
Abstract
Degradation of organic contaminants into less toxic substances is the best option to remove these compounds rather than using conventional techniques. The sulfate radical-based-advanced oxidation process is an effective strategy that degrades organic contaminants by activating peroxymonosulfate (PMS). Such a strategy generates singlet oxygen (1O2), hydroxyl (\(^ \bullet \!{{{\mathrm{OH}}}}\)), and sulfate (\({{{\mathrm{SO}}}}_4^{ \bullet\! - }\)) radicals. \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) is distinguished by its high oxidation selectivity and activity toward the degradation of organic contaminates compared to other radicals. Various catalysts are employed in PMS activation including layered doubled hydroxides (LDHs), which are characterized by their facile synthesis and high catalytic activity. This review article is the first attempt to compile the recent progress in the degradation of common organic pollutants including aromatic compounds, pharmaceutical residues, and dyes via the PMS activation using LDH-based catalysts. The degradation pathways, reaction parameters’ influence, stability of LDHs, and comparisons between different LDH-based catalysts are investigated in this work.
Similar content being viewed by others

Introduction
Recently, with the augmentation of urbanization and industrialization to meet human needs, a humongous amount of toxic wastes has been released into the environment1,2,3,4,5,6. Therefore, water pollution takes the priority worldwide over other issues because of the continual discharge of industrial waste containing different organic dyes, agrochemicals, personal care products, pharmaceuticals products, and aromatic compounds into water bodies7,8,9,10,11. Such detrimental contaminants constitute a major threat to the environment and human health owing to their low biodegradability, stability, and high toxicity12,13,14,15,16,17,18.
Several biological, physical, and chemical treatment techniques have been adopted to remove these wastes from water bodies. Biological processes such as bioreactors, biologically activated sludge, microbiological treatments, and lagoons possess many positive aspects including getting rid of biodegradable organic matter along with a significant capability of removing biological oxygen demand, and suspended solids19,20. On the other hand, they reveal several drawbacks such as slow rate of degradation, low biodegradability of dyes, and the possible formation of biological sludge21,22. While physical processes include membrane filtration, which is remarked as quite a straightforward process and convenient method, yet it is of a high operational cost23,24. Furthermore, electrodialysis is characterized by its high separation efficiency, but it is expensive because of the energy requirement25,26. Moreover, chemical treatments such as coagulation-flocculation depend on adding chemicals to convert suspended solid into sludge. Despite being a cost-effective technique, it results in the formation of secondary pollutants, requiring a further treatment27,28,29. Therefore, it could be concluded that these techniques are not capable of completely removing the organic residuals from wastewater30,31,32,33,34,35.
Over the past few decades, advanced oxidation processes (AOPs) have acquired a noticeable attention by virtue of their potent oxidizing ability, rapid reaction rate, simplicity, great efficiency, and extensive applications for removing the organic contaminants36,37,38,39,40,41. Simply, AOPs exploit the high reactive oxygen species (ROS) such as \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals to degrade the organic residuals into less hazardous compounds or completely mineralize them into carbon dioxide (CO2) and water vapor (H2O)42,43. Nonetheless, \(^ \bullet \!{{{\mathrm{OH}}}}\)-based AOPs comprising photo-Fenton, electro-Fenton, photo-sono-Fenton, photo-electro-Fenton, and sono-Fenton suffer some limitations including the necessary acidic conditions (pH 2–4) and the instability of hydrogen peroxide44,45,46. In the mid-2000′ s the progression of research resulted in arousing a sub-category of AOPs which is the sulfate radical-based advanced oxidation processes (SR-AOPs)47.
Recently, SR-AOPs have been considered one of the most effective methods in the degradation of organic residuals owing to their potent oxidizing ability and high efficiency. Besides, \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\) in comparison with \(^ \bullet \!{{{\mathrm{OH}}}}\) affords diverse merits. For instance, \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) acquires higher redox potential (Eo = 2.5–3.1 V) than \(^ \bullet \!{{{\mathrm{OH}}}}\) (Eo = 2.8 V). Interestingly, \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\) possesses magnificent oxidation selectivity and the ability to react with distinct organic compounds with a superior rate constant (range from 106 to 109 M−1 s−1) and a wide functioning pH range48. In addition, the longer half-lifetime of \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) (30–40 μs) compared to \(^ \bullet \! {\rm{OH}}\)(10− 3 μs) is considered another advantage of \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\)49. For producing \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\), a convenient oxidant is energized such as PMS that has attained the researchers’ interest owing to its remarkable features; i) PMS can be obtained in a solid form that could be easily carried, stored and transported; ii) PMS can generate a high yield of ROS; iii) Numerous materials could be employed in the activation of PMS50.
Ghanbari et al.51 and Wang et al.52 reported that the direct reaction of PMS on its own, even though being a thermodynamically potent oxidant, is sluggish when used with most of the toxic pollutants. Thus, an appropriate method is required for PMS activation and generation of sulfate radicals. PMS activation can be done by applying different approaches like ultrasound (US), ultraviolet (UV), heat, microwave, alkali, and transition metals. However, the activation using transition metals is more favorable as it is more economical in comparison with energy-based techniques such as UV, US, microwave, and heat53. Activation by alkali extends the duration of the treatment process as it needs pH adjustment before and after treatment, rendering the process more complex. Therefore, homogenous and heterogeneous catalysis have been employed to counteract the aforementioned limitations for PMS activation where the catalysts are either dissolved metal ions or solids, respectively54.
Notable homogenous catalysis exhibits rapid reaction and higher efficiency particularly at pH < 7. However, it causes secondary pollution through the leaching of their component metal ions into water, resulting in the difficulty of recovering the applied catalyst from treated water and raising the overall cost of the treatment process. Additionally, it could be resulting in detrimental human health effects. On the other hand, heterogeneous catalysis offers a lot of advantages including the catalyst’s easy recovery, durability in various conditions47,55, applicability over a wide pH range (2–9), and also it does not require a secondary treatment56.
The activation of PMS by transition metals is represented in Eqs. (1–4). Initially, the catalyst activates PMS, generating ROS such as \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) and \(^ \bullet \! {\rm{OH}}\) radicals, in addition to 1O2 engenders from the electron-transfer mechanism. The transition metal ions (Mn+) initiate the reaction through generation \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\), resulting in the oxidation of the metal to Mn+1. Interestingly, the weak redox potential of peroxymonosulfate radical (\({{{\mathrm{HSO}}}}_5^ -\)/\({{{\mathrm{SO}}}}_5^{ \bullet\! - }\)) (Eo = 1.1 V) facilitates the recovery of Mn+1 via the electron transfer. Moreover, the presence of two or more convenient transition metals leads to the synergistic enhancement of PMS activation57,58.
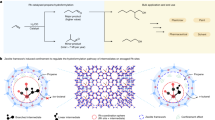
LDH is an ionic clay two-dimensional (2D) material with a hydrotalcite-like structure, having an individual ion-exchange characteristic owing to its anionic interlayer and cationic metal layers59. The structure of LDHs consists of divalent and trivalent cations that are neutralized by the commutable anions intercalated into the interlayer60. LDHs general formula is exemplified as [M1−x2+ Mx3+ (OH)2]x+(An−)x/n • mH2O, in which M2+ symbolizes the divalent metal cations while M3+ trivalent ones, An− shows the exchangeable anions among the layers (such as carbonates or nitrates), and x represents the molar ratio between metal ions (Fig. 1a)61. Moreover, LDHs are characterized by their outstanding chemical and physical properties, as well as their facile fabrication and cost-effectiveness62. Consequently, LDHs are applied for bountiful applications such as catalysis, batteries, cosmetics, adsorption, sensors, etc.63,64,65. In particular, the abundant choices of M2+/M3+ render LDHs as efficient heterogeneous catalysts possessing a high PMS activation capacity for degrading toxic organic residuals from water bodies66,67. Furthermore, the decomposition of LDHs via calcination from 400 to 600 °C forms mixed metal-oxides known as layered double oxides (LDOs). Interestingly, LDOs possess several advantages including extraordinary surface area, eco-friendly nature, easy preparation, and light-sensitive catalytic activity. In addition, LDOs have a higher catalytic activity in AOPs compared to LDHs due to their higher stability68. Table 1 summarized a comparison between LDHs and LDOs.

a The structure and bountiful applications of LDHs, and b Schematic illustration of the three superior preparation routes of LDHs.
LDHs have exhibited a promising performance in AOPs, which is most likely due to (a) LDHs pH-buffering extent acquiring the ability to retain the neutral pH levels; (b) LDH-based catalysts possess a steady structure that can boost the reusability and limits the metals’ leaching; (c) the simplicity of synthesizing LDHs and the capability of immobilizing distinct metal ions can enhance the catalytic functionality; (d) the presence of assorted ROS specifically 1O2 can diminish the negative influence of coexisting substrates as inorganic anions; (e) several LDH-based nanocomposites can be composed by paring LDHs or LDOs along with other materials such as carbonaceous and magnetic materials to enhance the catalytic performance, conductibility, and stability69.
Various review articles have recently highlighted the synthesis of LDH-based catalysts and their applications in wastewater treatment. For instance, Yang et al.70 investigated the controlling factors that affect the photodegradation of dyes using LDHs in 2016. Wu et al.71 has focused recently on the progression of the photocatalytic activities of LDH-based catalysts, in terms of energy and structural fabrication. In 2021, Kohantorabi et al.72 addressed the catalytic activity of many heterogeneous catalysts including metal/bimetallic oxides, supported noble/non-noble metals, carbonaceous-based materials, LDHs, metal-organic frameworks (MOFs), and zeolites towards the PMS activation. Moreover, Xie et al. highlighted the preparation and characterization approaches of LDHs as well as their application in AOPs69.
Based on the authors’ knowledge, the PMS activation by LDH for the degradation of organic contaminants was briefly discussed as one of the AOPs. However, the recent increase in research papers that addressed this hot spot topic needs a thorough and up-to-date assessment. In this connection, our review aimed to summarize the recent progress in the degradation of common organic pollutants including aromatic compounds, pharmaceutical residues, and dyes via the PMS activation using LDH-based catalysts. Importantly, the review focused on the various preparation methods of LDH and the controlling factors on the quality and quantity of the produced LDH. Furthermore, the degradation mechanism pathways were discussed based on quenching tests and advanced analyses like X-ray absorption fine structure (XAFS) spectroscopy, X-ray photoelectron spectroscopy (XPS), electron spin resonance (ESR) spectroscopy, etc. Notably, the durability of LDH-based catalysts during the PMS activation was evaluated from leaching and reusability tests. Ultimately, in light of our in-depth investigations, we concluded the progress, challenges, and recommend ations for inspiring researchers to further developments.
Synthesis routes of LDHs
The LDH fabrication is fulfilled by the combination of a considerable range of divalent and trivalent cations with diverse interlayer anions. In preparation of LDHs, many factors must be taken into consideration, for instance, the sort of metallic cations and interlayer anion, the M2+/M3+ ratio, pH condition, route of assemblage, and atmosphere control. Notably, such parameters control the main characteristic of LDHs including surface area, crystallinity, morphology, and particle size. Furthermore, it is greatly noticed that the various physical features of the fabricated LDHs make them possess propitious abilities to be utilized in industrial fields73. In the last few years, environmentally safe and cost-effective preparation techniques have been fostered including; co-precipitation, hydrothermal/solvothermal, urea hydrolysis, sol-gel, ion exchange, calcination-rehydration, etc.
Co-precipitation route
The co-precipitation method is one of the simplest, cost-effective, and most efficient techniques for fabricating of LDHs74,75. In addition, such an approach provides a direct preparation of LDHs from the diversified M2+ and M3+ and a wide scale of anions from inorganic anions (viz., carbonate (\({{{\mathrm{CO}}}}_3^{2 - }\)), nitrate (\({{{\mathrm{NO}}}}_3^ -\)) and chloride (Cl−)) to organic molecules and biomolecules76. The fabrication of LDHs via co-precipitation approaches involves the addition of anion solution into a solution from a proper ratio of M2+/M3+, then adding a base or urea hydrolysis for raising the solution pH (from 8 to 10) and prompting the precipitation of LDH77. Ultimately, the reaction mixture is stirred for one day at room temperature, filtered, washed with deionized water several times, and dried using vacuum or conical funnel75. While for attaining LDO, the synthesized LDHs are calcined at moderate temperature (from 450 to 600 °C), endowing them with plenty of active sites, high surface area, and uniform distributed composition73. On the other hand, the calcination temperature is not permitted to be too high since mixed metal oxides could be sintered and transformed into spinel phase, and diminishing both of the pore volume and surface area78.
Hydrothermal route
Unlike the co-precipitation route, LDHs’ synthesis via the hydrothermal route is an ideal route for the rapid procedure with a broad-scale industrialization potentiality. It is more commonly operated in autoclaves, generating autogenous pressure due to heating at temperatures ranging from 60 to 200 oC79,80,81. In this method, an alkali solution encompassing the metal salts that will compose the LDH is stirred well before pouring into an autoclave and the solution is maintained at hydrothermal conditions for a specific duration that could vary between hours to many days82,83. Then it is left for crystallization at room temperature, washing, and subsequently dried84. A higher yield of the hydrothermally fabricated LHDs becomes conspicuous by augmenting the temperature and the time of the synthesis process. Interestingly, the hydrothermal approach possesses special merits such as producing LDHs with higher crystalline morphology compared to that fabricated by the co-precipitation method as well as averting the toxic residuals that could be negatively influencing humans and the environment (i.e. Cl−, OH−, and \({{{\mathrm{NO}}}}_3^ -\))75,85.
Urea hydrolysis
Urea has been extensively applied as a precipitating agent for a plethora of metals into their metal hydroxides analogous as it is a weak Brønsted base (pKb = 13.8). Thereby, the resultant slow hydrolysis rate could control the degree of supersaturation and precipitation. In general, the urea hydrolysis takes place via two reaction phases: (1) the yielding of ammonium cyanate (NH4CNO) which is considered as a rate-determining step (Eq. 1); and (2) the fast hydrolysis of cyanate to form carbonate (Eq. 2)86.
Moreover, the rate of urea hydrolysis can be simply dominated by adjusting the reaction temperature, where it is noticeable that the rate constant is boosted 200 times by raising the process temperature from 60 to 100 °C84. Notably, the urea hydrolysis route is distinguished by the easiness of rinsing the precipitate rather than the co-precipitation that needs reiterated rinsing to eliminate the alkali-metal ions87. However, the urea hydrolysis approach suffers a major shortcoming, which is the presence of carbonate anions in the fabricated LDHs88.
