyses of the OSIL samples from this study and Goring-Harford et al. (2018), we apply 2% and ± 0.06‰ as an estimate of external reproducibility for Cr and δ53Cr, respectively, for all samples in this study.
As OSIL is a salinity standard and different batches do not have identical Cr concentrations and δ53Cr values (Rickli et al., 2019), we undertook a cross-calibration exercise with the University of Saskatchewan in efforts to better assess the accuracy of our Cr and δ53Cr data. Both groups measured three seawater samples collected by the University of Saskatchewan in the Beaufort Sea. δ53Cr data for all samples were identical within the analytical uncertainty (Supplementary Info. 1; Table S2); Cr concentrations measured in our laboratory were 12-14% lower than those measured in Saskatchewan (Table S2). We have not been able to pinpoint the source of this difference, although we note that it is within the range of the uncertainty of Cr concentrations reported in a recent inter-laboratory comparison of the trace metal composition of NASS seawater (Yang et al., 2018).
2.3 Analysis of dissolved Fe concentrations and other ancillary parameters
Filtered (at 0.2 μm) samples of seawater for analysis of dissolved Fe (dFe) were acidified on board and analyzed using flow injection analysis with chemiluminescence detection (FIA-CL) inside a Class 1000 clean laboratory either onboard or at the University of Southampton, as discussed in Kunde et al. (2019). The accuracy of the method was assessed by repeat quantification of dFe in reference samples (SAFe; Johnson et al., 2007). Filtered (at 0.2 μm) seawater for analysis of soluble Fe (sFe, i.e., truly dissolved Fe) was additionally filtered in-line through 0.02 μm syringe filters (Anotop, Whatman) before it was acidified (Kunde et al., 2019). Concentrations of sFe were then determined using the same method as dFe. Concentrations of colloidal Fe (cFe, operationally defined as Fe in the size fraction 0.02 - 0.2 μm; e.g., Fitzsimmons et al., 2015) were derived from the difference between dFe and sFe. These dissolved and colloidal Fe data have been previously published in Kunde et al. (2019) and are given in Supplementary Info. 1 (Tables S1; S3).
A Seabird 911 plus conductivity, temperature and depth (CTD) profiler system together with additional sensors was attached to the titanium frame during the seawater sampling. Sensors were cross-calibrated with analyses of discrete seawater samples on board. Salinity was calibrated using an Autosal 8400B salinometer (Guildline). Chlorophyll-a (Chl-a) was measured by a fluorescence sensor that was calibrated using a fluorimeter (Turner Designs Trilogy). Dissolved oxygen (O2) was measured by a Seabird SBE43 sensor, calibrated against a photometric automated Winkler titration system. Turbidity was monitored using the WETLabs BBRTD light scattering sensor.
Macronutrient concentrations were analyzed on board using a 5-channel (nitrate, nitrite, phosphate, silicic acid, and ammonium) segmented flow auto-analyzer (Woodward and Rees, 2001). The macronutrient data from this cruise are discussed in Artigue et al. (2020).
3 Results
Profiles of Chl-a, O2 concentration and turbidity at Stations 1, 4 and 6 are shown in Figure 2; vertical distributions of the macronutrients phosphate (PO4) and silicic acid (Si) at these stations are illustrated in Supplementary Info. 2 (Figure S1). Overall, there was no significant inter-site variation in these biogeochemical properties. At each station, Chl-a peaked at approximately 140 m water depth, with concentrations up to 0.4 μg L-1. This so-called deep chlorophyll maximum (DCM) is a common feature in oligotrophic sub-tropical regions where the nutrient supply to the euphotic zone is minimal and the phytoplankton biomass and rate of primary production are low throughout the year (e.g., Pérez et al., 2006; Mignot et al., 2014). The concentrations of PO4 and Si were the lowest within the upper ~150 m waters, and then increased with depth; PO4 reached a concentration maximum at approximately 800 to 1000 m water depth whereas Si concentrations were highest close to the seabed. Oxygen concentrations showed minimum values of 140 μmol/kg at depths between ~700 and ~900 m. Deeper waters were well oxygenated with O2 concentrations of >230 μmol kg-1 below ~2000 m water depth. Levels of turbidity were highest immediately below the surface (and above the DCM), which can be attributed to input of lithogenic particles of North African dust (Ye and Völker, 2017; Kunde et al., 2019), and gradually decreased with depth. Relatively high levels of turbidity were also found close to seafloor, which is likely due to resuspension of seabed sediments (Ohnemus and Lam, 2015). At Station 4 there was a local increase in turbidity between 3300 and 3500 m water depth that delineates the particle-rich hydrothermal plume (Kunde et al., 2019) above the Snake Pit hydrothermal vent field (Beaulieu, 2015) on the Mid-Atlantic Ridge.
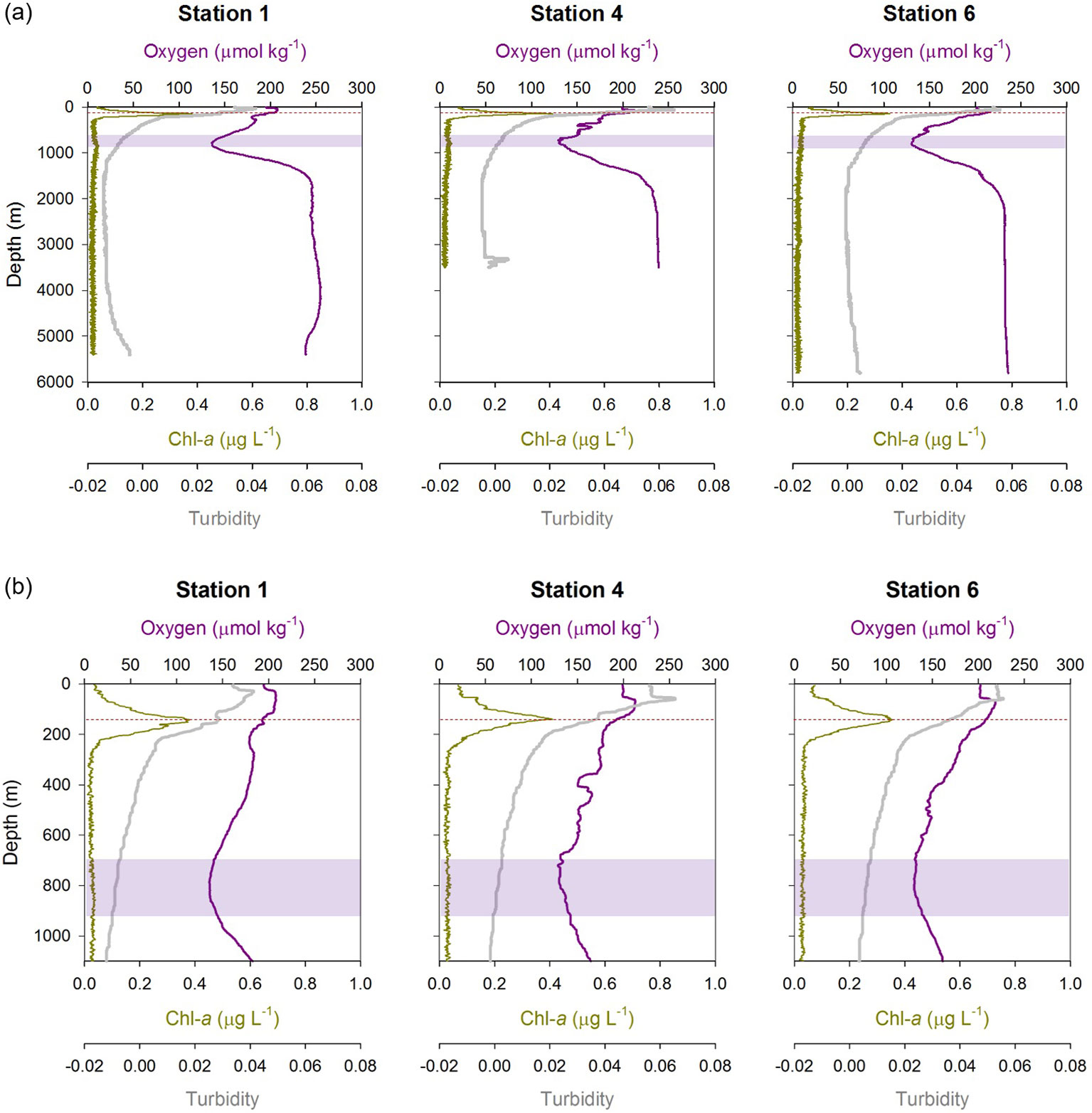
Figure 2 Biogeochemical properties (dissolved oxygen, Chl-a concentration and turbidity) at Stations 1, 4, and 6: (A) full water column depth profiles; (B) upper 1100 m of the water column. The deep Chl-a maximum is marked by the red horizontal dashed line, and the oxygen minimum is highlighted by the purple band at each station.
Full water column depth profiles for dissolved Fe, Cr and δ53Cr at the three stations are shown in Figure 3A (data given in Supplementary Info. 1). Concentrations of dissolved Cr ranged between 1.84 and 2.63 nmol kg-1; the vertical distribution of Cr resembled that of dissolved Fe, notably in the euphotic (up to 200 m water depth) and mesopelagic (~200 to 1100 m water depth) zones. Measured δ53Cr values varied from 1.06 to 1.42‰ and the δ53Cr profiles appear to mirror the shape of the Cr concentration profiles. Lowest Cr concentrations (1.84 to 2.04 nmol kg-1) and highest δ53Cr values (1.36 to 1.42‰) coincided with lowest dFe concentrations (down to ~0.4 nM) between approximately 100 to 400 m water depth. Overall, subsurface waters (between the base of the surface mixed layer and 700 m water depth) were very slightly depleted in Cr and very slightly enriched in heavy Cr isotopes (average Cr = 2.14 ± 0.15 nmol kg-1, δ53Cr = 1.36 ± 0.04‰, 1SD, n=16), relative to intermediate (Cr = 2.31 ± 0.18 nmol kg-1, δ53Cr = 1.26 ± 0.06‰, 1SD, n=6) and deep (average Cr = 2.44 ± 0.18 nmol kg-1, δ53Cr = 1.19 ± 0.11‰, 1SD, n=9) waters.