Anion-exchange route
The anion-exchange route is an indirect synthesis route as the used precursor in this technique is the LDH that has robust anion-exchange characteristics. The facility of monovalent ions exchange is arranged as follows: \({{{\mathrm{NO}}}}_3^ -\) > Br− > Cl− > F− > OH−. Therefore, it is obvious that nitrate ions are displaced quite easier, hence they are the utmost proper precursors for the fabrication of LDHs via anion-exchange route89. Furthermore, this technique involves the synthesis of LDHs with host anions using a co-precipitation approach, then exchanging with the guest anions. This approach is only applied when the co-precipitation approach is improper, like the unstable di- or tri-valent metal cations or anions in basic conditions90. In general, the mechanism of the anion-exchange reaction can be expressed as follows:
Where A is the host anions while X expresses as the guest having negative charges of m and n, sequentially85.
It is worth noting that the anion-exchange process is primarily governed by the electrostatic interaction between the brucite sheets and the exchanging anions. Moreover, the anion-exchange approach has strict conditions77;
-
(a)
Depending on the chemical composition of the host LDH layers.
-
(b)
The convenient solvent fosters the anion-exchange route.
-
(c)
The pH medium should be higher than 4.0 since LDH could be destroyed at a lower pH level.
-
(d)
Anion-exchange requires high temperatures.
On the other hand, anion-exchange has some drawbacks, including (a) that for the anion-exchange reaction, the LDH precursor should be obtained first by another approach. Therefore, two steps are needed to reach the final product, which results in increasing the process’s complexity and cost. (b) Neutral species are not able to be intercalated into LDH through this method91.
Reconstruction (rehydration) route
The reconstruction route primarily depends on making use of the memory effect of LDHs, where calcination of the LDHs is accomplished to detach the interlayer hydroxyl groups, anions, and water, producing mixed metal oxides92. Then, the produced mixed metal oxides (MMOs) are rehydrated through their dispersion in the target anions solution, ensuing the reestablishment of LDHs while the anionic species are inserted in the interlayer part of LDHs93. The temperature of calcination is the key parameter in this route since the increment in the calcination temperature directly diminishes the memory effect94. It is noteworthy to mention that the LDHs’ calcination temperature should not be over 550; the elevated temperatures will collapse the LDH structure. Nevertheless, the low calcination temperature affects the crystal phase95.
The preeminent merits of the reconstruction route are summarized in the following points;
-
1.
Both anions and neutral species can be incorporated into LDH.
-
2.
Due to the possibility to eliminate the incorporated anions within the calcination operation, thus LDH incorporated with any sort of anions can be exploited as the original reactant.
On the other hand, the reconstruction route suffers some drawbacks such as;
-
(a)
The recovery of LDH essentially relies on the calcined metal-oxides activity in the solutions of the reconstruction.
-
(b)
Time-consuming and laborious since it requires two steps or more of the entire process encompassing calcination in addition to reconstruction91.
Sol-gel route
Sol-gel approach has earned a great reputation owing to its cost-effectiveness and ability to produce LDHs with a superior purity96. Generally speaking, a number of variables should be taken into account when fabricating LDHs using the sol-gel method, including adjusting the temperature, changing the composition of the host anions, and adding or removing the species of reactant. Additionally, the fabrication process of the sol-gel route proceeds as follows: the components are continuously stirred to create sol, and the reaction mixture is aged to allow for the phase transition from sol to gel97. The solvent, pH, concentration of the metal precursors, and synthesis temperature are some of the parameters that can be adjusted to control the characteristics of the fabricated LDHs using this method. These parameters also affect the rate of hydrolysis and condensation of the metal precursor98. LDHs produced by the sol-gel procedures have higher thermal stability and lower crystallinity compared to those formed by the co-precipitation route93.
Overall, several studies concluded that co-precipitation, anion exchange, and reconstruction were superior to other LDH preparation methods. Schematic representations of these LDH synthesis methods are shown in Fig. 1b. It was determined by contrasting the three preparation techniques that co-precipitation stands out for its high yield, which can be up to three times that of other LDH preparation techniques. While anion exchange produces LDHs with a larger anion size, endowing them with further advantages in several pharmaceutical applications. Furthermore, reconstruction produces poor crystalline and partially intercalated LDHs99.
Environmental applications of PMS–based LDH systems
Degradation of aromatic compounds
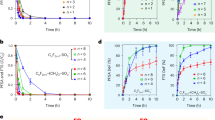
Fazli et al.100 reported the photocatalytic activity of Fe3O4@CuCr-LDH nanocomposite towards the degradation of diethyl phthalate (DEP) through PMS activation. As shown in Fig. 2a, the fabrication of CuCr-LDH was successful because even in the purest CuCr-LDH or magnetic composite, the associated metal hydroxide (M-OH) absorption peaks were clearly visible below 1000 cm−1. Since pH has the greatest influence on the PMS activation process, its impact was studied over a wide pH range from 3 to 12 (Fig. 2b). The optimal pH was determined to be 8 because there is plenty of H+ in a highly acidic medium that forms H-bonds with the peroxy bond of \({{{\mathrm{HSO}}}}_5^{2 - }\). Zeta potential measurements revealed that the pHPZC of Fe3O4@CuCr-LDH was 8.9, indicating that the surface was positively charged in both neutral and acidic media. Thence, the H-attached \({{{\mathrm{HSO}}}}_5^{2 - }\) could not reach to the positive surface of Fe3O4@CuCr-LDH, so the catalytic degradation of DEP was directly declined. Additionally, Jawad et al.‘s101 study looked into how high acidic medium (4.32 mg/L at pH 3) caused the leaching of Cu ions in CuOx@Co-LDH to peak, hindering the phenol’s degradation. Moreover, there was a significant diminution in the DEP degradation efficiency at a high basic medium, which may be ascribed to the interaction between PMS and \({{{\mathrm{SO}}}}_5^{2 - }\). As a result, 1O2 is formed, which possesses a lower oxidizing potential than \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals. On the other hand, the PMS concentration strongly controlled the degradation aptitude of DEP at which a synchronic amelioration in the DEP degradation efficiency was observed with raising the PMS concentration from 1 to 14 mM (Fig. 2c). This finding may be anticipated by the abundance of the reactive \({{{\mathrm{SO}}}}_4^{ \bullet - }\). Nonetheless, the increase in the PMS concentration from 8 to 14 mM caused a slight enhancement in the DEP, which is concomitant with sang et al. study102. This common behavior may be assigned to the self-quenching reaction between \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and the extra PMS103,104.

a Fourier transform infrared (FTIR) spectra, b Effect of pH and c Effect of PMS concentration on the degradation efficiency of DEP adapted with permission from ref. 100, Copyright, 2021, Elsevier. d The schematic diagram of the PMS activation by BC-LDH for the removal of DEP, e Normalized Co K-edge, and f Ni K-edge X-ray absorption near edge structure (XANES) spectra of BC-LDH adapted with permission from ref. 105, Copyright, 2022, Elsevier.
In another attempt, Yang et al.105 reported a quite smart and economical approach to stabilize the adsorbed Co2+ and Ni2+ onto a biochar (BC) surface to fabricate BC-LDH composite, which was utilized for the catalytic degradation of DEP (Fig. 2d). BC-LDH showed enhanced catalytic behavior towards the PMS activation since the degradation efficiency of DEP reached 90% within 240 min. XPS spectra of BC-LDH before and after the catalytic degradation of DEP were used to interpret the degradation mechanism. XPS spectrum of Co2p after the degradation reaction revealed a slight decrease in the Co2+ proportion from 50.1 to 49.8%, accompanied by an increase in the Co3+ proportion from 48.9 to 50.2%. This observation was accredited to the participation of Co2+ in the PMS activation, resulting in partial oxidation of Co2+ to Co3+, which was in line with Li et al. observation106. Furthermore, the C‒C proportion in the C1s spectrum increased from 21.8 to 29.3% compared to pure BC, indicating the oxidation of the aromatic moieties (C=C→C-C) during DEP oxidation107. Moreover, Co and Ni K-edge XAFS spectroscopy of BC-LDH (Fig. 2e, f) revealed a slight shift of Co K-edge to the high energy after DEP degradation, reflecting the partial oxidation of Co2+ to Co3+. Contrariwise, there was no change in the Ni K-edge of BC-LDH after the catalytic reaction, signifying that Ni did not participate in the reaction.
In another investigation, Zhao et al.108 highlighted the activation of PMS by Fe-Al LDH to efficiently degrade Bisphenol A (BPA). Notably, Mӧssbauer spectroscopy was used to explain the surface transformation of Fe-Al LDH after the catalytic reaction. It was observed from the Mӧssbauer spectrum of Fe-Al LDH that there were two species of Fe2+ (Fe–O–Fe and Fe-O-Al)109,110. Unequivocally, the electronegativity of Fe2+ in Fe–O–Fe is lower than Fe-O-Al owing to the higher electron-withdrawing property of Fe than Al. Furthermore, the proportions of Fe–O–Fe and Fe-O-Al in Fe-Al LDH were 15% and 62%, respectively, with a small proportion of Fe3+ that may be due to the sample oxidation throughout the measurement. The Mӧssbauer spectrum of the transformed Fe-Al LDH after the BPA degradation showed that the proportion of Fe2+ in both species decreased with an increase in the formed Fe3+. This finding designated the participation of Fe2+ in both reactive species which may be attributed to the good conductivity of the whole metal layer. Moreover, to scrutinize the impact of time on the active species during the activation of PMS, Fe-Al LDH/PMS was examined by an in-situ electron paramagnetic resonance (EPR) test. It was observed that there were three active species (\(^ \bullet \!{{{\mathrm{OH}}}}\), \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) and superoxide radical (\({{{\mathrm{O}}}}_2^{ \bullet \!- }\))) and the distinctive signal peaks of \(^ \bullet \! {\rm{OH}}\) were the dominant compared to \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) signals over time. In addition, the intensity of both active species increased with time and reached its peak at around 5 min. Then, the signals of \(^ \bullet \!{{{\mathrm{OH}}}}\) and \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) disappeared after 20 min, but the \({{{\mathrm{O}}}}_2^{ \bullet \!- }\) signals were almost still with their high intensity. On the contrary, the high intensity of \(^ \bullet \! {\rm{OH}}\) and \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) signals was observed for a long time in some catalyst/PMS systems, indicating that PMS could be slowly and continuously activated111. Besides, a quenching test was conducted to define the dominant reactive radical in the BPA degradation. Several scavengers were utilized including methanol (MeOH) for \(^ \bullet \!{{{\mathrm{OH}}}}\) and \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\), isopropanol for \(^ \bullet \!{{{\mathrm{OH}}}}\), p-benzoquinone (p-BQ) for \({{{\mathrm{O}}}}_2^{ \bullet\! - }\) and sodium azide (NaN3) for 1O2. The results clarified that there was a decline in the degradation efficiency in the presence of methanol, reflecting the participation of both \(^ \bullet \!{{{\mathrm{OH}}}}\) and \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) in the BPA degradation. While \({{{\mathrm{O}}}}_2^{ \bullet\! - }\) is the controlling reactive radical since the BPA degradation significantly dwindled in the presence of p-BQ. Consequently, the radical pathway of the BPA degradation was suggested based on these results and it was further confirmed when the quenching of 1O2 slightly decreased the degradation efficiency.
Continuing with BPA removal studies, Shahazad et al. successfully synthesized a hexagonal crystalline Cu-rGO LDH nanohybrid for activating PMS to degrade BPA. The scanning electron microscope (SEM) image (Fig. 3a) revealed the hexagonal morphology of Cu-CO3 LDH. While Fig. 3b indicated that the hexagonal structure remained intact after the ion exchange and the Cu-NO3 LDH formation. The Tyndall light scattering effect showed the dispersion of the exfoliated graphene oxide (GO) and Cu-LDH nanosheets in dimethyl formamide and formamide, respectively, as shown in Fig. 3c. The high-resolution transmission electron microscopy (HRTEM) of Cu-rGO LDH (Fig. 3d, e) showed the periodic assembly of reduced graphene oxide (rGO) and Cu-LDH. In addition, the selected area electron diffraction (SAED) of Cu-rGO LDH (Fig. 3f) revealed that the interlayer spacing between two LDH layers was 1.03 nm. The SAED of Cu-rGO LDH (Fig. 3g) inferred the 2D structure of both Cu-LDH and rGO, implying the 2D order of Cu-rGO LDH. Figure 3h elucidates the different pathways of the 1O2 formation. Furthermore, EPR (Fig. 3i, j) and chemical quenching tests confirmed that the BPA degradation followed the non-radical pathway through the PMS activation by Cu-rGO LDH. Also, it was proposed that inner-sphere complexation between Cu-rGO LDH and PMS may be responsible for boosting the PMS decomposition. The plausible mechanism of generating 1O2 is clarified in Eqs. 7–11. Besides, it was found that the PMS concentration declined after the interaction with Cu-rGO LDH in the absence of BPA. This observation signified the strong bond between PMS and Cu-rGO LDH, forming a complex that is responsible for the formation of 1O2 through \({{{\mathrm{O}}}}_2^{ \bullet \!- }\) (Eqs. 11–13). Such a finding was in line with Shahzad et al.112 study that suggested the phenol degradation through the PMS activation by Fe-rGO LDH followed a ternary system as the electrons were transferred from phenol (donor) to PMS (acceptor) via Fe-rGO LDH (mediator). Subsequently, the formation of non-radical ROS agreed with the quenching test that inferred the domination of 1O2.

a, b SEM images of Cu-rGO LDH, c Tyndall light scattering effect, d, e TEM of Cu-rGO LDH, f, g SAED of Cu-rGO LDH, h the mechanism of 1O2 formation in the Cu-rGO LDH/PMS system, i, j ESR spectra of TMP-1O2 and DMPO-\({{{\mathrm{O}}}}_2^{ \bullet \!- }\) and k, l Fe Mössbauer spectra FeAl-LDH and DOM-LDH, and m the proposed degradation mechanism adapted with permission from ref. 115 Copyright, 2020, Elsevier.
Notably, the inner-sphere complexation was confirmed by recording a red shift in the distinguishing peak of PMS using the attenuated total Reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) spectrum after the interaction between Cu-rGO LDH and PMS. Such a result was concomitant with Yang et al.113. Furthermore, the OH groups might be replaced when PMS bonded with the catalyst active sites during the complexation. In addition, the different pathways of the 1O2 generation could be attributed to the uncharged oxygen in \({{{\mathrm{HSO}}}}_5^ -\) involving in the PMS/Cu-rGO LDH inner-sphere complexation114.
In that respect, Ye et al.115 improved the degradation efficiency of BPA from 60% to 93% within 60 min using FeAl-LDH after the introduction of dissolved organic matter (DOM) into the reaction116. The SEM images displayed the platelets-like structure of FeAl-LDH with a size range of 20–60 nm, which tended to aggregate and disperse randomly, as it is typical of LDHs117. After the introduction of DOM, the morphology changed into a flower-like morphology with a diameter of 50 nm and a thickness of 3 nm. Furthermore, a significant enhancement in the specific surface area, pore width, and pore volume of DOM-LDH (70.67 m2 g−1, 11.84 nm, and 0.181 cm3 g−1) was observed compared to the pure FeAl-LDH (56.18 m2 g−1, 1.725 nm, and 0.069 cm3 g−1), suggesting a better interaction between DOM-LDH, PMS, and BPA. Mössbauer spectrum of FeAl-LDH (Fig. 3k) represented the trans-coordinated octahedral Fe (II) species with 32.9% and both cis and trans-coordinated octahedral Fe (III) with 67.1%, whereas the spectrum of DOM-LDH (Fig. 3l) demonstrated trans-coordinated octahedral Fe (II) species with 61.2% and both cis and trans-coordinated octahedral Fe (III) with 19.2%. This result denoted the rapid conversion of Fe (III) to Fe (II) via the reduction by humic acid-like compound, which accelerated the PMS activation. Therefore, the superior catalytic degradation efficacy was achieved as elucidated in Fig. 3m.
In another study, Hou et al.118 highlighted the PMS activation by CuFeAl-LDO to degrade tetrabromobisphenol A (TBBPA). CuFeAl-LDO was fabricated by the calcination of CuFeAl-LDH at 600 °C. SEM images revealed a significant change in the morphology of CuFeAl-LDH after the calcination at which it has nanosheet morphology, while CuFeAl-LDO particles are globular. The experimental results implied the efficient degradation behavior of CuFeAl-LDO since the degradation rate of TBBPA reached 99.91% within 60 min using 0.1 g CuFeAl-LDO, 0.5 mM PMS, 15 mg/L TBBPA, and pH 8.5. Additionally, the successful regeneration of CuFeAl-LDO was confirmed as the degradation efficiency of TBBPA decreased around 13% after the 5th cycle. It is worth mentioning that this decrease in the catalytic performance of CuFeAl-LDO could be attributed to the loss or aggregation of the catalyst during the reaction. In addition, the possibility of producing an intermediate that could be adsorbed on the catalyst surface and cover the active sites resulted in the deactivation of CuFeAl-LDO119,120. Moreover, the leaching of Fe and Cu ions from CuFeAl-LDO was detected by inductively coupled plasma mass spectrometry (ICP-MS). It was found that the leaching amount of Fe and Cu were 0.003 and 0.064 mg/L, respectively, which are lower than the allowed limits by the United States Environmental Protection Agency (USEPA).
In this perspective, Lu et al.121 fabricated CuCoFe-LDH to activate PMS for the degradation of nitrobenzene (NB). SEM image displayed the stacked thin slices of CuCoFe-LDH, while the SAED pattern revealed the planes (012) and (113) where d (012) = 0.2629 nm and d(113) = 0.1528 nm. It was found that the NB degradation by CuCoFe-LDH/PMS generated intermediate products at which ultra-performance liquid chromatography-time-of-flight (UPLC-TOF) analysis clarified the formation of hydroxyl-NB. Furthermore, time-of-flight mass spectrometry (TOF-MS) depicted the extremely low intensity of the NB signal and the presence of propanedioic acid (negative ion mode)122. Also, the presence of these intermediates was confirmed by the continuous decrease in the pH of the system during the NB degradation process. Based on headspace-gas chromatography/mass spectrometry (HS-GC-MS) and UPLC-TOF analysis, it was proposed that the degradation pathway of NB by CuCoFe-LDH/PMS was driven by the substitution reactions between \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals and NB This suggested degradation process confirmed the domination of \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals for the NB degradation process123,124. Also, Ali et al. confirmed the formation of intermediates during the degradation of para hydroxyl benzoic acid (PHB) using FeMgAl-MoS4/PMS. The degradation pathway of PHB took place through decarboxylation or dehydration reactions and 2-hydroxypropanoic acid, 2-hydroxyacedic acid, and fumaric acid were generated. Eventually, these intermediate molecular acids were converted to CO2 and H2O67.
In one more attempt, Zhu et al.125 synthesized Ni15Cu-LDHs and introduced oxygen vacancies (OVs) through Cu doping, which are anionic surface defects contributing to the positively charged surfaces of catalysts. Such OVs can enhance the adsorption of reactants, shorten the electron-transfer distances, and accelerate the degradation process without releasing radicals into the liquid phase126. Consequently, Ni15Cu-LDHs, in which OVs were introduced, achieved an outstanding degradation efficacy of almost 99.07% against methyl 4-hydroxybenzoate (MeP) within just 10 minutes. Interestingly, Ye et al.49 achieved 100% degradation efficiency of dimethyl phthalate within 60 min using BC-modified CoFe-LDH since BC played the role of catalyst carrier and electron mediator which enhanced the physicochemical characteristics of CoFe-LDH and improved the redox cycle of transition metals.
In another study, Zhao et al.127 reported the degradation efficiency of 2,4-dichlorophenoxyacetic acid (2,4-D) using Fe3O4–CsxWO3/NiAl LDH Composite. It was figured out that the presence of co-existing anions such as \({{{\mathrm{CO}}}}_3^{2 - }\), bicarbonate (\({{{\mathrm{HCO}}}}_3^ -\)), and Cl− reduced the degradation efficiency of 2,4-D from 90.53% to 69.80% and 26.81% by increasing the concentration of \({{{\mathrm{CO}}}}_3^{2 - }\) and \({{{\mathrm{HCO}}}}_3^ -\) from 0 to 16 ppm, respectively. This result could be ascribed to the significant disrupting of \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \({\!}^ \bullet \!{{{\mathrm{OH}}}}\) by \({{{\mathrm{CO}}}}_3^{2 - }\) and \({{{\mathrm{HCO}}}}_3^ -\) to produce a weak oxidant (\({{{\mathrm{CO}}}}_3^{ \bullet - }\))128. In contrast, the degradation efficiency enhanced from 90.53% to 99.14% by increasing the Cl− concentration from 0 to 16 ppm in 180 min, which could be attributed to the enhancement of the \({{{\mathrm{SO}}}}_4^{ \bullet - }\) production by the chlorine free radicals129. The reusability of Fe3O4–CsxWO3/NiAl LDH was not that good as it decreased to 75% after four consecutive cycles. On the other hand, a quite promising regeneration ability of CuCoFe-LDH (93%) was obtained after five cycles of p-nitrophenol (P-NP) degradation, signifying a high degree of CuCoFe-LDH stability130.
Notably, the catalytic degradation mechanism of 2,4-D by Fe3O4-CsxWO3/NiAl-LDH/PMS was proposed as follows; Fe3O4-CsxWO3/NiAl-LDH formed electron/hole pairs (e−/h+) as the produced e− at the conduction band reacted with O2 and generated \({{{\mathrm{O}}}}_2^{ \bullet - }\) (Eq. 14). Meanwhile, h+ at the valence band resulted in the formation of \({\!}^ \bullet \!{{{\mathrm{OH}}}}\) when reacting with water molecules (Eq. 15).
Consequently, the generated \({{{\mathrm{O}}}}_2^{ \bullet\! - }\) began to decompose PMS and released \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\) as explained in Eq. (16) which in turn initiated the degradation of 2,4-D with the aid of other free radicals. Additionally, the produced e−s were suggested to be responsible for releasing \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) and \({\!}^{\bullet} \!{\mathrm{OH}}\) from PMS activation as elucidated by Eq. (17). It should be noted that the intermediate products resulting from the degradation of 2,4-D were further degraded by \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\), \({{{\mathrm{O}}}}_2^{ \bullet\! - }\), \({\!}^{\bullet} \!{\mathrm{OH}}\), and also h+ into CO2 and H2O (Eq. 18). However, the degradation performance of \({{{\mathrm{O}}}}_2^{ \bullet - }\) and h+ was not significant as it was confirmed by investigating the effect of the four reactive radicals (tertiary butyl alcohol (TBA), MeOH, potassium iodide (KI) and p-BQ), where TBA is a scavenger for \({\!}^{\bullet} \!{\mathrm{OH}}\), MeOH for both \({\!}^{\bullet} \!{\mathrm{OH}}\) and \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\), KI for h+, and p-BQ for \({{{\mathrm{O}}}}_2^{ \bullet \!- }\). As a result, \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\) and \({\!}^{\bullet}\!{\rm{OH}}\) were identified as the primary controlling radicals in the degradation of 2,4-D.
Interestingly, the synergistic effect of the catalyst (ZnCo−LDH@CeO2), PMS, and light resulted in extraordinary degradation efficiency of phenol (99.8%), which was reported by Gao et al.131. Such a superb activity was assigned to the high content of cerium oxide (CeO2) that resulted in the introduction of more active sites being exposed on the catalyst surface. Moreover, metal ions including Co2+ and Ce3+ showed excellent performance in the activation of PMS and generating \({{{\mathrm{SO}}}}_4^{ \bullet - }\) for the degradation of phenol in another study132. Likewise, Ye et al.133 achieved 100% degradation of p-arsanilic acid (p-ASA) using CoMnFe-LDH/PMS and CoMnFe-LDO/PMS within 30 min. PMS activation was based on the direct electron transfer from adsorbed p-ASA on the surface of CoMnFe-LDO/PMS. While in the LDH system, the PMS activation depended on the coupling with surface hydroxyl groups in CoMnFe-LDH/PMS system. However, it was noticed that CoMnFe-LDH was shown to be superior to CoMnFe-LDO in terms of PMS activation owing to its significant affinity for PMS via surface \(^ \bullet \!{{{\mathrm{OH}}}}\) complexation and multiple-metal redox cycle49,134.
In one word, this section summarizes the recent progression in aromatic compounds’ degradation through the PMS activation using LDHs. In light of the aforementioned studies, the most used approaches for LDHs’ preparation are hydrothermal and co-precipitation methods. Furthermore, the fabricated LDHs are modified by forming composites with carbonaceous materials (BC, and GO) that can act as carriers and electron mediators to enhance the physicochemical characteristics of LDHs and improve the redox cycle of the transition metals. Also, mixing LDHs with metal oxides/sulfides (Fe3O4, CeO2, WO3, MoS4) enhances the redox cycle, enriches the catalytic medium with electrons, and facilitates the catalyst separation and reusability by employing the magnetic metal oxides. Besides, the calcination of LDH to form LDO can be considered one of the most impeccable modification ways to LDHs since it could improve the morphology, surface area, and degradation activity of LDHs, along with decreasing the metal leaching during the catalytic reaction. However, such a modification technique is not commonly used. Therefore, further studies in this aspect are highly recommended.
Interestingly, LDH-based catalysts modified forms revealed an excellent degradation ability toward a number of aromatic compounds like DEP, BPA, TBBPA, NB, 2,4-D, and p-ASA since the degradation percent attained almost 100% in some cases. Although some LDH-based catalysts revealed good reusability, others showed a decline in the degradation percentage ranging from 15 to 20% after the 3rd or the 4th cycle. This finding implied the metals leaching during the catalytic reaction which is a serious challenge restricting LDHs’ applications in industry. In addition, the substantial metal leaching in a highly acidic medium (pH ≤ 3) was recorded in some studies to be more than four-fold of the acceptable limit by EPA.
More importantly, the degradation mechanism of aromatic compounds via the PMS activation followed two paths; a radical pathway dominated by \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) as well as the contribution of \({{{\mathrm{O}}}}_2^{ \bullet - }\) in some cases. While in the non-radical pathway \({{{\mathrm{O}}}}_2^{ \bullet - }\) is the dominant ROS that forms a complex with PMS to generate 1O2. In this pathway, PMS acts as an acceptor, aromatic compounds as a donor, and LDHs as a mediator.
Unexpectedly, when the interfering ions’ effect on the degradation efficiency of the aromatic compounds was investigated, the presence of Cl− ions improved the efficiency owing to their capability of enhancing the \({{{\mathrm{SO}}}}_4^{ \bullet - }\) production via the chlorine free radical. Such findings regarding the degradation of the noxious organic pollutants in wastewater via the PMS activation using LDH-based composites are summarized in Table 2.
Degradation of pharmaceutical residues
In one study, Caoa et al.135 fabricated AlCo-LDH through an unconventional approach using Co-ZIF owing to the high crystallinity and porosity of metal-organic frameworks (MOFs), rendering them a propitious precursor to synthesize LDHs. The as-fabricated AlCo-LDH was examined by X-ray diffraction (XRD) technique, revealing that the characteristic peaks of Co-ZIF completely changed and became identical to the amorphous Al(OH)3. In addition, the decrease in the pH of the aluminum solution after the etching process is another evidence to confirm the hydrolysis of the metal ions as clarified by the following equations:
Besides, the low concentration of Co ions in AlCo-LDH also indicated the replacement of Co2+ by Al3+ in Co-ZIF to form AlCo-LDH. The catalytic performance of PMS, Co-ZIF-D/PMS, and AlCo-LDH/PMS were scrutinized by the degradation of tetracycline (TC). The degradation efficacy of TC was only 26.4% and 37.43% using PMS and Co-ZIF/PMS, respectively. However, the efficacy boosted to 96.1% when AlCo-LDH/PMS was used. The promising degradation efficiency of TC by AlCo-LDH/PMS system is most likely due to the high surface area of AlCo-LDH. The impact of different quenching reagents was examined, clarifying that the reactive species responsible for the degradation of TC were \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and 1O2 species. These results were confirmed via EPR by detecting the characteristic peaks of DMPO-\({{{\mathrm{SO}}}}_4^{ \bullet - }\) and TMP-1O2 upon introducing 5,5-dimethyl- pyrroline-oxide (DMPO) and 2,2,6,6-tetramethyl-4-piperidinol (TMP), respectively, into the reaction. Due to the high electronegativity of Co, it was concluded to be responsible for the electrons’ transfer to PMS and further generating the reactive species, which in turn attacked TC and degraded it into CO2 and H2O (Fig. 4a).