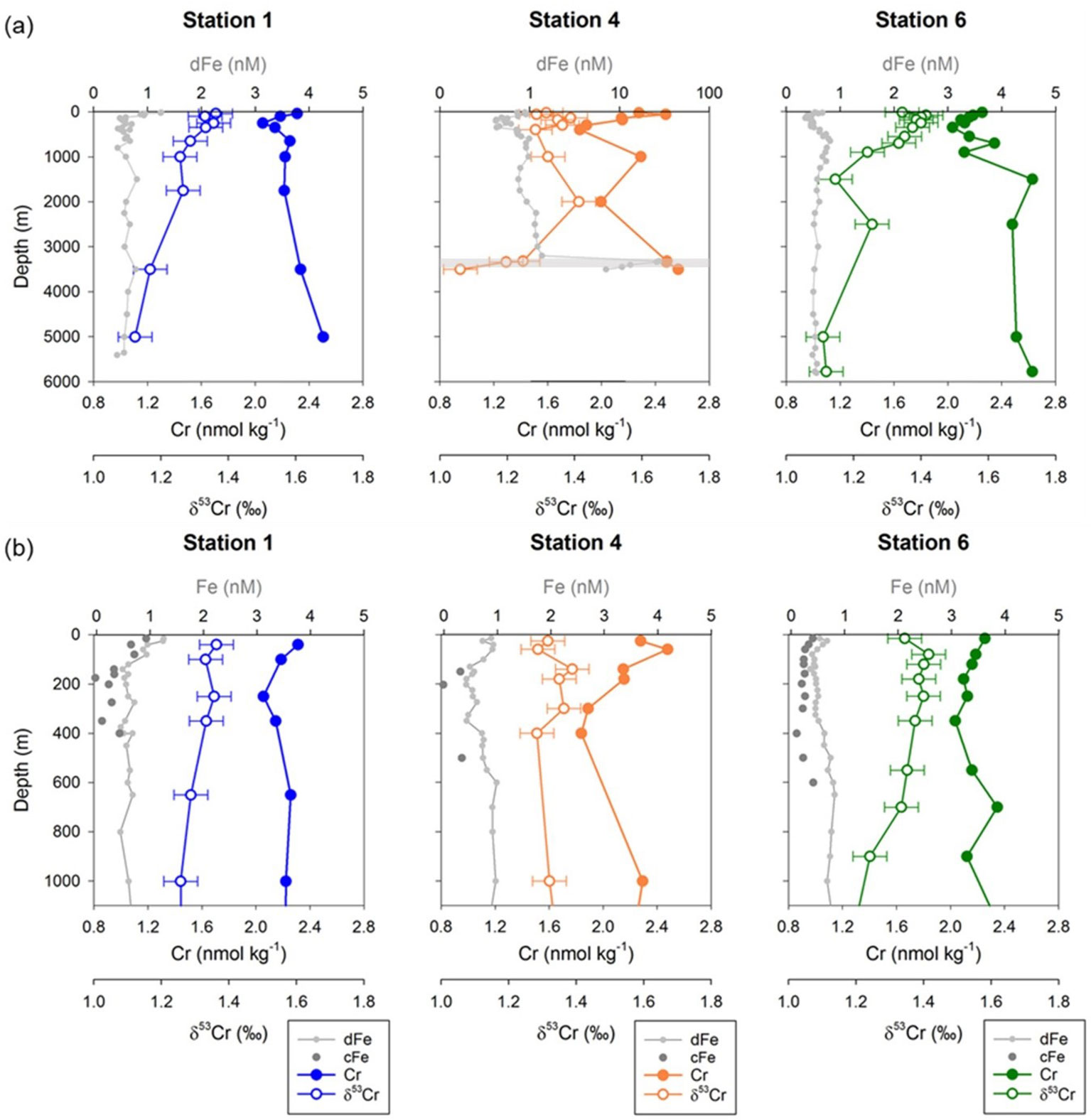
Figure 3 (A) Full water column depth profiles of Cr concentration, δ53Cr values, and dissolved Fe concentrations at Stations 1, 4, and 6. (B) Profiles of Cr concentrations, δ53Cr values, and dissolved and colloidal Fe concentrations for the upper 1100 m of the water column at Stations 1, 4 and 6. Location of the hydrothermal plume at Station 4 is highlighted by the grey horizontal band. Error bars on Cr and Fe concentrations are smaller than the size of symbols. Error bars on δ53Cr represent the external reproducibility of the measurements (± 0.06‰, 2SD) based on the repeat analyses of OSIL Atlantic seawater salinity standard. Fe data are from Kunde et al. (2019). All Cr data are given in Supplementary Info. 1 (Table S1).
Seawater Cr concentrations and δ53Cr values measured in deep waters (>2000 m water depth) in this study are generally consistent with those reported for deep waters at a site further to the south (~12 °N) in the eastern sub-tropical north Atlantic (Cr = 2.5 to 2.9 nmol kg-1, δ53Cr = 1.08 to 1.20‰: Goring-Harford et al., 2018; Supplementary Info. 2; Figure S2). By contrast, Cr concentrations and δ53Cr values in waters between the base of the surface mixed layer and 2000 m water depth were respectively, generally lower and higher at our sites at ~22 °N than they were at ~12 °N (Figure S2).
Although no other Cr isotope data have been published to date for seawater samples collected within ± 10° latitude of our sites, Cr concentration data have been reported by a number of authors (Jeandel and Minster, 1987; Mugo and Orians, 1993; Sirinawin et al., 2000; Connelly et al., 2006) and are highly variable, ranging from ~2 nmol kg-1 to almost 5 nmol kg-1. Our data occupy the lower end of this range (Figure S2). There are numerous potential explanations for the differences between the data sets. For example: (1) The sampling stations span almost the full width of the Atlantic Ocean, crossing many oceanographic boundaries. (2) These earlier data were obtained on samples that were not collected following GEOTRACES trace metal protocols that have been established more recently. While seawater samples collected using standard Niskin bottles from a conventional rosettes do not appear to be affected by Cr contamination (e.g., Scheiderich et al., 2015; Janssen et al., 2020; Moos et al., 2020), some types of filter can be associated with high and variable Cr blanks (e.g., Yiğiterhan et al., 2011). (3) Differences in sample treatment and analysis. Chromium concentrations were determined using various different techniques that may capture different forms of Cr. The Fe(II) co-precipitation used in this study (and also by Jeandel and Minster, 1987; Connelly et al., 2006) is unlikely to capture organically-bound dissolved Cr (Goring-Harford et al., 2020), as this fraction is thought to be resistant to reduction and adsorption (Nakayama et al., 1981). By contrast, the Mg(OH)2 co-precipitation technique is expected to capture organically-bound Cr (Moos and Boyle, 2019; Davidson et al., 2020; Huang et al., 2021) and techniques that determine Cr through complexation with various ligands may capture the more labile organic-bound Cr (Mugo and Orians, 1993; Sirinawin et al., 2000). Note however that concentrations of organically-bound Cr are usually very low (e.g., Kaczynski and Kieber, 1994). In addition, most of the data reported in Jeandel and Minster (1987) were from analysis of unfiltered water samples; a handful of samples were filtered and Cr concentrations of filtered samples were between 0.15 and 1.1 nmol kg-1 (mean 0.62 nmol kg-1) lower than unfiltered samples. We also note that there are wide variations between more recent and earlier Cr concentration data from other locations. For example, Mugo and Orians (1993) reported total Cr data from 3000-4000 m water depth at Station P26 in the North Pacific of between 3.53 and 4.18 nmol kg-1 (data converted from reported values in nM to nmol kg-1 assuming salinity = 34.67; Janssen et al., 2021), whereas Janssen et al. (2021) measured values of between 4.52 and 4.54 nmol kg-1 for the same station over the same depth range. While there is a clear need for more concerted inter-laboratory assessments of Cr and Cr isotope data (see also Moos et al., 2020 and Huang et al., 2021), we nevertheless believe our data are analytically robust as we have carried out inter-laboratory calibration exercises as discussed in Section 2.2 together with other tests (reported in Goring-Harford et al., 2018) to validate the accuracy and precision of our data.
4 Discussion
4.1 Potential sources of Cr to the North Atlantic
Dust supply from North African deserts directly affects the subtropical North Atlantic Ocean (Jickells et al., 2005). Total (wet + dry) deposition of lithogenic particles to surface waters ranged from ~1 × 10-5 kg m-2 d-1 at Station 6 to ~4 × 10-6 kg m-2 d-1 at Station 1 (Artigue et al., 2021), which may dominate over biogenic particles (Ohnemus and Lam, 2015). Input of Fe and aluminum (Al) from dust deposition across the GEOTRACES GApr08 transect is evidenced by elevated concentrations of dissolved Fe and Al in the surface mixed layer (SML), which extends to 20 m, 50 m and 60 m water depth respectively, for Stations 1, 4 and 6 (Kunde et al., 2019; Artigue et al., 2021). Only two samples were collected for Cr analysis from within the SML (at Stations 4 and 6); these had Cr concentrations of 2.28 nmol kg-1 (Station 4) and 2.25 nmol kg-1 (Station 6) (Figure 3B), similar to the Cr concentration for samples collected from the upper layer of intermediate waters at these stations (2.29 nmol kg-1 at 1001 m of Station 4 and 2.12-2.35 nmol kg-1 at 701-901 m of Station 6). The δ53Cr values of dissolved Cr from the SML were 1.32‰ (Station 4) and 1.34‰ (Station 6). The δ53Cr value of atmospheric dust is expected to be similar to that of crustal rocks (−0.12 ± 0.10‰; Schoenberg et al., 2008), although dissolution promoted by strong ligands may enrich heavier Cr isotopes in the dissolved phase (δ53Cr up to 1.23‰; Saad et al., 2017). For total (wet + dry) deposition fluxes of ~8 × 10-6 kg m-2 d-1 (Station 4) and ~1 × 10-5 kg m-2 d-1 (Station 6; Artigue et al., 2021), with a Cr loading equivalent to average upper continental crust (~100 ppm; e.g., Rudnick and Gao, 2003), and assuming 10% of this Cr is soluble (Chester and Murphy, 1990), then the annual input of dust-derived Cr to the surface mixed layer at these stations can be expected to increase the Cr concentration by ~0.01 nM. This is within the analytical uncertainty of the measured Cr concentrations in the SML. By contrast, the expected increases in concentrations of dissolved Fe and Al due to dust deposition calculated in the same way are of the order of >1 nM, which is consistent with observations (Kunde et al., 2019; Artigue et al., 2021). It thus appears that input of dissolved Cr from atmospheric dust deposition to the surface ocean was not significant, at least at the time of sampling. In this connection, it is important to note that there can be considerable variability in dust transport and deposition in this region (Artigue et al., 2021) and potentially, variations in the Cr content of aerosol particles (Rädlein and Heumann, 1995), that may lead to increased input of dust derived Cr to surface waters at other times of the year.
The neutrally buoyant hydrothermal plume above the Snake Pit vent field was identified by a turbidity anomaly at water depths of between 3300-3500 m at Station 4 (Figure 2). Dissolved Fe concentrations reached 10.7 to 27.7 nM within the plume (Table S3), much higher than background seawater (~0.67 nM; Kunde et al., 2019), suggesting a substantial contribution of dFe from the Snake Pit vent fluids that have an end-member Fe concentration of ~3.5 mM (Findlay et al., 2015). The particulate Fe content in the plume was also high, up to ~20 nM (Kunde et al., 2019). Seawater samples from within the hydrothermal plume exhibit Cr concentrations of 2.48 to 2.57 nmol kg-1 and δ53Cr values of 1.06 to 1.25‰ (Figure 3A). These Cr concentrations are slightly higher (by 0.14 - 0.23 nmol kg-1) than a single sample collected from a similar water depth within the same water mass (3500 m, NEADW) at Station 1; the δ53Cr value of the sample from Station 1 (1.17‰) is within the range of the plume samples. By contrast, seawater samples collected from a site of diffuse flow in the North Fiji basin were observed to be strongly enriched in total dissolved Cr (up to 20 nmol kg-1) up to 400 m above the seabed relative to background values (~10 nmol kg-1; Sander and Koschinsky, 2000). However, as discussed in Janssen et al. (2023), these results may not offer unequivocal support for hydrothermally sourced Cr. We cannot make a direct comparison between our Snakepit data and those from the North Fiji basin hydrothermal site due to differences in analytical methods, but we nevertheless assert that the hydrothermal input to the Snakepit plume can be expected to be small, for the following reasons. Firstly, reduced species of Cr in high temperature hydrothermal fluids have limited solubility (Huang et al., 2019) and recent analyses of high temperature (up to 311°C) hydrothermal fluids suggest that Cr concentrations are <30 nmol kg-1 (Janssen et al., 2023). Secondly, the highest Fe content (dissolved + particulate) sampled in the Snakepit plume was 47.7 nM (Kunde et al., 2019), versus a background Fe content of 0.67 nM (Kunde et al., 2019). Assuming that ~25% of the Fe in the Snakepit vent fluids precipitates immediately as Fe-sulfide as it is expelled at the seabed (James and Elderfield, 1996), then the total (dissolved + particulate) Fe data indicate that the seawater:vent fluid mixing ratio is ~56,000 at plume height. Even assuming that none of the vent fluid Cr precipitated as the fluids were expelled at the seabed (cf., German et al., 1991), the expected increase in Cr in the plume due to vent fluid input would be <5 × 10-4 nmol kg-1, which is smaller than the analytical uncertainty of our measurements.