a The TC degradation by AlCo-LDH/PMS system, adapted with permission from ref. 135, Copyright, 2022, Elsevier, b the IBP degradation by Carbon-ZnFe LDH/PMS system, adapted with permission from ref. 136 Copyright, 2021, Elsevier, c the MTZ degradation using 3D Co3Mn-LDH/rGA/PMS system, adapted with permission from ref. 137 Copyright, 2021, Elsevier and d the possible mechanism of SMX and SCP degradation by ultrathin-NiAl-LDH/PMS system, adapted with permission from ref. 138 Copyright, 2022, Elsevier.
Concomitantly with the above-mentioned study, Naderi et al.136 concluded that \({{{\mathrm{SO}}}}_4^{ \bullet - }\) was majorly driving the degradation process of Ibuprofen (IBP) when Carbon-ZnFe LDH was used to activate PMS and generate sulfate free radicals, as shown in Fig. 4b. The role of LDH compound was emphasized in this study by showing that the degradation efficiency was just 24% using PMS compared to 60.6% upon adding ZnFe-LDH to the PMS solution. Such a substantial increase in the degradation efficacy was attributed to the hydrotalcite structure of ZnFe-LDH, which results in providing a high surface area and generating more degradation sites, along with the enhanced electron transfer owing to the presence of electron-rich Fe active sites. Also, it is worth mentioning that when the LDH compound was modified with carbon nanoparticles, the IBP degradation efficacy increased to 75.4%, and the bio-toxicity of the released wastewater was largely diminished to 15.8%. Such a result was accredited to the carbonaceous activating role in improving the rate of electron transfer.
In the same line, He et al.137 highlighted the effective role of carbonaceous materials in improving the electron transfer process by scrutinizing the effect of three-dimensional (3D) graphene aerogel in enhancing the degradation efficiency of Metronidazole (MTZ) that reached 93.7% using 3D Co3Mn-LDH/rGA as a PMS activator under visible light irradiation (Fig. 4c). Not only did 3D Co3Mn-LDH/rGA perform as a PMS activator, but it also served as a photocatalyst, which was confirmed by recording a degradation efficiency of only 56.4% using Co3Mn-LDH/rGA in the absence of light. Moreover, the electron transfer from 3D Co3Mn-LDH/rGA to PMS was indicated by the oxidation of Co2+ to Co3+ and Mn3+ to Mn4+ with 12.2% and 13.6%, respectively. Also, when different radical scavenging experiments were conducted, it was revealed that \(^ \bullet \!{{{\mathrm{OH}}}}\), \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\), and 1O2 are the main species responsible for MTZ degradation.
In this perspective, Wang et al.138 investigated the synergistic effect between the super-adsorption and the catalytic PMS degradation of sulfamethoxazole (SMX) and sulfachloropyridazine (SCP) using ultrathin-NiAl-LDH. Firstly, the presence of a positive charge on the surface of ultrathin-NiAl-LDH electrostatically attracted the negatively-charged SMX and SCP, and led to their super-adsorption on the surface of the LDH compound. Secondly, the PMS was activated by ultrathin-NiAl-LDH and resulted in the formation of \(^ \bullet \!{{{\mathrm{OH}}}}\) and \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) that led to the degradation of both SMX and SCP into CO2, H2O, and other byproducts, as displayed in Fig. 4d. Additionally, it is worth mentioning that the super-adsorption effect of ultrathin-NiAl-LDH was responsible for minimizing the migration distance between the reactive radicals and the two antibiotic compounds that resulted in degradation efficiencies of 98.8% and 100% for SMX and SCP, respectively.
In another investigation, Wanga et al.139 also adopted the etching process to fabricate FeCo-LDH from zeolitic imidazolate framework-67 (ZIF-67). As the Fe/Co ratio of 1.5 revealed the highest TC degradation efficiency, reaching 92%, it was reported that the molar ratio between Fe and Co significantly affected the catalytic performance of FeCo-LDH/PMS. This superb degradation aptitude of FeCo-LDH/PMS towards TC may be attributed to the contribution of both Fe and Co ions in producing \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) (Eqs. 21 and 22);
The 3D-Excitation Emission Matrix Fluorescence Spectrophotometer (3D-EEMs) was utilized to define the degradation pathway of TC by FeCo-LDH-1.5/PMS (Fig. 5a–f). The characteristic peak of humic acids-like matter was observed at the range of Ex/Em (325–375 nm)/(475–550 nm), indicating the partial decomposition of TC. In addition, a noticeable increase in the peak was observed by increasing the time range from 5 to 10 min, which may be accredited to the complete decomposition of TC. Subsequently, a different fluorescence peak appeared at Ex/Em = (300–350 nm)/(375–450 nm), inferring that humic acid-like matter decomposed into CO2 and H2O. The efficiency of FeCo-LDH-1.5/PMS was not affected by changing the pH level. Thus, it could be demonstrating high degradation efficiencies upon being used in various aquatic environments such as medical wastewater, tap water, and rivers. The leaching of Co from FeCo-LDH-1.5/PMS after three cycles led to decreasing the concentration of Co from 11.61 to 8.85% and thereby the degradation efficiency decreased from 92% to 84%.

a–f The 3D-EEMs of the TC degradation by FeCo-LDH-1.5, adapted with permission from ref. 139, Copyright, 2022, Elsevier.
In another study, Li et al.140 prepared a series of CoFeLa-LDH using different molar ratios between Co, Fe, and La for the degradation of TC. The XRD patterns of CoFe-LDHs showed that the augmentation in the La concentration decreased the intensity of the characteristic peaks of CoFe-LDH. Although the peak of La-O was not detected by the XRD at low concentrations of La, it was identified by the FTIR spectrum. From SEM and Brunauer–Emmett–Teller (BET) analyses of CoFeLa-LDHs, it was noticed that CoFeLa-LDH2 with Co/Fe/La ratios (20:8:2) had a better crystallinity with an average particle size of 497 nm and a higher surface area (122.1 m2 g−1) compared to the other ratios. More importantly, it was observed that CoFeLa-LDH2 without PMS had a low degradation efficiency of just 10.5% against TC, and there was no such a significant difference when PMS was used on its own, as the efficiency was only 5.5%. Moreover, CoFeLa-LDH2/PMS showed a significant degradation efficiency of 90.1%. Moreover, the highest degradation efficiency was obtained at 0.05 g/L CoFeLa-LDH2, whereas increasing the CoFeLa-LDH2 dosage to 0.1 g/L led to quenching the reactive radical species, and decreasing the efficiency. In addition, the maximum TC degradation was detected at pH 5 as the amount of \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) diminished by their reaction with H+ at lower pH levels as elucidated by Eqs. 23 and 24. While at high pH levels, the concentration of \(^ \bullet \!{{{\mathrm{OH}}}}\) was higher than \({{{\mathrm{SO}}}}_4^{ \bullet - }\), which resulted in minimizing the degradation performance of CoFeLa-LDH2/PMS since \(^ \bullet \!{{{\mathrm{OH}}}}\) is less reactive than \({{{\mathrm{SO}}}}_4^{ \bullet - }\). Nonetheless, NiFeLa-LDH/PMS demonstrated excellent catalytic efficiency toward TC over a wide pH range (3–10) because the controlling reactive radicals were \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\), which were generated at pH < 7 and pH > 9, respectively. Moreover, a high degradation efficacy was observed at pH < 7. Such an observation was accredited to the generation of \({{{\mathrm{HSO}}}}_5^ -\) in acidic and neutral media that could be attracted to the positively charged NiFeLa-LDH and the enhancement of free radicals’ generation. This result was in line with Chena et al.50 who elucidated the constant degradation performance of MgMn-LDO towards TC over a wide pH range from 4 to 1050.
Quenching experiments and EPR indicated that both \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) participated in the TC degradation, but \({{{\mathrm{SO}}}}_4^{ \bullet - }\) was the predominant species. In addition, the contribution of the non-radical species to the catalytic degradation process was observed during the reaction. Moreover, the adsorption mechanism was explained by XPS where the decline in the binding energy of Co (II) and the increment of Co (III) indicated the conversion of Co (II) to Co (III). Likewise, the binding energy of both Fe and La changed, which evinced that Co (II), La (III), and Fe (II) contributed to the generation of \({{{\mathrm{SO}}}}_4^{ \bullet - }\). Notably, the influence of the presence of interfering anions like \({{{\mathrm{NO}}}}_3^ -\), Cl–, and \({{{\mathrm{HCO}}}}_3^ -\) was investigated, revealing a significant decrease in the degradation efficacy of TC in the presence of both Cl– and \({{{\mathrm{HCO}}}}_3^ -\). Such behavior is most likely due to the scavenging of the produced \(^ \bullet \!{{{\mathrm{OH}}}}\) and \({{{\mathrm{SO}}}}_4^{ \bullet - }\) by these co-existing anion species. These results agreed with Zhang et al.141 who reported a notable decline in the degradation efficiency of carbamazepine (CBZ) utilizing Ni/Co-LDH/PMS from 91 to 21% in the presence of Cl– or \({{{\mathrm{HCO}}}}_3^ -\). Nevertheless, it was concluded that \({{{\mathrm{NO}}}}_3^ -\) had a negligible effect on the degradation efficiency of both CBZ and TC. Also, Chen et al.142 confirmed the negative effect of a high concentration of Cl− on the degradation efficiency of sulfamethoxazole (SMX) by MgMnLDH-Cu-4/PMS. Such an effect could be attributed to the generation of \({\rm{Cl}}^{\bullet}\) from the interaction between \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and Cl−, which then combined with Cl− to produce \({{{\mathrm{Cl}}}}_2^{ \bullet - }\) that has a lower degradation ability than \({{{\mathrm{SO}}}}_4^{ \bullet - }\). In contrast, Zeng et al. study143 elucidated the positive impact of F− and Cl− on the SMX degradation since F− enhanced the release of more \(^ \bullet \!{{{\mathrm{OH}}}}\) at acidic pH, resulting in an amelioration in the degradation efficiency of SMX. However, there was a significant inhibition effect of F− on the degradation process at pH 9. The co-existing Cl− reacted with \(^ \bullet \!{{{\mathrm{OH}}}}\) to produce \({\rm{Cl}}^{\bullet}\) which had the same degradation ability as \(^ \bullet \!{{{\mathrm{OH}}}}\) as follows
In another investigation, Yang et al.61 fabricated a series of ComFen-LDHs with different molar ratios between Co and Fe through the combination of in situ exfoliation and co-precipitation methods. Such an innovative technique resulted in the formation of a thinner structure of ComFen-LDHs with high stability, prevented the leaching of Co and Fe, and increased the active sites. The XRD patterns showed an enhancement in the crystallinity of ComFen-LDHs with raising the Co2+ content. On the contrary, the increase in the Fe3+ ratio diminished the peak intensity of ComFen-LDHs, which may be attributed to the ability of Fe3+ of inhibiting the crystallization in acidic conditions. Furthermore, the low interlayer spacing (7.63) of Co1Fe1-LDHs compared to other fabricated LDHs reflected its thinner structure. Noteworthy, the increase in the Co2+ content improved the degradation efficiency of ciprofloxacin (CIP) up to 86.9%. Furthermore, it was found that the optimum pH of the CIP degradation by Co1Fe1-LDHs/PMS was in the range of 5–9. This finding could be explained by the leaching of Co2+ and Fe3+ at pH < 3, whereas at pH 11, there was an electrostatic repulsion force between the dominant species of PMS (\({{{\mathrm{SO}}}}_5^{2 - }\)) and the negatively charged surface of Co1Fe1-LDHs. Conversely, Zeng et al. study143 exhibited an enhancement in the SMX degradation at a pH range of 3–5, which may be attributed to the strong attraction between the positively charged surface of CoAlLDH@CoFe-PBA and the anionic species of PMS (\({{{\mathrm{HSO}}}}_5^ -\)). However, at pH ranging from 5 to 6.7, there was repulsion between SMX−, \({{{\mathrm{HSO}}}}_5^ -\), and the negatively charged surface of the catalyst. Also, the possibility of the reactive species’ inhibition (\({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\)) at high pH levels retards the degradation process of SMX. However, the improved degradation efficiency was recorded at higher pH, which indicated that there were other dominant reactive species. Consequently, it was essential to determine the controlling reactive species at different pH levels. It was concluded from the quenching experiments that 1O2 and \(^ \bullet \!{{{\mathrm{OH}}}}\) were responsible for the degradation process in an acidic medium, while \({{{\mathrm{SO}}}}_4^{ \bullet - }\) along with 1O2 were playing the major role in SMX degradation in an alkaline medium.
In one more attempt, Guo et al.144 fabricated Calotropis gigantean fiber (CGF) and polyvinylidene fluoride (PVDF) membraned-decorated CoCu-LDH to degrade SMX via the PMS activation. In the case of CoCu-LDH@CGF composite, the tubular structure of pure CGF did not change but the surface texture became rougher, while the pure PVDF showed a highly porous surface compared to CoCu-LDH@PVDF composite. The change in the porosity between LDH@PVDF composite and pure PVDF was also confirmed by the permeability test where the permeability of PVDF decreased from 170.61 to 62.39 L−1 h−1 m−2 bar−1 and also the porosity decreased from 37.42 to 21.41% after the combination with CoCu-LDH. It was monitored that PVDF/PMS system exhibited a degradation efficiency that was almost equal to adsorption efficiency, indicating that PVDF had not the ability to activate PMS. The CoCu-LDH@PVDF composite membrane’s stability was tested by reusing the membrane for ten cycles. Interestingly, it was found that the diminution in the SMX degradation efficiency was insignificant, and the changes in XRD pattern, the porosity, and the permeability of the used CoCu-LDH@PVDF after the 10th cycle were negligible. Notably, the Co2+ leaching percent from CoCu-LDH@CGF was larger than CoCu-LDH@PVDF, which may be attributed to the high concentration of the doped CoCu-LDH in CoCu-LDH@CGF (100 mg/L) compared to CoCu-LDH@PVDF (17.5 mg/L). Furthermore, CoCu-LDH was completely covered by a PVDF membrane, preventing Co2+ leaching. The degradation mechanism of SMX using CoCu-LDH@PVDF is summarized in Fig. 6a. In addition, the XPS spectra of CoCu-LDH before and after (Fig. 6b-f) the activation process showed a slight change in peak intensity and peak areas of C, O, Co, and Cu confirming the oxidation/reduction reactions on the surface of CoCu-LDH.

a The PMS activation b XPS survey, c C1s, d O1s e Cu2p and f Co2p, adapted with permission from ref. 144, Copyright, 2022, Elsevier.
In another study, Zeng et al.145 examined the catalytic activity of CoFe-LDH for the degradation of CBZ using the incorporation of Prussian blue analogues (PBA). The SEM and TEM images (Fig. 7a, b) of PBA showed its uniform cubic morphology. Furthermore, the SEM image Fig. 7c revealed the sheet-like morphology of PBA/CoFe-LDH, agreeing with what observed in the TEM image Fig. 7d. Furthermore, the energy dispersive spectroscopy (EDS) (Fig. 7e) indicated the presence of Co, Fe, N, and C in PBA/CoFe-LDH. The formation of the PBA/CoFe-LDH nanosheet may be attributed to electrostatic forces between the tightly bound regions of the LDH layers, which inhibited PBA growth.

a SEM of PBA, b TEM of PBA, c SEM of PBA/CoFe-LDH, d TEM of PBA/CoFe-LDH and e EDS of PBA/CoFe-LDH, adapted with permission from ref. 145, Copyright, 2022, RSC.
Firstly, PMS was used for the degradation of CBZ and there was no obvious effect on the concentration of CBZ. Similarly, PBA/CoFe-LDH was used in the absence of PMS, resulting in a quite low degradation efficiency of CBZ (<2%), but the efficiency increased to 31.8% using PBA/CoFe-LDH/PMS. Nevertheless, the degradation of CBZ by PBA/CoFe-LDH/PMS was slow, which could be accredited to the necessity of entering \({{{\mathrm{HSO}}}}_5^ -\) into the interlayers of PBA/CoFe-LDH to be activated. Whereas in most catalytic activated systems, \({{{\mathrm{HSO}}}}_5^ -\) could react with active sites on the surface of the catalyst, leading to a rapid generation of the active species. The high stability of PBA/CoFe-LDH was confirmed by its recycling for more than six cycles with no obvious change in the degradation efficiency. In addition, the XRD patterns of PBA/CoFe-LDH before and after the catalytic reaction showed a subtle difference. Such a high stability of PBA/CoFe-LDH is most likely due to the slight leaching percent of both Co (8.1 μg/L) and Fe ions (5.4 μg/L). Notably, different intermediates were produced throughout the degradation of CBZ, so the toxicity of CBZ and all intermediates were tested on different aquatic organisms including fish and daphnia. Some intermediates showed acute toxicity compared to CBZ towards fish, while others were quite toxic to daphnia.
In another investigation, He et al. studied the degradation mechanism of secnidazole (SNZ) by Co3Mn–LDH/PMS. It was reported that SNZ was hardly decomposed by the Co3Mn–LDH catalyzed PMS process in the presence of FFA, indicating that 1O2 played a significant role in the SNZ degradation process. The reaction was slightly repressed by TBA, while MeOH demonstrated a main inhibitory effect on the degradation process, denoting that both \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) were involving in the degradation reaction. The possible generation of \({{{\mathrm{O}}}}_2^{ \bullet - }\) was investigated by using p-BQ, and it was shown that \({{{\mathrm{O}}}}_2^{ \bullet - }\) had an essential role in the degradation process. Furthermore, when potassium dichromate (K2Cr2O7) was utilized as an e– quencher, the degradation dwindled by 78.8%, showing that the e– generated by Co3Mn–LDH was critical in attacking the peroxy bond of PMS for the production of the aforementioned ROS. Consequently, it was revealed that the degradation process was driven by both of the radical and the non-radical pathways. Besides, XPS spectra demonstrated that 8.3% of Co2+ and 8.4% of Mn3+ converted to Co3+ and Mn4+, respectively after the reaction, demonstrating their contribution in the production of \({{{\mathrm{SO}}}}_4^{ \bullet - }\), \(^ \bullet \!{{{\mathrm{OH}}}}\), \({{{\mathrm{O}}}}_2^{ \bullet - }\) and 1O2. However, it has to be noticed that high-valence Co3+ and Mn4+ are difficult to be reverted to a low-valence state, resulting in lower degradation efficiency in the recycling experiments. Consequently, He et al.137 endeavored to investigate the reusability of Co3Mn-LDH by the fabrication of 3D Co3Mn-LDH/rGA. Unexpectedly, the degradation efficiently of metronidazole (MTZ) by 3D Co3Mn-LDH/rGA/PMS was found to be above 67.0% after the 10th cycle. The formation of 3D Co3Mn-LDH/rGA was demonstrated by the abundant 3D pore network of rGA, in which abundant pores having an average size of about 2.0 nm were observed. This network could have provided broad pathways for the transfer of electrons. Additionally, it can be seen from the HRTEM image that the nanosized Co3Mn-LDH was collected on the rGA sheets. The pristine Co3Mn-LDH showed a weak photocurrent response (0.445 mA cm−2) under visible light irradiation. However, the photocurrent response increased obviously to 0.632 and 1.02 mA cm−2 after loading Co3Mn-LDH on 2D rGO and 3D rGA, respectively, indicating the improved separation efficiency of photo-induced carriers146. In addition, the photocurrent response of Co3Mn-LDH/rGA with 3D graphene structure was figured out to be higher than that of Co3Mn-LDH/rGO, which may be ascribed to the abundance of e− transfer channels in 3D graphene aerogels.
In another investigation, Ma et al.147 prepared a composite of Mg-Fe LDH@biochar for the degradation of doxycycline (DOX). The distinctive peaks of metal-oxygen linkages (MO and O-MO, where M = Mg and Fe) manifested clearly between 1000 cm−1 and 450 cm−1148. These metal–oxygen linkages of the LDH and oxygen-containing functional groups on the biochar surface may play an essential role in DOX degradation. Additionally, a highly graphitized carbon structure with a number of lamellar could increase charge transfer and enhance DOX degradation that reached 88.76% in 120 min at pH 7 through the non-radical pathway. Moreover, Guo et al.149 investigated the degradation of Lomefloxacin (LOM) utilizing Co-Cu-LDH composite, and the efficiency was 96.19% in 30 min at pH 6.67. Such a result was attributed to the presence of many active sites on the surface of Co-Cu-LDH. Furthermore, CBZ was totally degraded (100%) in just 20 min at pH 5.8 by CoMgFe-LDO catalyst48. Such an excellent efficiency was assigned to the synergistic effect between Co and Fe active sites in the PMS solution. Also, it has to be noticed that \({{{\mathrm{SO}}}}_4^{ \bullet - }\), 1O2, and \(^ \bullet \!{{{\mathrm{OH}}}}\) were responsible for LOM degradation, while in the case of DOX removal, the non-radical pathway was predominant.
To sum up, this section discussed the recent research articles investigating the degradation of pharmaceuticals via the activation of PMS by LDH-based catalysts. The commonly adopted preparation approaches for preparing LDH are hydrothermal, and co-precipitation methods, followed by modification methods like transition metals immobilization, calcination, and in situ etching by sodium sulfide or ZIF-67.
Surprisingly, it was found that the degradation efficacy of the most common residual contaminants; TC, DOX, IBA, SMX, SCP, CBZ, LOM, and CNZ was in the range of 86–100%. To fulfill such propitious results, several modification techniques have been applied including the formation of carbonaceous substances/LDH composites, which provide more electrons in the catalyst/PMS system. Furthermore, the preparation of layered triple hydroxides (LTH) such as MgCuFe, CoFeNi, and CoFeLa widely provides more electrons, enhances the generated ROS, and improve the oxidation/reduction cycle. Also, the incorporation of transition metals (Co, Cu, etc.) into LDHs enhanced the catalytic activity of LDHs toward pharmaceuticals’ degradation. In addition, LDO can be considered an easy, simple, costless, and effective modification method for LDHs as it was mentioned in the previous section. Notably, the calcined LTH exhibited advanced degradation efficiency toward the pharmaceutical residues and good reusability. On the other hand, the fabrication of LDH membranes showed auspicious enhancements in the reusability, durability, and catalytic activity of LDHs. Such a conclusion is based on the fact that the degradation efficiency of the pharmaceutical contaminants exceeded 92% along with a subtle decrease in the efficiency in the range of 0.5–3.1% after 10 cycles of regeneration.
Regarding the degradation mechanism of the pharmaceutical contaminants via the activation of PMS by LDHs-based catalysts, it was observed that the radical pathway is the most dominant mechanism. However, in rare cases, it was found that the degradation was driven by both radical and non-radical pathways, since the applied characterization tools and quenching test clarified the contribution of \({{{\mathrm{SO}}}}_4^{ \bullet - }\), \(^ \bullet \!{{{\mathrm{OH}}}}\), \({{{\mathrm{O}}}}_2^{ \bullet - }\), and 1O2 in the degradation process. The recent studies regarding the degradation of pharmaceuticals through the PMS activation process by LDH composites are listed in Table 2.
Degradation of organic dyes
Unlike the traditional Fenton oxidation process, PMS activation has been considered a powerful and cost-effective approach for the degradation of persistent complex structures such as organic dyes owing to the strong oxidizing power of \({{{\mathrm{SO}}}}_4^{ \bullet - }\), its long lifetime, and excellent activity of the ROS over a wide pH range150,151. Recently, transition metal-based LDHs, particularly, cobalt-based LDHs showed a thermodynamically favorable behavior in the PMS activation. Such a behavior was based on the suitable redox potential position of Co (II)\Co (III) (1.8 V) between \({{{\mathrm{HSO}}}}_5^ -\)\\({{{\mathrm{SO}}}}_5^{ \bullet - }\) (1.1 V) and \({{{\mathrm{HSO}}}}_5^ -\)\\({{{\mathrm{SO}}}}_4^{ \bullet - }\) (2.6–3.1 V), resulting in a remarkable degradation efficiency of various dyes152,153. For instance, a NiCo-LDH derived from the in situ etching of Ni-MOF template was tested for the PMS activation63. TEM images confirmed the successful synthesis of NiCo-LDH sheets from MOF microspheres. An optimal concentration of PMS (10−5 M) allowed NiCo-LDH to degrade reactive red-120 dye (RR-120) with an astounding efficiency of 89% within 10 minutes. Notably, the radical quencher experiment showed that both \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) free radicals were mainly responsible for the effective degradation of RR-120. Moreover, NiCo-LDH accomplished an excellent degradation efficiency of 97% under neutral conditions compared with acidic and alkaline conditions. This behavior was attributed to the protons scavenging for both \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) under the acidic conditions154. In addition, the insufficient contact between NiCo-LDH surface and PMS due to the large number of OH− formed surface complexes with LDH under alkaline conditions36. Similarly, Zhao et al. demonstrated a rapid and a complete degradation of acid orange G (AOG) within 240 seconds under natural conditions (pH = 6.87) using Co-Mn LDH36. Such a result was primarily attributed to the generation of \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) free radicals, which had a strong oxidizing power towards the AOG decomposition. This assumption was further verified through the radical scavenger experiment that showed a significant inhibition of the AOG degradation below 50% after the addition of 170 mM of ethanol (EtOH) and TBA, confirming that \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) were the main active species. More importantly, the XPS analysis exhibited an obvious shift of the Co2p1/2 and Co2p3/2 peaks from 796.8 eV and 781.1 eV to 796.6 eV and 780.5 eV, respectively, after the catalytic process. This finding was assigned to the coexistence of Co2+ and Co3+ due to the thermodynamically suitable standard reduction potential of Co3+\Co2+ (1.81 V) relative to that of \({{{\mathrm{HSO}}}}_5^ -\)\\({{{\mathrm{SO}}}}_5^{ \bullet - }\) (1.1 V), resulting in a feasible reduction of Co2+ into Co3+ by \({{{\mathrm{HSO}}}}_5^{ \bullet - }\). In addition, the atomic ratio of Mn3+/Mn4+ revealed a significant decline from 71.1: 28.9 to 55.2: 44.8 with a slight shift of the Mn 2p3/2 peaks from 641.9 eV and 643.7 eV to 641.6 eV and 643.1 eV, respectively, confirming the generation of Mn4+ through the catalytic reaction155. Thus, based on the XPS investigation and the radical scavenger experiments, the plausible mechanism of PMS activation was postulated as shown in the following equations: Firstly, the metal cations (Co2+ and Mn3+) converted \({{{\mathrm{HSO}}}}_5^ -\) into \({{{\mathrm{SO}}}}_4^{ \bullet - }\). Subsequently, \({{{\mathrm{SO}}}}_4^{ \bullet - }\) oxidized H2O molecules and produced \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals. Afterwards, the generated ROS (\({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\)) mineralized AOG into CO2 and H2O. In addition, \({{{\mathrm{SO}}}}_5^{ \bullet - }\) contributed to the AOG degradation reaction during the recovery of high valence state metals (Mn4+ and Co3+).
According to the above reactions, the higher valence species of Co3+ and Mn4+ could be regenerated into Co2+ and Mn3+ through the reduction process by \({{{\mathrm{HSO}}}}_5^ -\), propagating the redox chain reactions156. Also, MnFe-LDH accomplished a fascinating degradation efficiency of acid orange 7 (AO7) of 97.56% in 30 min under optimum conditions of pH = 6.1, catalyst dosage = 0.2 g\L, and PMS dosage = 0.2 g\L157. Such an outstanding result was accompanied with the electronic synergy between the redox couples of Fe3+\Fe2+ (0.77 V) and Mn3+\Mn2+ (1.51 V) through the efficient charge transfer from Fe2+ to Mn3+ to generate Fe3+ and Mn2+. Consequently, \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals were generated and employed in the efficient degradation of AO7. MnFe-LDH exhibited a low degradation efficiency of 41.13% in an acidic medium due to the observable leaching of the metal active sites (i.e., Mn = 4.49 mg/L, Fe = 0.11 mg/L). Thus, the PMS activation process was suppressed as indicated by Eqs. 37 and 38.