At all three stations, the highest Cr concentrations (and the lowest δ53Cr values) were recorded in samples taken from closest to the seabed. Slightly elevated Cr concentrations in deep waters have been reported in other studies and are generally attributed to release from sediments (e.g., Murray et al., 1983; Jeandel and Minster, 1987). In support of this, Cr concentrations in pore waters of oxic sediments can be up to an order of magnitude higher than Cr concentrations in overlying bottom waters (Shaw et al., 1990; Janssen et al., 2021). Our data suggest that Cr released by sediments is isotopically light relative to overlying bottom waters, which is consistent with release of Cr from lithogenic material (that has δ53Cr = −0.12 ± 0.10 ‰; Schoenberg et al., 2008) and/or from authigenic sediment phases that have δ53Cr values that range from −1.2 ‰ (Bauer et al., 2019) to 0.61 ‰ (Gueguen et al., 2016). Similarly, mass balance calculations based on bottom vs. deep water Cr concentrations and δ53Cr values in the Tasman Sea have shown that oxic pore waters seem to be a source of relatively isotopically light Cr to bottom waters (δ53Cr = 0.34 ± 0.25 ‰; Janssen et al., 2021).
A rough estimate of the Cr flux from sediments can be obtained from the gradient of the Cr concentration in deep waters and estimated rates of turbulent mixing above the seabed (Equation 2):
where Δ[Cr] is change in Cr concentration between the deepest water sample and the next deepest water sample, Δz is depth interval separating the deepest from the next deepest water sample and D is diapycnal diffusivity. Discounting Station 1 (as no samples were collected within <30 m above the seabed), then the estimated Cr flux from sediments at Station 6 is ~5 × 10-3 nmol cm-2 yr-1 (for a D value of 0.1 cm2 s-1; Polzin et al., 1997), while the estimated Cr flux from sediments at Station 4 is considerably higher, ~2 nmol cm-2 yr-1. Note for Station 4, we use a higher value for D (5 cm2 s-1), because mixing rates have been shown to be much higher above the Mid-Atlantic Ridge than they are above the seabed in the abyssal ocean (Polzin et al., 1997). If these fluxes are applied to the areal extent of all oxic oceanic sediments (ca. 108 km2), then the estimated global benthic flux of Cr would range from 5 × 106 mol yr-1 (Station 6) to 3 × 109 mol yr-1 (Station 4), equivalent to <0.3% to ~10 times the estimated river flux (Table 1). The value for FCr we calculate for Station 4 is comparable to that calculated based on pore water measurements in calcareous ooze in the Tasman Sea (up to ~ 3.2 nmol cm-2 yr-1; Janssen et al., 2021), supporting the suggestion that sediment inputs of Cr can be important at the local scale (Janssen et al., 2021). In the case of Station 4, situated above the Snakepit hydrothermal vent field, loss of Cr from the sediments is likely fueled by oxidation of Cr(III) associated with metalliferous Fe-(oxyhydr)oxides derived from the hydrothermal plume, followed by diagenetic remobilization of the Cr(VI) that forms (Bauer et al., 2019).
4.2 Removal of Cr in subsurface waters
Lowest Cr concentrations were found in subsurface waters at between ~100 and ~500 m water depth (Figure 3B). The most Cr-depleted samples in subsurface waters had Cr concentrations of as low as 1.84 nmol kg-1, between ~9 and 20% lower than the samples in the surface mixed layer (2.25-2.28 nmol kg-1) and ~8 to 18% lower than the uppermost intermediate waters (2.25 ± 0.10 nmol kg-1, 1SD, n=4). δ53Cr values of the most Cr-depleted samples were also very slightly higher, by up to ~0.14‰ than the uppermost intermediate waters, which is greater than our analytical uncertainty of ± 0.06‰ (2SD). As this interval of very slightly higher δ53Cr is a feature for all 3 stations, we tentatively assert that it may be real. In support of this, Jeandel and Minster (1987) also noted a weak but non-negligible subsurface Cr minimum at water depths between ~100 and 500 m in the North Atlantic Ocean (24 °N to 39 °N).
Removal of Cr does not appear to be associated with biological uptake in these oligotrophic waters. Lowest Cr concentrations occurred below the DCM (and below where macronutrient concentrations were the lowest; Figure S1), and there is no significant correlation (at p<0.05) between δ53Cr and Chl-a concentration in the subsurface waters (Figure 4A). Low productivity waters in the North Pacific also showed no evidence for Cr removal (Janssen et al., 2020). Additionally, there is no significant (at p<0.05) correlation between δ53Cr values and O2 concentrations, or Cr and O2 concentrations in the subsurface waters (Figure 4B), suggesting that there is no redox control on Cr isotope behavior in this slightly O2 deficient depth interval (O2 concentrations >130 μmol kg-1). Recent studies on oxygen deficient waters (with O2 concentrations down to ~13.2 μmol kg-1) in the eastern sub-tropical Atlantic and the North Pacific have drawn the same conclusion (Goring-Harford et al., 2018; Moos and Boyle, 2019; Janssen et al., 2020) and reduction of Cr(VI) has only been demonstrated in waters with extremely low levels of oxygen (e.g., < 2 μmol kg-1; Murray et al., 1983; Rue et al., 1997; Moos et al., 2020; Nasemann et al., 2020; Huang et al., 2021).
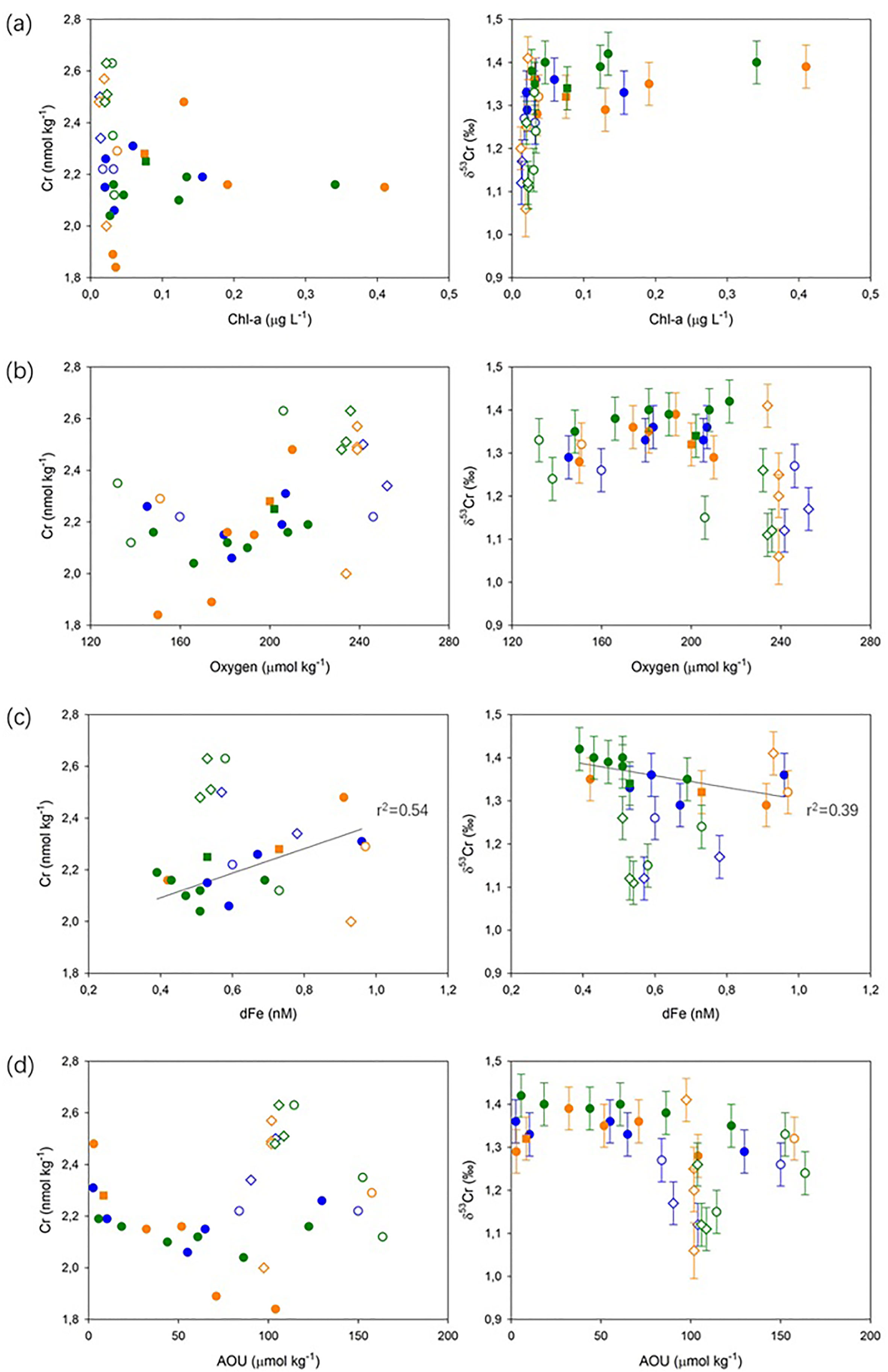
Figure 4 Cr and δ53Cr as a function of (A) Chl-a concentration; (B) dissolved oxygen concentration; (C) dFe concentration; and (D) apparent oxygen utilization. SML (■), subsurface (●), intermediate (○), and deep (◊) waters are identified by the different symbols. Blue, orange, and green colors represent Stations 1, 4 and 6, respectively, as for Figure 3. Solid lines show significant correlations (p<0.05) between Cr and dFe, and δ53Cr and dFe data in subsurface waters.
Figure 3 shows that the lowest Cr waters coincide with lowest dissolved and colloidal Fe concentrations, suggesting that cycling of Cr and Fe may be coupled in the subsurface waters. In this study area, depletion of dFe has been shown to be associated with removal of cFe from the water column and is not exclusively a result of biological uptake (Kunde et al., 2019). This is because: (1) experimental studies have indicated that sFe is biologically preferred over cFe (Chen and Wang, 2001; Hurst and Bruland, 2007); (2) lowest cFe concentrations occur below the DCM (~140 m water depth) which is, although not exclusively, an indicator of biomass maximum (Figure 3B); and (3) an enrichment of particulate Fe (pFe), which would capture the cellular Fe pool, was not observed (Kunde et al., 2019). The simultaneous minima of pFe and cFe (Kunde et al., 2019) suggests that cFe is not simply transferred from the dissolved to the particulate phase, but both phases are removed together as they sink through the DCM. The subsurface deficit of dFe may therefore be explained by active aggregation of colloidal Fe (e.g., Honeyman and Santschi, 1989), in the form of Fe-(oxyhydr)oxides, into filterable particles and/or scavenging of cFe onto settling dust-derived particles (Kunde et al., 2019). A similar Fe removal mechanism has also been proposed to explain changes in Fe size partitioning in other parts of the North Atlantic Ocean (Fitzsimmons et al., 2015; Ohnemus and Lam, 2015).
As Cr concentrations and δ53Cr values only show a significant correlation (p<0.05) with dFe concentrations in the subsurface waters (and show no significant correlations with any of the other ancillary parameters that were measured; Figure 4), we suggest that removal of Cr proceeds via scavenging by newly-formed colloidal aggregates that subsequently fall out of the water column, as discussed above. This process has not been recognized previously, but is likely enhanced in this area of exceptionally high dust deposition (Section 4.1; Jickells et al., 2005), as the rate of colloidal aggregation is predicted to increase with particle loading due to so-called ‘colloidal pumping’ (e.g., Honeyman and Santschi, 1989). These aggregates ‘shuttle’ rapidly through the SML and into the sub-surface waters, where they are predicted to scavenge trace metals (Ohnemus and Lam, 2015), including presumably Cr. To date, only a handful of measurements of Cr concentrations in the colloidal and particulate fractions have been reported (Klun et al., 2019; GEOTRACES Intermediate Data Product Group, 2021), but these are clearly critical for proper assessment of the role of colloid-particulate interactions in Cr cycling.
Adsorption of Cr(VI) onto Fe-oxides (Pettine, 2000) or SiO2-Al2O3 minerals (Frank et al., 2019) is limited in seawater because high concentrations of sulfate and/or chloride compete with chromate for adsorption sites (Pettine, 2000; Frank et al., 2019). However, Cr(III) species can be effectively scavenged by Fe-(oxyhydr)oxides (Frei et al., 2009), clay minerals and sand (Richard and Bourg, 1991) as well as biogenic particles (Semeniuk et al., 2016). Therefore, it seems likely that removal of Cr occurs mainly via scavenging of Cr(III). Cr(III) may be sourced from biologically (Connelly et al., 2006; Janssen et al., 2020; Huang et al., 2021) and/or photochemically (Kieber and Helz, 1992; Kaczynski and Kieber, 1993; Achterberg and van den Berg, 1997; Li et al., 2009) mediated reduction of seawater Cr(VI), or possibly from regeneration of Cr (III) from particles exported from the deep chlorophyll maximum (Janssen et al., 2020). If real, then the very small increase in δ53Cr in the Cr-depleted waters would lend support to this idea. Cr(III) has been widely shown to be isotopically light relative to Cr(VI), hence removal of Cr(III) can be expected to leave the residual dissolved Cr pool isotopically heavy (Ellis et al., 2002; Døssing et al., 2011; Kitchen et al., 2012; Janssen et al., 2020; Huang et al., 2021). Species-specific Cr and Cr isotope measurements of water samples, as well as the particulate phase, will be essential for validating these ideas.
4.3 Regeneration of Cr in deeper waters
While there is some evidence for scavenging of Cr in subsurface waters (Section 4.2), concentrations of total dissolved Cr were highest in intermediate and deep waters, suggesting that Cr may be re-released. Although concentrations of Cr and Fe (and δ53Cr and Fe) were correlated in the subsurface waters (Figure 4C), the cycling of Cr and Fe is apparently decoupled in intermediate and deep waters. In intermediate waters (700-2000 m water depth), there is a positive correlation between dFe concentrations and apparent oxygen utilization (AOU) respectively at Stations 4 and 6 (p<0.05; Supplementary Info. 2; Figure S3) that reflects remineralization of sinking organic material within the water column (Kunde et al., 2019), as also observed in other areas (Hatta et al., 2015). While such a correlation is not observed at Station 1, this is likely due to the small number of data points, the influence of atmospheric dust (Ye and Völker, 2017; Kunde et al., 2019), and/or other additional external Fe sources (Artigue et al., 2021). By contrast, there is no positive relationship between Cr concentrations and AOU, or δ53Cr values and AOU, within the same depth interval at any of our sampling stations (Figure 4D). Consistent with our observation that there is no obvious biological control on Cr concentrations and δ53Cr in subsurface waters, and recent observations of Cr behavior in intermediate and deep water masses in the Southern, Pacific and Atlantic oceans (Janssen et al., 2021), regeneration of Cr in intermediate waters via organic matter respiration does not appear to be an important control on Cr distributions in this part of the North Atlantic Ocean; thus, the release of Cr appears to be mechanistically different from Fe.
As removal of Cr in subsurface waters at our study sites, where lithogenic suspended particles are abundant, appears to proceed via scavenging of Cr(III) by colloid aggregates that consist of authigenic Fe-(oxyhydr)oxides and/or dust particles (Section 4.2), increased concentrations of dissolved Cr in intermediate waters could be due to re-oxidation of Cr(III) to the less particle reactive Cr(VI), potentially driven by reduction (and dissolution) of manganese(III, IV) oxides (MnOx) that are present throughout the oxygenated water column (Jones et al., 2020). However, as concentrations of particulate Mn are very low (sub-nanomolar) in oceanic environments (Jones et al., 2020; Xiang et al., 2021), the predicted oxidation rate of Cr(III) is extremely slow, ~2 × 10-5 nmol kg-1 yr-1 (van der Weijden and Reith, 1982). The residence time of fine lithogenic mineral particles (~1 to 5 μm diameter) in the upper 2000 m of the North Atlantic Ocean water column is months to years (Ohnemus and Lam, 2015; Ohnemus et al., 2019), over which time oxidation of Cr(III) can be expected to increase the Cr concentration of seawater by approximately 10-5 to 10-4 nmol kg-1 in the intermediate waters. Clearly, this is too small to account for the observed 0.12 to 0.47 nmol kg-1 increase in dissolved Cr (Section 3) in intermediate waters relative to the lowest Cr subsurface waters (i.e., those between 100 and 400 m water depth).
It thus seems more likely that regeneration of Cr occurs either via dissolution of authigenic Fe-(oxyhydr)oxides and/or dust particles and/or desorption of Cr(III) from these particles. The latter would mean that the scavenging of Cr from subsurface waters is reversible. In deeper waters, concentrations of particulate matter are lower (Figure 2), so the equilibrium between Cr(III) in the dissolved and particulate phases will shift towards the dissolved phase (e.g., Bacon and Anderson, 1982). Reversible exchange between dissolved and particulate phases has been shown to successfully reconcile the oceanic distributions of dissolved copper and zinc whose concentrations also generally increase with depth (e.g., Little et al., 2013; John and Conway, 2014; Weber et al., 2018), although unlike Cr, copper and zinc are directly influenced by biological uptake and are strongly complexed with organic ligands in seawater (e.g., Little et al., 2013).
Regeneration of Cr is unlikely to fractionate Cr isotopes to any great extent. Cr isotope fractionation caused by oxidation of Cr(III) by MnOx minerals is, as yet poorly constrained (Bain and Bullen, 2005; Ellis et al., 2008), but recent laboratory studies suggested that differences in δ53Cr values between Cr(VI) and Cr(III) (of up to 0.39‰) cannot be sustained over time (Ansari and Johnson, 2022). Adsorption and desorption processes also do not appear to fractionate Cr isotopes due to rapid isotopic exchange on particle surfaces (Ellis et al., 2004; Wang et al., 2015); thus, the isotopic compositions of dissolved and scavenged Cr(III) are generally assumed to be identical (Huang et al., 2021). Consistent with our observations, the δ53Cr value of regenerated Cr, either in the form of Cr(VI) or Cr(III), can be expected to be isotopically light compared to dissolved Cr(VI) that remains in the subsurface waters.
4.4 Accumulation of Cr in deep waters and effects of water mass mixing
Excluding samples collected from within 30 m of the seabed, deep waters (>2000 m water depth) at our stations in the sub-tropical North Atlantic have lower Cr concentrations (2.40 ± 0.19 nmol kg-1; n=7) and higher δ53Cr values (1.22 ± 0.10‰; n=7) than Pacific waters (average Cr = 4.75 ± 0.42 nmol kg-1 and δ53Cr = 0.77 ± 0.10‰, 1SD, n=25; Moos and Boyle, 2019; Nasemann et al., 2020; Huang et al., 2021; Janssen et al., 2021) from >2000 m depth (including PDW, UCDW and LCDW water masses). Previous studies have suggested that Cr may accumulate in deep waters along the thermohaline flow path from the Atlantic to the Pacific Ocean (Jeandel and Minster, 1987; Janssen et al., 2021), and this behaviour has also been predicted by a model of the ocean Cr cycle (Pöppelmeier et al., 2021). Comparing our data with others from the literature (Goring-Harford et al., 2018; Moos and Boyle, 2019; Nasemann et al., 2020; Huang et al., 2021; Janssen et al., 2021), it is further apparent that Cr concentrations increase as the conventional radiocarbon (14C) age of the deep waters (Supplementary Info. 1; Table S4; Supplementary Info. 2; S1) increases, whereas the δ53Cr value of total dissolved Cr decreases (Figure 5). Thus, isotopically light Cr accumulates in deep waters presumably via release from sinking particles as the deep water masses age as they are transported laterally from the Atlantic Ocean to the Southern Ocean, the South Pacific and finally into the North Pacific Ocean (DeVries and Holzer, 2019).
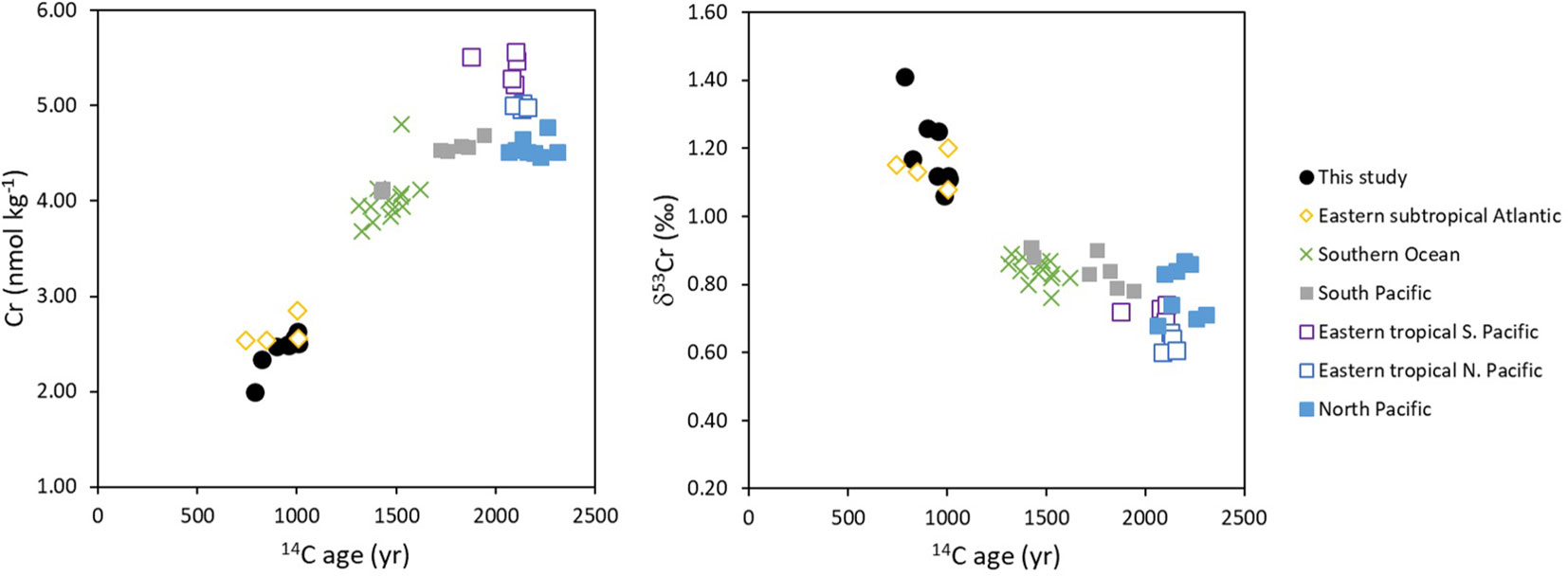
Figure 5 Cr and δ53Cr vs radiocarbon (14C) age of deep water masses (≥ 2000 m water depth). Cr data are from: this study (sub-tropical North Atlantic); Goring-Harford et al. (2018) (eastern subtropical Atlantic); Rickli et al. (2019) (Southern Ocean); Moos and Boyle (2019) (North Pacific); Janssen et al. (2021) (Southern Ocean, North Pacific, eastern tropical South Pacific and South Pacific); Nasemann et al. (2020) (eastern tropical South Pacific); Huang et al. (2021) (eastern tropical North Pacific). 14C age calculated based on Δ14C data extracted from de Lavergne et al. (2017), as described in Supplementary Info. 2 (S1). Note that the 14C age of newly-formed North Atlantic Deep Water is ~400 years (Matsumoto, 2007). Data are provided in Supplementary Info. 1 (Table S4).