In another attempt, a magnetically separable Fe3O4@CoFe-LDH was prepared by the co-precipitation method for the degradation of AO7158. The magnetic core served as an electron donor to boost the transferred charges to the metal active sites, activating the Co(II)/Co(III) redox chain, and accelerating the generation of \({{{\mathrm{SO}}}}_4^{ \bullet - }\) radicals for better catalytic degradation of AO7. Therefore, the observed result was an excellent degradation performance of 95.1% after 15 minutes. Furthermore, Fe3O4@CoFe-LDH demonstrated potential heterogeneous catalytic applicability in real wastewater, inferring its outstanding stability with a degradation efficiency of 88.7% after 5 cycles of regeneration.
Within the same aspect, Li et al.159 designed an innovative and scalable strategy for the efficient catalytic degradation of methylene blue (MB). Their work was based on an in situ facile hydrothermal growth of CoFeLDH onto PVDF foam assisted by the addition of NH4F forming 3D porous foam. Notably, SEM images depicted the formation of needle-like CoFeLDH nanoarrays onto PVDF after the addition of NH4F due to the formation of F-metal coordinate bonds, providing myriad numbers of active sites to accelerate the PMS activation. Accordingly, CoFeLDH(F)/PVDF nanoarrays achieved catalytic degradation efficiency against methylene blue (MB) two times higher than CoFeLDH/PVDF nanosheets. This spectacular result could be attributed to the high porosity and super hydrophilicity that facilitated the mass transfer and the interaction of MB with the LDH surface. Additionally, the F-Metal coordinate interaction accelerated the valence state changes of the metal active sites. Consequently, the efficient generation of \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\) free radicals was promoted for a rapid de-colorization of wastewater. Interestingly, CoFeLDH(F)/PVDF nanoarrays revealed enhanced ESR signals corresponding to \({{{\mathrm{SO}}}}_4^{ \bullet\! - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals comparable to CoFeLDH/PVDF nanosheets, indicating their effective role in generating free radicals. According to the EPR measurements and the radical scavenging experiments, \({{{\mathrm{SO}}}}_4^{ \bullet \!- }\) and 1O2 were concluded to be the main degrading species as confirmed by the reduced degradation efficiencies to 45.7% and 53.5% after the addition of FFA and methanol, respectively. The PMS catalytic activation mechanism was suggested in the following equations. Firstly, sulfate radicals were produced via the redox reactions of Fe (II) and Co (II), as elucidated in Eqs. 40 and 41. Secondly, a sequential generation of ROS including 1O2, \(^ \bullet \!{{{\mathrm{OH}}}}\), and \({{{\mathrm{O}}}}_2^{ \bullet\! - }\) occurred (Eqs. 44–47) during the reduction of high valence state cations Fe (III) and Co (III).
Surprisingly, under optimal conditions, CoFeLDH(F)/PVDF nanoarrays demonstrated a fantastic catalytic degradation efficiency of 99.56% in a dynamically packed column reaction system after the 10th cycle in 5 minutes. Furthermore, CoFeLDH(F)/PVDF achieved good catalytic degradation efficiencies against several organic pollutants such as Rhodamine B (RhB) (100%), TC (98.59%), BPA (86.65%), and atrazine (ATZ) (45.84%), verifying its remarkable catalytic efficacy and applicability in real wastewater treatment.
In this respect, Cheng et al.153 also used FeCo-LDH to degrade RhB via PMS activation. FeCo-LDH was fabricated in a metal molar ratio of 1:2 for Fe: Co. Different quenching experiments were established by using two radical scavengers with different concentrations: TBA for \(^ \bullet \!{{{\mathrm{OH}}}}\) and ethanol for \(^ \bullet \!{{{\mathrm{OH}}}}\), \({{{\mathrm{SO}}}}_4^{ \bullet - }\), and \({{{\mathrm{SO}}}}_5^{ \bullet - }\). It was concluded that the degradation process mainly depends on sulfate radicals, whereas increasing the concentration of TBA had no significant effect on the RhB degradation efficiency. Such a result was further confirmed by observing a substantial drop in the degradation efficiency upon increasing the concentration of ethanol. As shown in Fig. 8a, \({{{\mathrm{SO}}}}_4^{ \bullet - }\) radicals were generated from the PMS activation by the oxidation of Fe (II) to Fe (III) and the reduction of Co (III) to Co (II). While \({{{\mathrm{SO}}}}_5^{ \bullet - }\) was produced by the activation of PMS in presence of Co (III), and \(^ \bullet \!{{{\mathrm{OH}}}}\) was generated from the interaction between \({{{\mathrm{SO}}}}_4^{ \bullet - }\) and water molecules. Finally, the produced free radicals attacked the RhB molecules and degraded them into CO2 and H2O as clarified in Fig. 8b. The feature that made PMS/FeCo-LDH highly efficient in the catalytic degradation process was the distinctive structure of LDH, which can keep the same structure due to the reversible oxidation and reduction of Co (II) and Fe (III).

a The degradation mechanism of RhB, b the PMS activation by CoFe-LDH adapted with permission from ref. 153, Copyright, 2022, Elsevier. c The schematic diagram of MgCoAl-LDH nanosheets and nanoscrolls, d TEM of MgCoAl-LDH nanosheets, e TEM of MgCoAl-LDH nanoscrolls, and f XRD of MgCoAl-LDH nanosheets and nanoscrolls adapted with permission from ref. 160, Copyright, 2022, Elsevier.
In another study, Sun et al.160 inspected the catalytic efficiency of both MgCoAl-LDH nanosheets and MgCoAl-LDH nanoscrolls towards the degradation of MB via the PMS activation (Fig. 8c). TEM images (Fig. 8d, e) revealed the hexagonal structure of MgCoAl-LDH nanosheets and the curl structure of MgCoAl-LDH nanoscrolls. While XRD patterns showed that there was no difference between the crystallite phases of LDH nanoscrolls and nanosheets in terms of the position and the intensity as illustrated in Fig. 8f. It was reported that MgCoAl-LDH nanoscrolls achieved a complete degradation of MB within 40 min compared to MgCoAl-LDH nanosheets that achieved only 72% under the same reaction conditions. The superior catalytic performance of MgCoAl-LDH nanoscrolls was clearly attributed to their high surface area (63.49 m2 g−1) compared to that of MgCoAl-LDH nanosheets (15.43 m2 g−1). It is noteworthy that the radical quenching experiments revealed that 1O2 was the main active species, showing a remarkable decline in kobs value by 90.2% after the addition of 1 mM L-histidine that was used as a trapping agent for 1O2. Additionally, the XPS spectrum survey confirmed the predominant role of Co3+/Co2+ redox cycle in the PMS activation. Such a role was described by the diminished intensity of the characteristic peak of Co3+ due to the reduction by PMS accompanied by a remarkable increase in the intensity of the peak corresponding to Co2+. On the contrary, the peaks corresponding to Mg1s and Al2p at 1303.8 eV and 74.3 eV, respectively, did not show any change after the catalytic reaction, indicating their insignificant role in the PMS activation. More importantly, the influence of co-existing anions such as \({{{\mathrm{NO}}}}_3^ -\), \({{{\mathrm{SO}}}}_4^{2 - }\), Cl−, \({{{\mathrm{HCO}}}}_3^ -\), \({{{\mathrm{CO}}}}_3^{2 - }\), and dihydrogen phosphate (H2PO4−) on the catalytic degradation of MB was inspected. Even at high concentrations of these interfering ions, the presence of \({{{\mathrm{NO}}}}_3^ -\) and \({{{\mathrm{SO}}}}_4^{2 - }\) had a minor effect on the degradation efficiency of MB. While the degradation efficacy significantly reduced by increasing the concentration of Cl− and \({{{\mathrm{HCO}}}}_3^ -\) due to their reaction with \({{{\mathrm{SO}}}}_4^{ \bullet - }\), forming \({{{\mathrm{Cl}}}}_2^{ \bullet - }\) and \({{{\mathrm{HCO}}}}_3^ \bullet\), respectively. These formed species had lower oxidizing power, which in turn suppressed the degradation efficiency of MB. Furthermore, it is worth mentioning that the concentration of \({{{\mathrm{CO}}}}_3^{2 - }\) obviously controlled the degradation efficiency of MB. The dramatic decrease in the degradation efficiency at a low concentration of \({{{\mathrm{CO}}}}_3^{2 - }\) (0.5 mM) was due to the change in the pH medium, which resulted in the conversion of \({{{\mathrm{CO}}}}_3^{ \bullet - }\) to \({{{\mathrm{HCO}}}}_3^ \bullet\) that had a low oxidation potential. However, at a high concentration of \({{{\mathrm{CO}}}}_3^{2 - }\) (10 mM), the degradation efficiency increased as a result of generating a large amount of \(^ \bullet \!{{{\mathrm{OH}}}}\) that had excellent oxidizing power. Such an assumption was further verified by EPR measurements, which showed a noticeable increase in the \(^ \bullet \!{{{\mathrm{OH}}}}\) signals with higher concentrations of \({{{\mathrm{CO}}}}_3^{ \bullet - }\) comparable to the 1O2 EPR signals that showed no change in the signals’ intensities with different concentrations. Additionally, EPR measurements manifested that the concentration of 1O2 increased gradually by raising the concentration of H2PO4−, which reduced the dissociation energies of O-O bonds in PMS. Therefore, the PMS activation was accelerated, leading to the enhancement of the degradation efficiency.
Shortly, this section summarizes the recent assessments of various anionic and cationic dyes’ degradation through the activation of PMS by LDH-based catalysts (Table 2). The most common methods for producing the used LDH-based catalysts were hydrothermal and co-precipitation. Notably, the investigated PMS/LDH-based catalysts exhibited an enhanced degradation efficiency toward RR-120, AO7, Congo red (CR), AOG, MB, and RhB dyes, with almost 100% in some cases. LDHs were modified using magnetic nanoparticles (Fe3O4), particularly to improve their regeneration. Additionally, the efficiency was further enhanced owing to the contribution of Fe ions in the PMS activation and the increased production of ROS. In some cases, the degradation efficacy declined by just 1% after 10 cycles of regenration. A conclusion that is in line with what was concluded in the previous sections of LDO and LTH, which revealed an advanced degradation performance toward multiple dyes along with a good reusability. Noteworthy, the variable morphology of the produced catalytic materials was quite effective on the degradation efficiency of dyes. For example, CoFeLDH(F)/PVDF nanoarrays showed a degradation percent of MB two times higher than CoFeLDH/PVDF nanosheets. In addition, the superior catalytic performance of MgCoAl-LDH nanoscrolls compared to nanosheets was based on the observed surface area of MgCoAl-LDH nanoscrolls, which was four times higher than that of MgCoAl-LDH nanosheets.
The investigation of the co-existing \({{{\mathrm{CO}}}}_3^{2 - }\) impact in the reaction system revealed a dramatic decrease in the degradation efficiency of dyes at low concentrations of \({{{\mathrm{CO}}}}_3^{2 - }\). Such a decrease was accredited to the changing pH levels, resulting in the conversion of \({{{\mathrm{CO}}}}_3^{ \bullet - }\) to \({{{\mathrm{HCO}}}}_3^ \bullet\) that had a low oxidation potential. In contrast, the degradation efficiency improved at high concentrations of \({{{\mathrm{CO}}}}_3^{2 - }\) as a result of generating a large amount of \(^ \bullet \!{{{\mathrm{OH}}}}\), which had an excellent oxidizing power.
Most of the reported studies clarified that the degradation of dyes using the activation of PMS by LDH-based catalysts proceeded via the radial pathway mechanism as follows;
-
Firstly, the transition metal cations converted \({{{\mathrm{HSO}}}}_5^ -\) into \({{{\mathrm{SO}}}}_4^{ \bullet - }\)
-
Secondly, \({{{\mathrm{SO}}}}_4^{ \bullet - }\) oxidized H2O molecules, releasing \(^ \bullet \!{{{\mathrm{OH}}}}\) radicals.
-
Finally, the generated ROS (\({{{\mathrm{SO}}}}_4^{ \bullet - }\) and \(^ \bullet \!{{{\mathrm{OH}}}}\)) mineralized dyes into CO2 and H2O.
In conclusion the recent progress of PMS-based AOPs that represent a revolution in the field of catalysis, focusing on LDH-based catalysts owing to their advanced catalytic performance, excellent stability, and reusability are summarized in this review. The remarkable catalytic activity of LDHs towards PMS activation resulted in an excellent degradation efficiency of various organic contaminants, which could be reaching 100% owing to the exceptional properties of LDH-based catalysts. In addition, the efficient reusability of LDH-based catalysts has been confirmed as the documented degradation efficiency was more than 80% after many cycles of regeneration in the majority of research studies. Noteworthy, it was concluded that the combination of LDHs with some materials like polyvinylidene fluoride, Prussian blue analogues, biochar, and Calotropis gigantean fiber could improve the chemical stability of LDHs and directly ameliorate its reusability. Most of the reported studies investigated the leaching amounts of the metal-containing LDHs after the PMS activation, and reported that the leaching amounts were less than the allowed limits. The common preparation routes were thoroughly discussed, clarifying the merits and demerits of each one, and elucidating some techniques to enhance the produced LDHs. Such a discussion was based on the crucial influence of the synthesis approach of LDHs on their characteristics and catalytic efficiency. The co-precipitation approach was determined to be suitable for the production of LDHs because of its simple processing, fast rate, and high yield. The ratio of M2+/M3+ is another important factor that affects the LDHs’ characteristics such as surface area. Moreover, the calcined LDHs were found to possess a higher surface area along with an enhanced catalytic activity and reusability compared to the non-calcined ones. In addition, LDOs are characterized with other advanced properties compared to LDHs, including well dispersion and easy separation, rendering them highly efficient catalysts for PMS activation and even more efficient than LDHs.
All in all, the degradation of organic contaminants through PMS activation by LDH-based catalysts offers many advantages to overcome the common limitations of PMS-AOPs including:
-
1.
LDHs can attract the negatively-charged PMS to its surface via the electrostatic interactions owing to its large pHPZC, promoting the generation of ROS and improving the degradation efficiency of organic contaminants.
-
2.
The remarkable stability of LDHs can solve one of the serious issues that hinder PMS-AOPs to be applied on an industrial scale, which is the low stability of the majority of catalysts. In general, the acidic medium in the PMS process could increase the metals’ leaching and collapse the structure of the catalyst. However, the large pHPZC of LDHs could offer a pH buffer that prevents the formation of an acidic medium.
Nonetheless, LDHs have more aspects to go through and still need to be further investigated, so the main recommendations are:
-
1.