The deep waters therefore act as a reservoir for Cr ‘raining’ down in particles descending from subsurface waters. While the close similarity between deep water Cr and macronutrient concentrations (Supplementary Info. 2; Figure S4) implies that deep water Cr concentrations are to a degree regulated by remineralization of organic material, especially in high productivity regions (Janssen et al., 2021), we show here that deep water Cr concentrations also reflect ‘reversible scavenging’ of Cr(III) taken up in subsurface waters in regions where lithogenic suspended particles are abundant (Section 4.2). Note that it is also likely that deep water Cr concentrations beneath OMZs are affected by Cr released by re-oxidation of Cr(III) (Nasemann et al., 2020; Huang et al., 2021; Janssen et al., 2021). Whilst sediments can represent an important source of Cr to the deep ocean (Janssen et al., 2021), their influence on a global scale remains uncertain (Pöppelmeier et al., 2021).
Water mass mixing can also influence the spatial distribution of Cr in deep waters at local (Rickli et al., 2019; Janssen et al., 2023) and ocean basin (Janssen et al., 2021) scales. Although there is some variation in the composition of deep water masses in our study area (Figure 1), to a first approximation they can be considered to consist of North Atlantic Deep Water (NADW) and Antarctic Bottom Water (AABW). The relative contributions of these end members can be estimated from the dissolved oxygen and phosphate concentrations of the seawater samples as follows (Broecker, 1991; Sarmiento et al., 2007):
where fNADW is the fraction of NADW, 1.59 and 0.74 are the PO4* values (in μmol kg-1) of, respectively, AABW and NADW (Artigue et al., 2020), and PO4* is given by:
where [PO4] and [O2] are, respectively, the PO4 and O2 concentrations of a seawater sample and 170 is the stoichiometric ratio of oxygen to phosphate (Anderson and Sarmiento, 1994).
Excluding one sample that has fNADW > 1 (due to input of Mediterranean seawater; Sarmiento et al., 2007) and samples collected from within 30 m of the seabed, there is a correlation (significant at p<0.1) between dissolved Cr and fNADW (r2 = 0.42) in sub-tropical Atlantic deep waters (>2000 m) (Figure S4), suggesting that water mass mixing can, to some degree, act to homogenize distinct Cr pools. However, there is no significant correlation between δ53Cr and 1/Cr for the same set of samples. From Figure S4 it appears likely that the NADW end-member has lower Cr and a higher δ53Cr value relative to the AABW end-member, but as no samples of ‘pure’ NADW (i.e., from ~45°N in the North Atlantic at ~4000 m water depth; Sarmiento et al., 2007) or ‘pure’ AABW (i.e., from ~35°S in the South Atlantic at ~4000 m water depth; Sarmiento et al., 2007) have been analyzed for Cr and δ53Cr to date, this cannot be confirmed.
Concentrations of dissolved Cr are also positively correlated to the 14C water mass age (Supplementary Info. 1) in Atlantic deep waters (significant at p<0.1), supporting the accumulation of Cr, but the correlation (r2 = 0.32) is weaker than it is for Cr- fNADW. The Atlantic Ocean is rapidly ventilated by NADW and high-latitude source waters and has developed a relatively weaker accumulation signal of dissolved metals, such as zinc, compared to other ocean basins (Weber et al., 2018; Middag et al., 2019). This also appears to be the case for Cr.
4.5 Global correlation between seawater Cr and δ53Cr
Data from this study confirm an inverse correlation between δ53Cr and logarithmic Cr concentration (r2 = 0.53, n=33; Figure 6A) and the slope of the linear regression is −0.79 ± 0.13‰. This is consistent with the previously proposed global δ53Cr-ln[Cr] relationship for the open ocean, which was considered to reflect a Rayleigh-type fractionation of Cr isotopes as Cr is removed from seawater (Scheiderich et al., 2015; Figure 6):
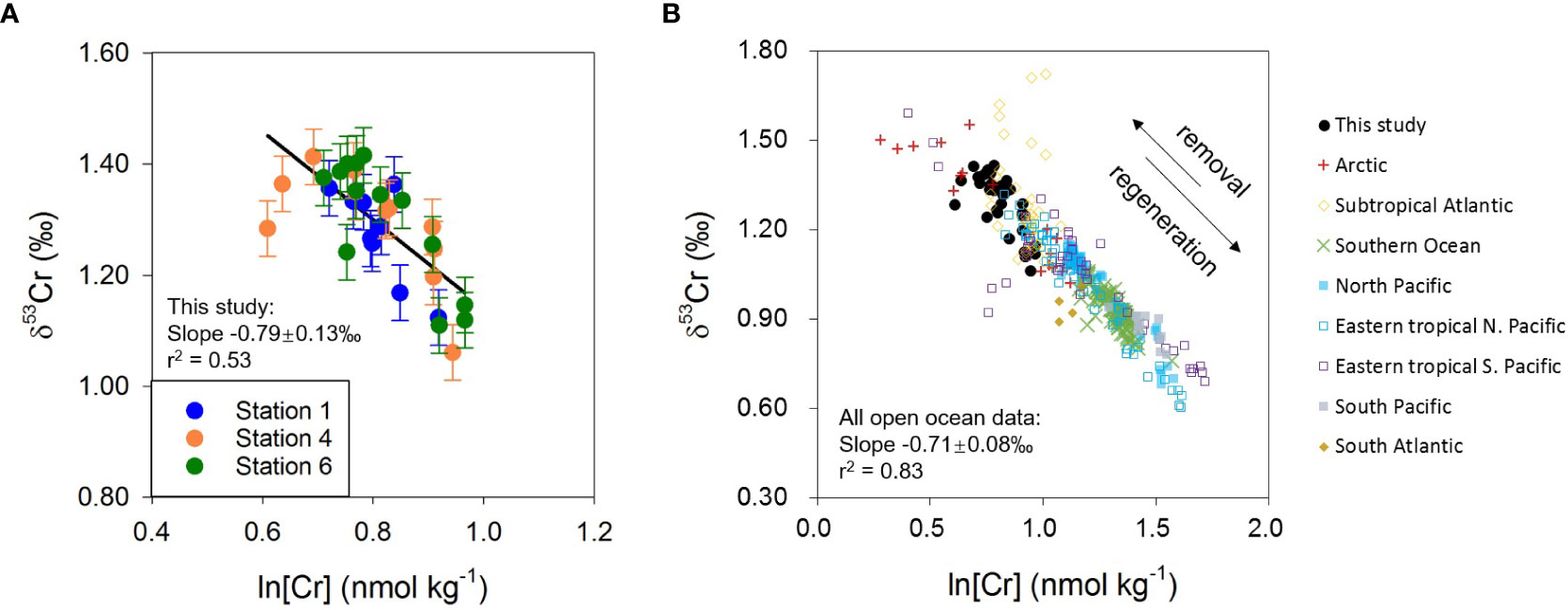
Figure 6 (A) Relationship between δ53Cr and ln[Cr] for new data from this study. (B) Relationship between δ53Cr and ln[Cr] for the global open ocean seawater. Literature data are from: Scheiderich et al. (2015) (Arctic); Goring-Harford et al. (2018) (eastern subtropical Atlantic); Rickli et al. (2019) (Southern Ocean); Moos and Boyle (2019) (North Pacific); Janssen et al. (2020) (North Pacific); Moos et al. (2020) (eastern tropical North Pacific); Huang et al. (2021) (eastern tropical North Pacific); Nasemann et al. (2020) (eastern tropical South Pacific); Janssen et al. (2021) (subtropical Atlantic, Southern Ocean, North Pacific, eastern tropical South Pacific, South Pacific, South Atlantic). Data sets from the South Atlantic (Bonnand et al., 2013) and the eastern Pacific OMZ (Bruggmann et al., 2019; Wang et al., 2019) are not shown as more recent studies from similar locations (Huang and Boyle, 2018; Moos et al., 2020; Nasemann et al., 2020) indicate that these data may not be representative of the true seawater signal or are likely not correct.
where δ53Cr0 represents the initial Cr isotope composition, f represents the fraction of Cr remaining in seawater, and ε is the isotope fractionation factor between Cr(III) and Cr(VI). The Cr isotope fractionation factor derived from all open ocean seawater samples reported in the literature to date, including the present study, is ε = −0.71 ± 0.08‰ (r2 = 0.83, n=347: Scheiderich et al., 2015; Goring-Harford et al., 2018; Moos and Boyle, 2019; Rickli et al., 2019; Janssen et al., 2020; Moos et al., 2020; Nasemann et al., 2020; Huang et al., 2021; Janssen et al., 2021; this study) (Table S5). This value is smaller than determined in the laboratory for Cr reduction by Fe(II) and/or organic matter (ϵ = −1.5 to −4.2‰: Døssing et al., 2011; Basu and Johnson, 2012; Kitchen et al., 2012), or biotic Cr reduction (ϵ = −1.6 to −4.3‰: Zhang et al., 2018; Zhang et al., 2019). It is now clear that although Cr is reduced in the euphotic zone due to biological and/or photochemical processes (e.g., Li et al., 2009; Janssen et al., 2020), as well as in the OMZs by organic matter, microbial activity, and possibly Fe(II) (Moos et al., 2020; Nasemann et al., 2020; Huang et al., 2021), the intrinsic Cr isotope fractionation is diminished as a portion of isotopically light Cr(III) remains in the dissolved phase (Moos et al., 2020; Nasemann et al., 2020; Huang et al., 2021; Wang, 2021). It is also possible that (1) the fractionation factor may be influenced by the rate of Cr reduction and removal (Jamieson-Hanes et al., 2014); and (2) scavenging/adsorption of Cr onto particles may cause a small (but as yet unconstrained) isotope fractionation (Ellis et al., 2004).
As discussed in Section 4.4, progressive regeneration of Cr that is removed from the upper water column in deep water causes accumulation of Cr with a lower δ53Cr value along the global ocean deep water flow path (Section 4.4). In theory, the Rayleigh-type δ53Cr-Cr curve requires that the δ53Cr difference between regenerated Cr and total dissolved Cr in the water column is approximately equal to ϵ = −0.71‰. However, simple mass balance modelling (Figure S5) shows that incremental addition of isotopically light Cr (with δ53Cr ≈ 0.08 to 0.36‰) to deep waters can partly reproduce the shape and slope of the global δ53Cr-ln[Cr] relationship. Given the relatively long seawater residence time of Cr (~3000 to 9500 years: Reinhard et al., 2013; McClain and Maher, 2016; Pöppelmeier et al., 2021) compared to the time scale of ocean ventilation (~1000 years: e.g., DeVries and Holzer, 2019), this implies that, while local Cr and Cr isotope variations caused by removal of Cr fit with the global δ53Cr-Cr relationship (e.g., Huang et al., 2021), regeneration of Cr from sinking particles and accumulation of isotopically light Cr as water masses age are also important processes that shape the large scale heterogeneity of Cr and Cr isotopes in the world’s major ocean basins and contribute to the systematic δ53Cr-Cr relationship discussed by other studies.
5 Conclusions
This study reports full water column depth profiles of dissolved Cr and δ53Cr at three stations in the sub-tropical North Atlantic. Considered together with ancillary data including dissolved, and colloidal Fe concentrations, turbidity, Chl-a, and dissolved oxygen as well as macronutrient concentrations, we have assessed the processes that regulate the behaviour of Cr and Cr isotopes in the modern sub-tropical North Atlantic Ocean. There is no clear evidence for significant inputs of Cr from atmospheric dust and hydrothermal vents in this area, but benthic inputs of Cr may be locally important, notably in the vicinity of hydrothermal vents. The distribution of dissolved Cr and δ53Cr rather appears to principally reflect internal cycling of Cr. Subsurface waters (above 700 m depth) are very slightly depleted in Cr, and very slightly enriched in heavy Cr isotopes, relative to deeper waters. Removal of Cr in subsurface waters is not directly controlled by levels of oxygen or biological uptake in this region where biological productivity is low and waters show only modest oxygen depletion, but does coincide with lowest concentrations of dFe and cFe. We suggest that Cr, most likely in the form of relatively isotopically light Cr(III), is taken up onto colloid aggregates of Fe-(oxyhydr)oxides and/or dust particles. Despite the concurrent removal of Cr and Fe in the upper 700 m water column, the cycling of Cr and Fe is apparently decoupled in deeper waters. While regeneration of Fe occurs via respiration of biogenic particles (Kunde et al., 2019), we show that subtly increased levels of Cr (with relatively low δ53Cr values) in intermediate waters most likely reflect reversible scavenging in this part of the North Atlantic Ocean. In deep waters, water mass mixing plays a role in controlling the Cr and δ53Cr distributions, but regeneration of Cr from particles is more obvious at least at the global scale.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
WW performed the chromium analyses and data interpretation and prepared the original draft of the manuscript. RJ designed the study, mentored WW and edited subsequent drafts. HG-H supported method validation, sample analysis, and data interpretation. KK collected samples in the field, performed the iron analyses, and edited the draft. EMW performed nutrient analysis in the field. ML acquired funding for the research cruise and led sample collection. DC supported the analytical work and data interpretation. All authors commented on drafts of the manuscript and have approved the submitted version.
Funding
This cruise was funded by the NERC-funded ZIPLOc project (NE/N001125/1). WW’s PhD studentship was funded by the Chinese Scholarship Council and the Graduate School of the National Oceanography Centre Southampton; KK’s PhD studentship was funded by the Graduate School of the National Oceanography Centre Southampton.
Acknowledgments
We thank the scientific team, captain, and crew of the RRS James Cook cruise JC150 for their invaluable help in the collection of the samples and for generously sharing ancillary data that have greatly assisted the interpretation of Cr data. We thank David Janssen for invaluable comments on a previous version of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1165304/full#supplementary-material
References
Achterberg E. P., van den Berg C. M. (1997). Chemical speciation of chromium and nickel in the western Mediterranean. Deep Sea Res. Part II: Topical Stud. Oceanography 44 (3-4), 693–720. doi: 10.1016/S0967-0645(96)00086-0
Albarède F., Beard B. (2004). Analytical methods for non-traditional isotopes. Rev. Mineralogy Geochemistry 55 (1), 113–152. doi: 10.2138/gsrmg.55.1.113
Anderson L. A., Sarmiento J. L. (1994). Redfield ratios of remineralization determined by nutrient data analysis. Global Biogeochemical Cycles 8 (1), 65–80. doi: 10.1029/93GB03318
Andronikov A. V., Novak M., Borodulina G. S., Efremenko N. A., Andronikova I. E., Chesalina G. L., et al. (2019). One river, two streams: chemical and chromium isotopic features of the Neglinka river (Karelia, northwest Russia). Hydrological Sci. J. 64 (8), 974–982. doi: 10.1080/02626667.2019.1617418
Ansari M. G., Johnson T. (2022). Chromium isotopic fractionation during oxidation of Cr(III)-bearing solids by manganese oxides at circum-neutral pH. Goldschmidt Abstract. doi: 10.46427/gold2022.11525
Artigue L., Lacan F., Van Gennip S., Lohan M. C., Wyatt N. J., Woodward E. M. S., et al. (2020). Water mass analysis along 22° N in the subtropical north Atlantic for the JC150 cruise (GEOTRACES, GApr08). Deep Sea Res. Part I: Oceanographic Res. Papers 158. doi: 10.1016/j.dsr.2020.103230
Artigue L., Wyatt N. J., Lacan F., Mahaffey C., Lohan M. C. (2021). The importance of water mass transport and dissolved-particle interactions on the aluminum cycle in the subtropical north Atlantic. Global Biogeochemical Cycles 35. doi: 10.1029/2020GB006569
Bacon M. P., Anderson R. F. (1982). Distribution of thorium isotopes between dissolved and particulate forms in the deep sea. J. Geophysical Research: Oceans 87 (C3), 2045–2056. doi: 10.1029/JC087iC03p02045
Bain D. J., Bullen T. D. (2005). Chromium isotope fractionation during oxidation of Cr(III) by manganese oxides. Geochimica et Cosmochimica Acta Supplement 69, A212.
Basu A., Johnson T. M. (2012). Determination of hexavalent chromium reduction using Cr stable isotopes: isotopic fractionation factors for permeable reactive barrier materials. Environ. Sci. Technol. 46 (10), 5353–5360. doi: 10.1021/es204086y
Bauer K. W., Cole D. B., Asael D., Francois R., Calvert S. E., Poulton S. W., et al. (2019). Chromium isotopes in marine hydrothermal sediments. Chem. Geology 529, 119286. doi: 10.1016/j.chemgeo.2019.119286
Beaulieu S. E. (2015) InterRidge global database of active submarine hydrothermal vent fields. prepared for InterRidge, version 3.3. Available at: http://vents-data.interridge.org.
Bonnand P., James R., Parkinson I., Connelly D., Fairchild I. (2013). The chromium isotopic composition of seawater and marine carbonates. Earth Planetary Sci. Lett. 382, 10–20. doi: 10.1016/j.epsl.2013.09.001
Bonnand P., Parkinson I. J., James R. H., Karjalainen A. M., Fehr M. A. (2011). Accurate and precise determination of stable Cr isotope compositions in carbonates by double spike MC-ICP-MS. J. Analytical Atomic Spectrometry 26 (3), 528–535. doi: 10.1039/c0ja00167h
Broecker W. S. (1991). The great ocean conveyor. Oceanography 4, 79–90. doi: 10.5670/oceanog.1991.07
Bruggmann S., Scholz F., Klaebe R. M., Canfield D. E., Frei R. (2019). Chromium isotope cycling in the water column and sediments of the Peruvian continental margin. Geochimica et Cosmochimica Acta 257, 224–242. doi: 10.1016/j.gca.2019.05.001
Campbell J. A., Yeats P. A. (1981). Dissolved chromium in the northwest Atlantic ocean. Earth Planetary Sci. Lett. 53 (3), 427–433. doi: 10.1016/0012-821X(81)90047-9
Chen M., Wang W. X. (2001). Bioavailability of natural colloid-bound iron to marine plankton: influences of colloidal size and aging. Limnology & Oceanography 46 (8), 1956–1967. doi: 10.4319/lo.2001.46.8.1956
Chester R., Hughes M. J. (1969). The trace element geochemistry of a north Pacific pelagic clay core. Deep Sea Res. Oceanographic Abstracts 16 (6), 639–654. doi: 10.1016/0011-7471(69)90064-3
Chester R., Murphy K. J. T. (1990). “Metals in the marine atmosphere,” in Heavy metals in the marine environment (Boca Raton, FL: CRC Press), 27–49.
Connelly D. P., Statham P. J., Knap A. H. (2006). Seasonal changes in speciation of dissolved chromium in the surface Sargasso Sea. Deep Sea Res. Part I: Oceanographic Res. Papers 53 (12), 1975–1988. doi: 10.1016/j.dsr.2006.09.005
Cranston R. E. (1983). Chromium in Cascadia Basin, northeast Pacific ocean. Mar. Chem. 13 (2), 109–125. doi: 10.1016/0304-4203(83)90020-8
Cranston R. E., Murray J. W. (1980). Chromium species in the Columbia river and estuary 1. Limnology & Oceanography 25 (6), 1104–1112. doi: 10.4319/lo.1980.25.6.1104
D’Arcy J., Babechuk M. G., Døssing L. N., Gaucher C., Frei R. (2016). Processes controlling the chromium isotopic composition of river water: constraints from basaltic river catchments. Geochimica et Cosmochimica Acta 186, 296–315. doi: 10.1016/j.gca.2016.04.027
Davidson A. B., Semeniuk D. M., Koh J., Holmden C., Jaccard S. L., Francois R., et al. (2020). A Mg(OH)2 coprecipitation method for determining chromium speciation and isotopic composition in seawater. Limnology & Oceanography: Methods 18 (1), 8–19. doi: 10.1002/lom3.10342
de Lavergne C., Madec G., Roquet F., Holmes R. M., McDougall T. J. (2017). Abyssal ocean overturning shaped by seafloor distribution. Nature 551, 181–186. doi: 10.1038/nature24472
DeVries T., Holzer M. (2019). Radiocarbon and helium isotope constraints on deep ocean ventilation and mantle-3He sources. J. Geophysical Research: Oceans 124 (5), 3036–3057. doi: 10.1029/2018JC014716
Døssing L. N., Dideriksen K., Stipp S. L. S., Frei R. (2011). Reduction of hexavalent chromium by ferrous iron: a process of chromium isotope fractionation and its relevance to natural environments. Chem. Geology 285 (1-4), 157–166. doi: 10.1016/j.chemgeo.2011.04.005
Eary L. E., Rai D. (1987). Kinetics of chromium (III) oxidation to chromium (VI) by reaction with manganese dioxide. Environ. Sci. Technol. 21, 1187–1193. doi: 10.1021/es00165a005
Elderfield H. (1970). Chromium speciation in sea water. Earth Planetary Sci. Lett. 9 (1), 10–16. doi: 10.1016/0012-821X(70)90017-8
Ellis A. S., Johnson T. M., Bullen T. D. (2002). Chromium isotopes and the fate of hexavalent chromium in the environment. Science 295, 2060–2062. doi: 10.1126/science.1068368
Ellis A. S., Johnson T. M., Bullen T. D. (2004). Using chromium stable isotope ratios to quantify Cr(VI) reduction: lack of sorption effects. Environ. Sci. Technol. 38 (13), 3604–3607. doi: 10.1021/es0352294
Ellis A., Johnson T., Villalobos-Aragon A., Bullen T. (2008). Environmental cycling of Cr using stable isotopes: kinetic and equilibrium effects. In AGU Fall Meeting Abstracts (Vol. 2008, pp. H53F–08).