Developing the green synthesis routes of LDHs using benign materials and consuming low energy. Although there are many studies reported the fabrication of LDHs in water as a benign solvent, the procedures were based on using toxic materials such as urea, ethylene glycol, dimethyl sulfoxide, etc.161,162. Consequently, several investigations should be conducted to find out alternative and eco-friendly materials.
-
2.
Improving the crystallinity of LDHs that constitutes a critical challenge to the researchers in characterizing LDHs. Recently-synthesized materials such as LDHs and MOFs have no standard and researchers depend on previous studies that have some differences. Notably, it was found that the increase in the pH of the metal precursors significantly enhances the crystallinity of the obtained LDHs at a highly basic medium (pH 10–13.2)163. Therefore, further assessments of the preparation controlling factors are highly recommended.
-
3.
Although several research studies confirmed the low amount of metals’ leaching, the accumulation of these metals in water bodies definitely affects human health and the aquatic environment. Meanwhile, some studies revealed a high metal leaching concentration at a highly acidic medium (pH ≤ 3)61,101. Thereby, producing LDHs with controlled metal leaching at highly acidic medium still needs to be further investigated.
-
4.
Determining the optimal ratio of M2+/M3+ as it is a missing factor in the majority of research articles. It was reported that the increase in the Cu concentration in NiCu-LDH, results in Jahn–Teller distortion164. Hence, adjusting the M2+/M3+ ratio is an essential factor that should be considered during the fabrication of LDHs.
-
5.
Targeting the utilization of real wastewater samples as there are few studies addressing this issue.
-
6.
Considering the economic aspect by determining the cost of LDHs’ synthesis to design a comprehensive feasibility study.
Data availability
Data available upon request and any requests for materials should be addressed to Abdelazeem S. Eltaweil.
References
Jiang, X. et al. Ultrasound and microwave-assisted synthesis of copper-activated carbon and application to organic dyes removal. Powder Technol. 338, 857–868 (2018).
Hosny, M., Fawzy, M. & Eltaweil, A. S. Green synthesis of bimetallic Ag/ZnO@ Biohar nanocomposite for photocatalytic degradation of tetracycline, antibacterial and antioxidant activities. Sci. Rep. 12, 1–17 (2022).
Abd El-Monaem, E. M. et al. Sustainable adsorptive removal of antibiotic residues by chitosan composites: An insight into current developments and future recommendations. Arab. J. Chem. 15, 103743 (2022).
Hassan, S. S., Abdel Rahman, E. M., El-Subruiti, G. M., Kamel, A. H. & Diab, H. M. Removal of Uranium-238, Thorium-232, and Potassium-40 from Wastewater via Adsorption on Multiwalled Carbon Nanotubes. ACS Omega 7, 12342–12353 (2022).
Ali, S. H., Emran, M. Y. & Gomaa, H. in Waste recycling technologies for nanomaterials manufacturing 541–588 (Springer, 2021).
Diab, M., Attia, N. F., Attia, A. & El-Shahat, M. Green synthesis of cost-effective and efficient nanoadsorbents based on zero and two dimensional nanomaterials for Zn2+ and Cr3+ removal from aqueous solutions. Synth. Met. 265, 116411 (2020).
Qin, X. et al. Preparation of double functional carbon-based ZnO derived from rape straw for dye wastewater treatment. J. Water Process. Eng. 52, 103588 (2023).
Sun, J. A., Wang, L., Wang, Y., Lv, W. & Yao, Y. Activation of peroxymonosulfate by MgCoAl layered double hydroxide: Potential enhancement effects of catalyst morphology and coexisting anions. Chemosphere 286, 131640 (2022).
Hosny, M., Fawzy, M. & Eltaweil, A. S. Phytofabrication of bimetallic silver-copper/biochar nanocomposite for environmental and medical applications. J. Environ. Manag. 316, 115238 (2022).
Fathalla, A. R. & Mohamed, E. Copper and lead ions removal from aqueous solutions case study: fly ash carbon as low-cost effective sorbent. Egypt. J. Chem. 65, 1–4 (2022).
Kassem, K. O. et al. Design of mesoporous ZnO@ silica fume-derived SiO2 nanocomposite as photocatalyst for efficient crystal violet removal: Effective route to recycle industrial waste. J. Clean. Prod. 326, 129416 (2021).
Eltaweil, A. S., Abdelfatah, A. M., Hosny, M. & Fawzy, M. Novel Biogenic Synthesis of a Ag@ biochar nanocomposite as an antimicrobial agent and photocatalyst for methylene blue degradation. ACS Omega 7, 8046–8059 (2022).
El-Maghrabi, N., El-Borady, O. M., Hosny, M. & Fawzy, M. Catalytic and medical potential of a phyto-functionalized reduced graphene oxide–gold nanocomposite using willow-leaved knotgrass. ACS Omega 6, 34954–34966 (2021).
Ghiyasiyan-Arani, M., Salavati-Niasari, M. & Naseh, S. Enhanced photodegradation of dye in waste water using iron vanadate nanocomposite; ultrasound-assisted preparation and characterization. Ultrason. Sonochem. 39, 494–503 (2017).
Eltaweil, A. S., Hashem, O. A., Abdel-Hamid, H., Abd El-Monaem, E. M. & Ayoup, M. S. Synthesis of a new magnetic Sulfacetamide-Ethylacetoacetate hydrazone-chitosan Schiff-base for Cr (VI) removal. Int. J. Biol. Macromol. 222, 1465–1475 (2022).
Abdelfatah, A. M., El-Maghrabi, N., Mahmoud, A. E. D. & Fawzy, M. Synergetic effect of green synthesized reduced graphene oxide and nano-zero valent iron composite for the removal of doxycycline antibiotic from water. Sci. Rep. 12, 1–19 (2022).
Gomaa, H. et al. A hybrid spongy-like porous carbon-based on azopyrazole-benzenesulfonamide derivative for highly selective Fe3+-adsorption from real water samples. Microporous Mesoporous Mater. 330, 111578 (2022).
Cheng, S. et al. Preparation of magnetic adsorbent-photocatalyst composites for dye removal by synergistic effect of adsorption and photocatalysis. J. Clean. Prod. 348, 131301 (2022).
Hussein, M. A., Motawea, M. M., Elsenety, M. M., El-Bahy, S. M. & Gomaa, H. Mesoporous spongy Ni–Co oxides@ wheat straw-derived SiO2 for adsorption and photocatalytic degradation of methylene blue pollutants. Appl. Nanosci. 12, 1519–1536 (2022).
Gomaa, H. et al. A hybrid mesoporous CuO@ barley straw-derived SiO2 nanocomposite for adsorption and photocatalytic degradation of methylene blue from real wastewater. Colloids Surf. A Physicochem. Eng. Asp. 644, 128811 (2022).
Crini, G. & Lichtfouse, E. Advantages and disadvantages of techniques used for wastewater treatment. Environ. Chem. Lett. 17, 145–155 (2019).
Sakr, A. K. et al. Removal of uranium from nuclear effluent using regenerated bleaching earth steeped in β–naphthol. Radiat. Phys. Chem. 200, 110204 (2022).
Li, Z. & Yang, P. in IOP Conference Series: Earth and Environmental Science. 012185 (IOP Publishing).
Basha, I. K. et al. Sulfonated graphene oxide impregnated cellulose acetate floated beads for adsorption of methylene blue dye: optimization using response surface methodology. Sci. Rep. 12, 1–17 (2022).
Rajendran, S., Shahbaz, K. & Walvekar, R. A fundamental study on solubility of heavy metal oxides in ammonium and phosphonium based deep eutectic solvents. J. Eng. Sci. Technol. 2, 85–86 (2016).
Eltaweil, A. S., Abdelfatah, A. M., Hosny, M. & Fawzy, M. Novel biogenic synthesis of a Ag@ Biochar nanocomposite as an antimicrobial agent and photocatalyst for methylene blue degradation. ACS Omega 7, 8046–8059 (2022).
Leong, S. et al. TiO2 based photocatalytic membranes: A review. J. Membr. Sci. 472, 167–184 (2014).
Samsami, S., Mohamadizaniani, M., Sarrafzadeh, M.-H., Rene, E. R. & Firoozbahr, M. Recent advances in the treatment of dye-containing wastewater from textile industries: Overview and perspectives. Process Saf. Environ. Prot. 143, 138–163 (2020).
Gomaa, H., El-Monaem, A., Eman, M., Eltaweil, A. S. & Omer, A. M. Efficient removal of noxious methylene blue and crystal violet dyes at neutral conditions by reusable montmorillonite/NiFe2O4@ amine-functionalized chitosan composite. Sci. Rep. 12, 1–16 (2022).
Liu, L. et al. Treatment of industrial dye wastewater and pharmaceutical residue wastewater by advanced oxidation processes and its combination with nanocatalysts: A review. J. Water Process. Eng. 42, 102122 (2021).
Eltaweil, A. S. et al. Recent developments in alginate-based adsorbents for removing phosphate ions from wastewater: a review. RSC Adv. 12, 8228–8248 (2022).
Monsef, R. & Salavati-Niasari, M. Electrochemical sensor based on a chitosan-molybdenum vanadate nanocomposite for detection of hydroxychloroquine in biological samples. J. Colloid Interface Sci. 613, 1–14 (2022).
Gholami, T., Salavati-Niasari, M. & Varshoy, S. Electrochemical hydrogen storage capacity and optical properties of NiAl2O4/NiO nanocomposite synthesized by green method. Int. J. Hydrog. Energy 42, 5235–5245 (2017).
Eltaweil, A. S. et al. Graphene oxide incorporated cellulose acetate beads for efficient removal of methylene blue dye; isotherms, kinetic, mechanism and co-existing ions studies. J. Porous Mater. 30, 1–12 (2022).
Seaf El-Nasr, T. A., Gomaa, H., Emran, M. Y., Motawea, M. M. & Ismail, A.-R. A. in Waste recycling technologies for nanomaterials manufacturing 325-362 (Springer, 2021).
Zhao, X. et al. Co-Mn layered double hydroxide as an effective heterogeneous catalyst for degradation of organic dyes by activation of peroxymonosulfate. Chemosphere 204, 11–21 (2018).
Du, X. & Zhou, M. Strategies to enhance catalytic performance of metal–organic frameworks in sulfate radical-based advanced oxidation processes for organic pollutants removal. Chem. Eng. J. 403, 126346 (2021).
Hosny, M. et al. Facile synthesis of gold nanoparticles for anticancer, antioxidant applications, and photocatalytic degradation of toxic organic pollutants. ACS Omega 7, 3121–3133 (2022).
Monsef, R., Ghiyasiyan-Arani, M. & Salavati-Niasari, M. Application of ultrasound-aided method for the synthesis of NdVO4 nano-photocatalyst and investigation of eliminate dye in contaminant water. Ultrason. Sonochem. 42, 201–211 (2018).
Amiri, M., Eskandari, K. & Salavati-Niasari, M. Magnetically retrievable ferrite nanoparticles in the catalysis application. Adv. Colloid Interface Sci. 271, 101982 (2019).
Hu, P. & Long, M. Cobalt-catalyzed sulfate radical-based advanced oxidation: a review on heterogeneous catalysts and applications. Appl. Catal. B-Environ. 181, 103–117 (2016).
Xiong, Z., Jiang, Y., Wu, Z., Yao, G. & Lai, B. Synthesis strategies and emerging mechanisms of metal-organic frameworks for sulfate radical-based advanced oxidation process: A review. Chem. Eng. J. 421, 127863 (2021).
Hosny, M., Fawzy, M., El-Badry, Y. A., Hussein, E. E. & Eltaweil, A. S. Plant-assisted synthesis of gold nanoparticles for photocatalytic, anticancer, and antioxidant applications. J. Saudi Chem. 26, 101419 (2022).
Zhao, Q. et al. Metal-free carbon materials-catalyzed sulfate radical-based advanced oxidation processes: a review on heterogeneous catalysts and applications. Chemosphere 189, 224–238 (2017).
Xia, X. et al. A review study on sulfate-radical-based advanced oxidation processes for domestic/industrial wastewater treatment: degradation, efficiency, and mechanism. Front. Chem. 8, 1092 (2020).
Yousefi, S. R. et al. Dy2BaCuO5/Ba4DyCu3O9. 09 S‐scheme heterojunction nanocomposite with enhanced photocatalytic and antibacterial activities. J. Am. Ceram. Soc. 104, 2952–2965 (2021).
Giannakis, S., Lin, K.-Y. A. & Ghanbari, F. A review of the recent advances on the treatment of industrial wastewaters by Sulfate Radical-based Advanced Oxidation Processes (SR-AOPs). Chem. Eng. J. 406, 127083 (2021).
Hong, Y. et al. Heterogeneous activation of peroxymonosulfate by CoMgFe-LDO for degradation of carbamazepine: Efficiency, mechanism and degradation pathways. Chem. Eng. J. 391, 123604 (2020).
Ye, Q. et al. Enhancing peroxymonosulfate activation by Co-Fe layered double hydroxide catalysts via compositing with biochar. Chem. Eng. J. 417, 129111 (2021).
Chen, M. et al. Environmental application of MgMn-layered double oxide for simultaneous efficient removal of tetracycline and Cd pollution: performance and mechanism. J. Environ. Manag. 246, 164–173 (2019).
Ghanbari, F. & Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants. Chem. Eng. J. 310, 41–62 (2017).
Wang, W., Chen, M., Wang, D., Yan, M. & Liu, Z. Different activation methods in sulfate radical-based oxidation for organic pollutants degradation: Catalytic mechanism and toxicity assessment of degradation intermediates. Sci. Total Environ. 772, 145522 (2021).
Bouzayani, B., Rosales, E., Pazos, M., Elaoud, S. C. & Sanromán, M. A. Homogeneous and heterogeneous peroxymonosulfate activation by transition metals for the degradation of industrial leather dye. J. Clean. Prod. 228, 222–230 (2019).
Wang, L., Jiang, J., Ma, J., Pang, S. & Zhang, T. A review on advanced oxidation processes homogeneously initiated by copper (II). Chem. Eng. J. 427, 131721 (2022).
Zheng, X. et al. Metal-based catalysts for persulfate and peroxymonosulfate activation in heterogeneous ways: A review. Chem. Eng. J. 429, 132323 (2022).
Sun, J. et al. Peroxymonosulfate activation induced by spinel ferrite nanoparticles and their nanocomposites for organic pollutants removal: A review. J. Clean. Prod. 346, 131143 (2022).