Feely R. A., Baker E. T., Marumo K., Urabe T., Ishibashi J., Gendron J., et al. (1996). Hydrothermal plume particles and dissolved phosphate over the superfast-spreading southern East Pacific rise. Geochimica et Cosmochimica Acta 60 (13), 2297–2323. doi: 10.1016/0016-7037(96)00099-3
Findlay A. J., Gartman A., Shaw T. J., Luther G. W. (2015). Trace metal concentration and partitioning in the first 1.5 m of hydrothermal vent plumes along the mid-Atlantic ridge: TAG, Snakepit, and Rainbow. Chem. Geology 412, 117–131. doi: 10.1016/j.chemgeo.2015.07.021
Fitzsimmons J. N., Carrasco G. G., Wu J., Roshan S., Hatta M., Measures C. I., et al. (2015). Partitioning of dissolved iron and iron isotopes into soluble and colloidal phases along the GA03 GEOTRACES north Atlantic transect. Deep Sea Res. Part II: Topical Stud. Oceanography 116, 130–151. doi: 10.1016/j.dsr2.2014.11.014
Frank A. B., Klaebe R. M., Frei R. (2019). Fractionation behavior of chromium isotopes during the sorption of Cr(VI) on kaolin and its implications for using black shales as a paleoredox archive. Geochemistry Geophysics Geosystems 20 (5), 2290–2302. doi: 10.1029/2019GC008284
Frei R., Gaucher C., Poulton S. W., Canfield D. E. (2009). Fluctuations in Precambrian atmospheric oxygenation recorded by chromium isotopes. Nature 461, 250–253. doi: 10.1038/nature08266
Frei R., Poiré D., Frei K. M. (2014). Weathering on land and transport of chromium to the ocean in a subtropical region (Misiones, NW Argentina): a chromium stable isotope perspective. Chem. Geology 381, 110–124. doi: 10.1016/j.chemgeo.2014.05.015
GEOTRACES Intermediate Data Product Group (2021). The GEOTRACES intermediate data product 2021 (IDP2021) (NERC EDS British Oceanographic Data Centre NOC). doi: 10.5285/cf2d9ba9-d51d-3b7c-e053-8486abc0f5fd
German C. R., Campbell A. C., Edmond J. M. (1991). Hydrothermal scavenging at the mid-Atlantic ridge: modification of trace element dissolved fluxes. Earth Planetary Sci. Lett. 107 (1), 101–114. doi: 10.1016/0012-821X(91)90047-L
Goring-Harford H. J., Klar J. K., Donald H. K., Pearce C. R., Connelly D. P., James R. H. (2020). Behaviour of chromium and chromium isotopes during estuarine mixing in the Beaulieu estuary, UK. Earth Planetary Sci. Lett. 536. doi: 10.1016/j.epsl.2020.116166
Goring-Harford H. J., Klar J. K., Pearce C. R., Connelly D. P., Achterberg E. P., James R. H. (2018). Behaviour of chromium isotopes in the eastern sub-tropical Atlantic oxygen minimum zone. Geochimica et Cosmochimica Acta 236, 41–59. doi: 10.1016/j.gca.2018.03.004
Gueguen B., Reinhard C. T., Algeo T. J., Peterson L. C., Nielsen S. G., Wang X., et al. (2016). The chromium isotope composition of reducing and oxic marine sediments. Geochimica et Cosmochimica Acta 184, 1–19. doi: 10.1016/j.gca.2016.04.004
Hatta M., Measures C. I., Wu J., Roshan S., Fitzsimmons J. N., Sedwick P., et al. (2015). An overview of dissolved Fe and Mn distributions during the 2010–2011 US GEOTRACES north Atlantic cruises: GEOTRACES GA03. Deep Sea Res. Part II: Topical Stud. Oceanography 116, 117–129. doi: 10.1016/j.dsr2.2014.07.005
Honeyman B. D., Santschi P. H. (1989). A Brownian-pumping model for oceanic trace metal scavenging: evidence from Th isotopes. J. Mar. Res. 47 (4), 951–992. doi: 10.1357/002224089785076091
Huang T., Boyle E. A. (2018). Is the chromium concentration profile in the Argentine basin anomalous? AGU Fall Meeting Abstracts, OS21F–O1624.
Huang J., Hao J., Huang F., Sverjensky D. A. (2019). Mobility of chromium in high temperature crustal and upper mantle fluids. Geochemical Perspective Lett. 12, 1–6. doi: 10.7185/geochemlet.1926
Huang T., Moos S. B., Boyle E. A. (2021). Trivalent chromium isotopes in the eastern tropical north Pacific oxygen-deficient zone. Proc. Natl. Acad. Sci. 118 (8). doi: 10.1073/pnas.1918605118
Hurst M. P., Bruland K. W. (2007). An investigation into the exchange of iron and zinc between soluble, colloidal, and particulate size-fractions in shelf waters using low-abundance isotopes as tracers in shipboard incubation experiments. Mar. Chem. 103 (3-4), 211–226. doi: 10.1016/j.marchem.2006.07.001
Izbicki J. A., Ball J. W., Bullen T. D., Sutley S. J. (2008). Chromium, chromium isotopes and selected trace elements, western Mojave desert, USA. Appl. Geochemistry 23 (5), 1325–1352. doi: 10.1016/j.apgeochem.2007.11.015
James R. H., Elderfield H. (1996). Chemistry of ore-forming fluids and mineral formation rates in an active hydrothermal sulfide deposit on the Mid-Atlantic Ridge. Geology 24(12), 1147–1150. doi: 10.1130/0091-7613(1996)024<1147:COOFFA>2.3.CO;2
Jamieson-Hanes J. H., Lentz A. M., Amos R. T., Ptacek C. J., Blowes D. W. (2014). Examination of Cr(VI) treatment by zero-valent iron using in situ, real-time X-ray absorption spectroscopy and Cr isotope measurements. Geochimica et Cosmochimica Acta 142, 299–313. doi: 10.1016/j.gca.2014.07.031
Janssen D. J., Gilliard D., Rickli J., Nasemann P., Koschinsky A., Hassler C. S., et al. (2023). Chromium stable isotope distributions in the southwest Pacific ocean and constraints on hydrothermal input from the Kermadec Arc. Geochimica et Cosmochimica Acta 342, 31–44. doi: 10.1016/j.gca.2022.12.010
Janssen D. J., Rickli J., Abbott A. N., Ellwood M. J., Twining B. S., Ohnemus D. C., et al. (2021). Release from biogenic particles, benthic fluxes, and deep water circulation control Cr and δ53Cr distributions in the ocean interior. Earth Planetary Sci. Lett. 574. doi: 10.1016/j.epsl.2021.117163
Janssen D. J., Rickli J., Quay P. D., White A. E., Nasemann P., Jaccard S. L. (2020). Biological control of chromium redox and stable isotope composition in the surface ocean. Global Biogeochemical Cycles. 34, e2019GB006397. doi: 10.1029/2019GB006397
Janssen D. J., Rickli J., Wille M., Sepúlveda Steiner O., Vogel H., Dellwig O., et al. (2022). Chromium cycling in redox-stratified basins challenges δ53Cr paleoredox proxy applications. Geophysical Res. Lett. 49 (21), e2022GL099154. doi: 10.1029/2022GL099154
Jeandel C., Minster J. F. (1987). Chromium behavior in the ocean: global versus regional processes. Global Biogeochemical Cycles 1 (2), 131–154. doi: 10.1029/GB001i002p00131
Jickells T. D., An Z. S., Andersen K. K., Baker A. R., Bergametti G., Brooks N., et al. (2005). Global iron connections between desert dust, ocean biogeochemistry, and climate. Science 308 (5718), 67–71. doi: 10.1126/science.1105959
John S. G., Conway T. M. (2014). A role for scavenging in the marine biogeochemical cycling of zinc and zinc isotopes. Earth Planetary Sci. Lett. 394, 159–167. doi: 10.1016/j.epsl.2014.02.053
Johnson K. S., Elrod V., Fitzwater S., Plant J., Boyle E., Bergquist B., et al. (2007). Developing standards for dissolved iron in seawater. Eos Trans. Am. Geophysical Union 88 (11), 131–132. doi: 10.1029/2007EO110003
Jones M. R., Luther G. W. III, Tebo B. M. (2020). Distribution and concentration of soluble manganese (II), soluble reactive Mn (III)-l, and particulate MnO2 in the Northwest Atlantic ocean. Mar. Chem. 226, 103858. doi: 10.1016/j.marchem.2020.103858
Kaczynski S. E., Kieber R. J. (1993). Aqueous trivalent chromium photoproduction in natural waters. Environ. Sci. Technol. 27, 1572–1576. doi: 10.1021/es00045a011
Kaczynski S. E., Kieber R. J. (1994). Hydrophobic C18 bound organic complexes of chromium and their potential impact on the geochemistry of Cr in natural waters. Environ. Sci. Technol. 28 (5), 799–804. doi: 10.1021/es00054a009
Kieber R. J., Helz G. R. (1992). Indirect photoreduction of aqueous chromium(VI). Environ. Sci. Technol. 26, 307–312. doi: 10.1021/es00026a010
Kitchen J. W., Johnson T. M., Bullen T. D., Zhu J., Raddatz A. (2012). Chromium isotope fractionation factors for reduction of Cr(VI) by aqueous Fe(II) and organic molecules. Geochimica et Cosmochimica Acta 89, 190–201. doi: 10.1016/j.gca.2012.04.049
Klun K., Falnoga I., Mazej D., Šket P., Faganeli J. (2019). Colloidal organic matter and metal(loid)s in coastal waters (Gulf of Trieste, northern Adriatic Sea). Aquat. Geochemistry 25, 179–194. doi: 10.1007/s10498-019-09359-6
Kunde K., Wyatt N. J., González-Santana D., Tagliabue A., Mahaffey C., Lohan M. C. (2019). Iron distribution in the subtropical north Atlantic: the pivotal role of colloidal iron. Global Biogeochemical Cycles. 33, 1532–1547. doi: 10.1029/2019GB006326
Li B., Liao P., Liu P., Wang D., Ye Z., Wang J., et al. (2022). Formation, aggregation, and transport of NOM-Cr(III) colloids in aquatic environments. Environ. Science: Nano 9 (3), 1133–1145. doi: 10.1039/D1EN00861G
Li S. X., Zheng F. Y., Hong H. S., Deng N. S., Lin L. X. (2009). Influence of marine phytoplankton, transition metals and sunlight on the species distribution of chromium in surface seawater. Mar. Environ. Res. 67 (4-5), 199–206. doi: 10.1016/j.marenvres.2009.02.001
Little S. H., Vance D., Siddall M., Gasson E. (2013). A modelling assessment of the role of reversible scavenging in controlling oceanic dissolved Cu and Zn distributions. Global Biogeochemical Cycles 27 (3), 780–791. doi: 10.1002/gbc.20073
Matsumoto K. (2007). Radiocarbon-based circulation age of the world oceans. J. Geophysical Research: Oceans 112, C09004. doi: 10.1029/2007JC004095
McClain C. N., Maher K. (2016). Chromium fluxes and speciation in ultramafic catchments and global rivers. Chem. Geology 426, 135–157. doi: 10.1016/j.chemgeo.2016.01.021
Middag R., de Baar H. J. W., Bruland K. W. (2019). The relationships between dissolved zinc and major nutrients phosphate and silicate along the GEOTRACES GA02 transect in the West Atlantic ocean. Global Biogeochemical Cycles 33, 63–84. doi: 10.1029/2018GB006034
Mignot A., Claustre H., Uitz J., Poteau A., d’Ortenzio F., Xing X. (2014). Understanding the seasonal dynamics of phytoplankton biomass and the deep chlorophyll maximum in oligotrophic environments: a bio-argo float investigation. Global Biogeochemical Cycles 28 (8), 856–876. doi: 10.1002/2013GB00478
Moos S. B., Boyle E. A. (2019). Determination of accurate and precise chromium isotope ratios in seawater samples by MC-ICP-MS illustrated by analysis of SAFe station in the north Pacific ocean. Chem. Geology 511, 481–493. doi: 10.1016/j.chemgeo.2018.07.027
Moos S. B., Boyle E. A., Altabet M. A., Bourbonnais A. (2020). Investigating the cycling of chromium in the oxygen deficient waters of the Eastern tropical north Pacific ocean and the Santa Barbara basin using stable isotopes. Mar. Chem. 221. doi: 10.1016/j.marchem.2020.103756
Mugo R. K., Orians K. J. (1993). Seagoing method for the determination of chromium (III) and total chromium in sea water by electron-capture detection gas chromatography. Analytica Chimica Acta 271 (1), 1–9. doi: 10.1016/0003-2670(93)80545-V
Murray J. W., Spell B., Paul B. (1983). “The contrasting geochemistry of manganese and chromium in the eastern tropical Pacific ocean,” in Trace metals in sea water (Boston, MA: Springer), 643–669.
Nakayama E., Kuwamoto T., Tsurubo S., Tokoro H., Fujinaga T. (1981). Chemical speciation of chromium in sea water: part 1. Effect of naturally occurring organic materials on the complex formation of chromium (III). Analytica Chimica Acta 130 (2), 289–294. doi: 10.1016/S0003-2670(01)93006-5
Nasemann P., Janssen D. J., Rickli J., Grasse P., Frank M., Jaccard S. L. (2020). Chromium reduction and associated stable isotope fractionation restricted to anoxic shelf waters in the Peruvian oxygen minimum zone. Geochimica et Cosmochimica Acta. 285, 207–224. doi: 10.1016/j.gca.2020.06.027
Ohnemus D. C., Lam P. J. (2015). Cycling of lithogenic marine particles in the US GEOTRACES north Atlantic transect. Deep Sea Res. Part II: Topical Stud. Oceanography 116, 283–302. doi: 10.1016/j.dsr2.2014.11.019
Ohnemus D. C., Torrie R., Twining B. S. (2019). Exposing the distributions and elemental associations of scavenged particulate phases in the ocean using basin-scale multi-element data sets. Global Biogeochemical Cycles 33 (6), 725–748. doi: 10.1029/2018GB006145
Paulukat C., Døssing L. N., Mondal S. K., Voegelin A. R., Frei R. (2015). Oxidative release of chromium from Archean ultramafic rocks, its transport and environmental impact - a Cr isotope perspective on the Sukinda Valley ore district (Orissa, India). Appl. Geochemistry 59, 125–138. doi: 10.1016/j.apgeochem.2015.04.016
Pérez V., Fernández E., Marañón E., Morán X. A. G., Zubkov M. V. (2006). Vertical distribution of phytoplankton biomass, production and growth in the Atlantic subtropical gyres. Deep-Sea Res. Part I: Oceanographic Res. Papers 53 (10), 1616–1634. doi: 10.1016/j.dsr.2006.07.008
Pettine M., D’Ottone L., Campanella L., Millero F. J., Passino R. (1998). The reduction of chromium (VI) by iron (II) in aqueous solutions. Geochimica et Cosmochimica Acta 62, 1509–1519. doi: 10.1016/S0016-7037(98)00086-6
Pettine M. (2000). Redox processes of chromium in sea water. Chemical Processes in Marine Environments, 281–296. doi: 10.1007/978-3-662-04207-6_16
Polzin K. L., Toole J. M., Ledwell J. R., Schmitt R. W. (1997). Spatial variability of turbulent mixing in the abyssal ocean. Science 276 (5309), 93–96. doi: 10.1126/science.276.5309.93
Pöppelmeier F., Janssen D. J., Jaccard S. L., Stocker T. F. (2021). Modeling the marine chromium cycle: new constraints on global-scale processes. Biogeosciences Discussions 18, 5447–5463. doi: 10.5194/bg-2021-106
Rädlein N., Heumann K. G. (1995). Size fractionated impactor sampling of aerosol particles over the Atlantic ocean from Europe to Antarctica as a methodology for source identification of Cd, Pb, Tl, Ni, Cr, and Fe. Fresenius’ J. Analytical Chem. 352, 748–755. doi: 10.1007/BF00323059
Rai D., Sass B. M., Moore D. A. (1987). Chromium (III) hydrolysis constants and solubility of chromium (III) hydroxide. Inorganic Chem. 26 (3), 345–349. doi: 10.1021/ic00250a002
Reinhard C. T., Planavsky N. J., Robbins L. J., Partin C. A., Gill B. C., Lalonde S. V., et al. (2013). Proterozoic ocean redox and biogeochemical stasis. Proc. Natl. Acad. Sci. 110 (14), 5357–5362. doi: 10.1073/pnas.1208622110
Reinhard C. T., Planavsky N. J., Wang X., Fischer W. W., Johnson T. M., Lyons T. W. (2014). The isotopic composition of authigenic chromium in anoxic marine sediments: a case study from the Cariaco Basin. Earth Planetary Sci. Lett. 407, 9–18. doi: 10.1016/j.epsl.2014.09.024
Richard F. C., Bourg A. C. (1991). Aqueous geochemistry of chromium: a review. Water Res. 25 (7), 807–816. doi: 10.1016/0043-1354(91)90160-R
Rickli J., Janssen D. J., Hassler C., Ellwood M. J., Jaccard S. L. (2019). Chromium biogeochemistry and stable isotope distribution in the Southern Ocean. Geochimica et Cosmochimica Acta 262, 188–206. doi: 10.1016/j.gca.2019.07.033
Rigaud S., Radakovitch O., Couture R. M., Deflandre B., Cossa D., Garnier C., et al. (2013). Mobility and fluxes of trace elements and nutrients at the sediment–water interface of a lagoon under contrasting water column oxygenation conditions. Appl. Geochemistry 31, 35–51. doi: 10.1016/j.apgeochem.2012.12.003
Roseburrough R., Wang X. (2021). Chromium stable isotope geochemistry in the Mobile Bay estuary. Chem. Geology 584, 120530. doi: 10.1016/j.chemgeo.2021.120530
Rudnick R. L., Gao S. (2003). “3.01 - composition of the continental crust,” in Treatise on geochemistry. Eds. Holland H. D., Turekian K. K. (Pergamon: Oxford), 1–64.
Rudnicki M. D., Elderfield H. (1993). A chemical model of the buoyant and neutrally buoyant plume above the TAG vent field, 26 degrees N, mid-Atlantic ridge. Geochimica et Cosmochimica Acta 57 (13), 2939–2957. doi: 10.1016/0016-7037(93)90285-5
Rue E. L., Smith G. J., Cutter G. A., Bruland K. W. (1997). The response of trace element redox couples to suboxic conditions in the water column. Deep Sea Res. Part I: Oceanographic Res. Papers 44 (1), 113–134. doi: 10.1016/S0967-0637(96)00088-X
Saad E. M., Wang X., Planavsky N. J., Reinhard C. T., Tang Y. (2017). Redox-independent chromium isotope fractionation induced by ligand-promoted dissolution. Nat. Commun. 8 (1), 1–10. doi: 10.1038/s41467-017-01694-y
Sander S., Koschinsky A. (2000). Onboard-ship redox speciation of chromium in diffuse hydrothermal fluids from the north Fiji basin. Mar. Chem. 71 (1-2), 83–102. doi: 10.1016/S0304-4203(00)00042-6
Sarmiento J. L., Simeon J., Gnanadesikan A., Gruber N., Key R. M., Schlitzer R. (2007). Deep ocean biogeochemistry of silicic acid and nitrate. Global Biogeochemical Cycles 21 (1). doi: 10.1029/2006GB002720
Scheiderich K., Amini M., Holmden C., Francois R. (2015). Global variability of chromium isotopes in seawater demonstrated by pacific, Atlantic, and Arctic ocean samples. Earth Planetary Sci. Lett. 423, 87–97. doi: 10.1016/j.epsl.2015.04.030
Schoenberg R., Zink S., Staubwasser M., von Blanckenburg F. (2008). The stable Cr isotope inventory of solid earth reservoirs determined by double spike MC-ICP-MS. Chem. Geology 249 (3-4), 294–306. doi: 10.1016/j.chemgeo.2008.01.009
Semeniuk D. M., Maldonado M. T., Jaccard S. L. (2016). Chromium uptake and adsorption in marine phytoplankton - implications for the marine chromium cycle. Geochimica et Cosmochimica Acta 184, 41–54. doi: 10.1016/j.gca.2016.04.021
Shaw T. J., Gieskes J. M., Jahnke R. A. (1990). Early diagenesis in differing depositional environments: the response of transition metals in pore water. Geochimica et Cosmochimica Acta 54 (5), 1233–1246. doi: 10.1016/0016-7037(90)90149-F
Sirinawin W., Turner D. R., Westerlund S. (2000). Chromium (VI) distributions in the Arctic and the Atlantic oceans and a reassessment of the oceanic Cr cycle. Mar. Chem. 71 (3-4), 265–282. doi: 10.1016/S0304-4203(00)00055-4
Trinquier A., Birck J. L., Allègre C. J. (2008). High-precision analysis of chromium isotopes in terrestrial and meteorite samples by thermal ionization mass spectrometry. J. Analytical Atomic Spectrometry 23 (12), 1565–1574. doi: 10.1039/b809755k
Trocine R. P., Trefry J. H. (1988). Distribution and chemistry of suspended particles from an active hydrothermal vent site on the mid-Atlantic ridge at 26 N. Earth Planetary Sci. Lett. 88 (1-2), 1–15. doi: 10.1016/0012-821X(88)90041-6
van der Weijden C. H., Reith M. (1982). Chromium (III)-chromium (VI) interconversions in seawater. Mar. Chem. 11 (6), 565–572. doi: 10.1016/0304-4203(82)90003-2
Wang X. (2021). The chromium isotope fractionation factor in seawater. Chem. Geology 579. doi: 10.1016/j.chemgeo.2021.120358
Wang X., Glass J. B., Reinhard C. T., Planavsky N. J. (2019). Species-dependent chromium isotope fractionation across the eastern tropical north Pacific oxygen minimum zone. Geochemistry Geophysics Geosystems 20 (5), 2499–2514. doi: 10.1029/2018GC007883
Wang X., Johnson T. M., Ellis A. S. (2015). Equilibrium isotopic fractionation and isotopic exchange kinetics between Cr(III) and Cr(VI). Geochimica et Cosmochimica Acta 153, 72–90. doi: 10.1016/j.gca.2015.01.003
Weber T., John S., Tagliabue A., DeVries T. (2018). Biological uptake and reversible scavenging of zinc in the global ocean. Science 361 (6397), 72–76. doi: 10.1126/science.aap8532
Woodward E. M. S., Rees A. P. (2001). Nutrient distributions in an anticyclonic eddy in the northeast Atlantic ocean, with reference to nanomolar ammonium concentrations. Deep Sea Res. Part II: Topical Stud. Oceanography. 48, 775–793. doi: 10.1016/S0967-0645(00)00097-7
Wu W., Wang X., Reinhard C. T., Planavsky N. J. (2017). Chromium isotope systematics in the Connecticut river. Chem. Geology 456, 98–111. doi: 10.1016/j.chemgeo.2017.03.009
Xiang Y., Lam P. J., Lee J. M. (2021). Diel redox cycle of manganese in the surface Arctic Ocean. Geophysical Research Letters 48(23), e2021GL094805. doi: 10.1029/2021GL094805
Yang L., Nadeau K., Meija J., Grinberg P., Pagliano E., Ardini F., et al. (2018). Inter-laboratory study for the certification of trace elements in seawater certified reference materials NASS-7 and CASS-6. Analytical Bioanalytical Chem. 410 (18), 4469–4479. doi: 10.1007/s00216-018-1102-y
Ye Y., Völker C. (2017). On the role of dust-deposited lithogenic particles for iron cycling in the tropical and subtropical Atlantic. Global Biogeochemical Cycles 31 (10), 1543–1558. doi: 10.1002/2017GB005663
Yiğiterhan O., Murray J. W., Tuğrul S. (2011). Trace metal composition of suspended particulate matter in the water column of the Black Sea. Mar. Chem. 126 (1-4), 207–228. doi: 10.1016/j.marchem.2011.05.006
Zhang Q., Amor K., Galer S. J., Thompson I., Porcelli D. (2018). Variations of stable isotope fractionation during bacterial chromium reduction processes and their implications. Chem. Geology 481, 155–164. doi: 10.1016/j.chemgeo.2018.02.004
Keywords: chromium isotopes, North Atlantic Ocean, particle scavenging, regeneration, GEOTRACES
Citation: Wang W, Goring-Harford H, Kunde K, Woodward EMS, Lohan MC, Connelly DP and James RH (2023) Biogeochemical cycling of chromium and chromium isotopes in the sub-tropical North Atlantic Ocean. Front. Mar. Sci. 10:1165304. doi: 10.3389/fmars.2023.1165304
Received: 13 February 2023; Accepted: 09 May 2023;
Published: 26 May 2023.
Edited by:
Hermano Melo Queiroz, University of São Paulo, BrazilReviewed by:
Ruifeng Zhang, Shanghai Jiao Tong University, ChinaPeter Leslie Croot, University of Galway, Ireland
Copyright © 2023 Wang, Goring-Harford, Kunde, Woodward, Lohan, Connelly and James. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenhao Wang, Wenhao.Wang@soton.ac.uk