Koo, P.-L., Jaafar, N. F., Yap, P.-S. & Oh, W.-D. A review on the application of perovskite as peroxymonosulfate activator for organic pollutant removal. J. Environ. Chem. Eng. 10, 107093 (2021).
Gao, Q. et al. Utilizing cobalt-doped materials as heterogeneous catalysts to activate peroxymonosulfate for organic pollutant degradation: a critical review. Environ. Sci. Water Res. Technol. 7, 1197–1211 (2021).
Jawad, A. et al. Fe-MoS4: an effective and stable LDH-based adsorbent for selective removal of heavy metals. ACS Appl. Mater. Interf. 9, 28451–28463 (2017).
Zeng, H. et al. Degradation of dyes by peroxymonosulfate activated by ternary CoFeNi-layered double hydroxide: Catalytic performance, mechanism and kinetic modeling. J. Colloid Interface Sci. 515, 92–100 (2018).
Yang, Z. et al. Facile synthesis of cobalt-iron layered double hydroxides nanosheets for direct activation of peroxymonosulfate (PMS) during degradation of fluoroquinolones antibiotics. J. Clean. Prod. 310, 127584 (2021).
Shahzad, A. et al. Interlayered modified hydroxides for removal of graphene oxide from water: Mechanism and secondary applications. Sep. Purif. Technol. 284, 120305 (2022).
Ramachandran, R. et al. Efficient degradation of organic dye using Ni-MOF derived NiCo-LDH as peroxymonosulfate activator. Chemosphere 271, 128509 (2021).
Ali, J. K. et al. Enhanced removal of aqueous phenol with polyimide ultrafiltration membranes embedded with deep eutectic solvent-coated nanosilica. Chem. Eng. J. 408, 128017 (2021).
Li, D. et al. Persulfate coupled with Cu2+/LDH-MoS4: A novel process for the efficient atrazine abatement, mechanism and degradation pathway. Chem. Eng. J. 436, 134933 (2022).
Dung, N. T., Thao, V. D. & Huy, N. N. Decomposition of glyphosate in water by peroxymonosulfate activated with CuCoFe‐LDH material. Vietnam J. Chem. 59, 813–822 (2021).
Ali, J. et al. Regulating the redox centers of Fe through the enrichment of Mo moiety for persulfate activation: A new strategy to achieve maximum persulfate utilization efficiency. Water Res 181, 115862 (2020).
Azalok, K. A., Oladipo, A. A. & Gazi, M. UV-light-induced photocatalytic performance of reusable MnFe-LDO–biochar for tetracycline removal in water. J. Photochem. Photobiol. A: Chem. 405, 112976 (2021).
Xie, Z.-H. et al. Synthesis, application and catalytic performance of layered double hydroxide based catalysts in advanced oxidation processes for wastewater decontamination: A review. Chem. Eng. J. 414, 128713 (2021).
Yang, Z. et al. Utilization of LDH-based materials as potential adsorbents and photocatalysts for the decontamination of dyes wastewater: a review. RSC Adv. 6, 79415–79436 (2016).
Wu, M. et al. A review on fabricating heterostructures from layered double hydroxides for enhanced photocatalytic activities. Catal. Sci. Technol. 8, 1207–1228 (2018).
Kohantorabi, M., Moussavi, G. & Giannakis, S. A review of the innovations in metal-and carbon-based catalysts explored for heterogeneous peroxymonosulfate (PMS) activation, with focus on radical vs. non-radical degradation pathways of organic contaminants. Chem. Eng. J. 411, 127957 (2021).
Mallakpour, S., Hatami, M. & Hussain, C. M. Recent innovations in functionalized layered double hydroxides: Fabrication, characterization, and industrial applications. Adv. Colloid Interface Sci. 283, 102216 (2020).
Chubar, N. et al. Layered double hydroxides as the next generation inorganic anion exchangers: Synthetic methods versus applicability. Adv. Colloid Interface Sci. 245, 62–80 (2017).
Mallakpour, S. & Hatami, M. Condensation polymer/layered double hydroxide NCs: preparation, characterization, and utilizations. Eur. Polym. J. 90, 273–300 (2017).
Evans, D. G. & Slade, R. C. Structural aspects of layered double hydroxides. Layered double hydroxides 119, 1–87 (2006).
Mohapatra, L. & Parida, K. A review on the recent progress, challenges and perspective of layered double hydroxides as promising photocatalysts. J. Mater. Chem. A 4, 10744–10766 (2016).
Shakeel, M., Arif, M., Yasin, G., Li, B. & Khan, H. D. Layered by layered Ni-Mn-LDH/g-C3N4 nanohybrid for multi-purpose photo/electrocatalysis: morphology controlled strategy for effective charge carriers separation. Appl. Catal. B-Environ. 242, 485–498 (2019).
Clark, I., Dunne, P. W., Gomes, R. L. & Lester, E. Continuous hydrothermal synthesis of Ca2Al-NO3 layered double hydroxides: The impact of reactor temperature, pressure and NaOH concentration on crystal characteristics. J. Colloid Interface Sci. 504, 492–499 (2017).
Panahi-Kalamuei, M., Alizadeh, S., Mousavi-Kamazani, M. & Salavati-Niasari, M. Synthesis and characterization of CeO2 nanoparticles via hydrothermal route. J. Ind. Eng. Chem. 21, 1301–1305 (2015).
Yousefi, S. R., Masjedi-Arani, M., Morassaei, M. S., Salavati-Niasari, M. & Moayedi, H. Hydrothermal synthesis of DyMn2O5/Ba3Mn2O8 nanocomposite as a potential hydrogen storage material. Int. J. Hydrog. Energy 44, 24005–24016 (2019).
Chatterjee, A., Bharadiya, P. & Hansora, D. Layered double hydroxide based bionanocomposites. Appl. Clay Sci. 177, 19–36 (2019).
Aregay, G. G., Jawad, A., Du, Y., Shahzad, A. & Chen, Z. Efficient and selective removal of chromium (VI) by sulfide assembled hydrotalcite compounds through concurrent reduction and adsorption processes. J. Mol. Liq. 294, 111532 (2019).
Conterosito, E. et al. Facile preparation methods of hydrotalcite layered materials and their structural characterization by combined techniques. Inorg. Chim. Acta 470, 36–50 (2018).
Ge, L. et al. Layered double hydroxide based materials applied in persulfate based advanced oxidation processes: Property, mechanism, application and perspectives. J. Hazard. Mater. 424, 127612 (2022).
Karmakar, A. et al. A vast exploration of improvising synthetic strategies for enhancing the OER kinetics of LDH structures: a review. J. Mater. Chem. A 9, 1314–1352 (2021).
Dewangan, N. et al. Recent progress on layered double hydroxide (LDH) derived metal-based catalysts for CO2 conversion to valuable chemicals. Catal. Today 356, 490–513 (2020).
Baig, N. & Sajid, M. Applications of layered double hydroxides based electrochemical sensors for determination of environmental pollutants: a review. Trends Environ. Anal. Chem. 16, 1–15 (2017).
Aregay, G. G. et al. Application of layered double hydroxide enriched with electron rich sulfide moieties (S2O42−) for efficient and selective removal of vanadium (V) from diverse aqueous medium. Sci. Total Environ. 792, 148543 (2021).
Gautam, R. K., Singh, A. K. & Tiwari, I. Nanoscale layered double hydroxide modified hybrid nanomaterials for wastewater treatment: A review. J. Mol. Liq. 350, 118505 (2022).
Cao, Y., Zheng, D., Zhang, F., Pan, J. & Lin, C. Layered double hydroxide (LDH) for multi-functionalized corrosion protection of metals: A review. J. Mater. Sci. Technol. 102, 232–263 (2022).
Goh, K.-H., Lim, T.-T. & Dong, Z. Application of layered double hydroxides for removal of oxyanions: a review. Water Res 42, 1343–1368 (2008).
Karim, A. V., Hassani, A., Eghbali, P. & Nidheesh, P. Nanostructured modified layered double hydroxides (LDHs)-based catalysts: A review on synthesis, characterization, and applications in water remediation by advanced oxidation processes. Curr. Opin. Solid State Mater. Sci. 26, 100965 (2022).
Jijoe, P. S., Yashas, S. R. & Shivaraju, H. P. Fundamentals, synthesis, characterization and environmental applications of layered double hydroxides: a review. Environ. Chem. Lett. 19, 2643–2661 (2021).
Feng, X. et al. A review on heavy metal ions adsorption from water by layered double hydroxide and its composites. Sep. Purif. Technol. 284, 120099 (2022).
Mir, N. & Salavati-Niasari, M. Preparation of TiO2 nanoparticles by using tripodal tetraamine ligands as complexing agent via two-step sol–gel method and their application in dye-sensitized solar cells. Mater. Res. Bull. 48, 1660–1667 (2013).
Ding, J. et al. Synthesis and textural evolution of mesoporous Si3N4 aerogel with high specific surface area and excellent thermal insulation property via the urea assisted sol-gel technique. Chem. Eng. J. 382, 122880 (2020).
Mittal, J. Recent progress in the synthesis of Layered Double Hydroxides and their application for the adsorptive removal of dyes: A review. J. Environ. Manag. 295, 113017 (2021).
Mishra, G., Dash, B. & Pandey, S. Layered double hydroxides: A brief review from fundamentals to application as evolving biomaterials. Appl. Clay Sci. 153, 172–186 (2018).
Fazli, A., Brigante, M., Khataee, A. & Mailhot, G. Synthesis of a magnetically separable LDH-based S-scheme nano-heterojunction for the activation of peroxymonosulfate towards the efficient visible-light photodegradation of diethyl phthalate. Appl. Surf. Sci. 559, 149906 (2021).
Jawad, A. et al. Activation of persulfate by CuOx@ Co-LDH: a novel heterogeneous system for contaminant degradation with broad pH window and controlled leaching. Chem. Eng. J. 335, 548–559 (2018).
Sang, W. et al. Enhanced transition metal oxide based peroxymonosulfate activation by hydroxylamine for the degradation of sulfamethoxazole. Chem. Eng. J. 383, 123057 (2020).
Yin, Z. et al. Peroxymonosulfate enhancing visible light photocatalytic degradation of bezafibrate by Pd/g-C3N4 catalysts: The role of sulfate radicals and hydroxyl radicals. Chem. Eng. J. 390, 124532 (2020).
Cai, C., Zhang, H., Zhong, X. & Hou, L. Ultrasound enhanced heterogeneous activation of peroxymonosulfate by a bimetallic Fe–Co/SBA-15 catalyst for the degradation of Orange II in water. J. Hazard. Mater. 283, 70–79 (2015).
Yang, Q. et al. In situ stabilization of the adsorbed Co2+ and Ni2+ in rice straw biochar based on LDH and its reutilization in the activation of peroxymonosulfate. J. Hazard. Mater. 15, 126215 (2021).
Li, W. et al. Highly efficient activation of peroxymonosulfate by cobalt sulfide hollow nanospheres for fast ciprofloxacin degradation. J. Hazard. Mater. 389, 121856 (2020).
Pi, Z. et al. Persulfate activation by oxidation biochar supported magnetite particles for tetracycline removal: performance and degradation pathway. J. Clean. Prod. 235, 1103–1115 (2019).
Zhao, J. et al. Insight into in-situ chemical oxidation by Fe (II)-containing minerals: The role of inherent Fe (II)-OH in Fe (II)-Al LDHs. Chem. Eng. J. 433, 133835 (2021).
Starcher, A. N., Li, W., Kukkadapu, R. K., Elzinga, E. J. & Sparks, D. L. Fe (II) sorption on pyrophyllite: Effect of structural Fe (III)(impurity) in pyrophyllite on nature of layered double hydroxide (LDH) secondary mineral formation. Chem. Geol. 439, 152–160 (2016).
Sideris, P. J., Nielsen, U. G., Gan, Z. & Grey, C. P. Mg/Al ordering in layered double hydroxides revealed by multinuclear NMR spectroscopy. Science 321, 113–117 (2008).
Qin, W., Fang, G., Wang, Y. & Zhou, D. Mechanistic understanding of polychlorinated biphenyls degradation by peroxymonosulfate activated with CuFe2O4 nanoparticles: Key role of superoxide radicals. Chem. Eng. J. 348, 526–534 (2018).
Shahzad, A., Jawad, A., Ifthikar, J., Chen, Z. & Chen, Z. The hetero-assembly of reduced graphene oxide and hydroxide nanosheets as superlattice materials in PMS activation. Carbon 155, 740–755 (2019).
Yang, J.-C. E., Yuan, B., Cui, H.-J., Wang, S. & Fu, M.-L. Modulating oxone-MnOx/silica catalytic systems towards ibuprofen degradation: insights into system effects, reaction kinetics and mechanisms. Appl. Catal. B-Environ. 205, 327–339 (2017).
Shahzad, A. et al. Non-radical PMS activation by the nanohybrid material with periodic confinement of reduced graphene oxide (rGO) and Cu hydroxides. J. Hazard. Mater. 392, 122316 (2020).
Ye, Q. et al. Enhancing peroxymonosulfate activation of Fe-Al layered double hydroxide by dissolved organic matter: Performance and mechanism. Water Res. 185, 116246 (2020).
Chen, W., Teng, C.-Y., Qian, C. & Yu, H.-Q. Characterizing properties and environmental behaviors of dissolved organic matter using two-dimensional correlation spectroscopic analysis. Environ. sci. Technol. 53, 4683–4694 (2019).
Yuan, X. et al. Calcined graphene/MgAl-layered double hydroxides for enhanced Cr (VI) removal. Chem. Eng. J. 221, 204–213 (2013).
Hou, X. et al. A novel magnetic CuFeAl-LDO catalyst for efficient degradation of tetrabromobisphenol a in water. Chem. Eng. J. 430, 133107 (2021).
Tan, Y. et al. Natural diatomite mediated spherically monodispersed CoFe2O4 nanoparticles for efficient catalytic oxidation of bisphenol A through activating peroxymonosulfate. Chem. Eng. J. 388, 124386 (2020).
Xu, Y., Ai, J. & Zhang, H. The mechanism of degradation of bisphenol A using the magnetically separable CuFe2O4/peroxymonosulfate heterogeneous oxidation process. J. Hazard. Mater. 309, 87–96 (2016).
Lu, H., Sui, M., Yuan, B., Wang, J. & Lv, Y. Efficient degradation of nitrobenzene by Cu-Co-Fe-LDH catalyzed peroxymonosulfate to produce hydroxyl radicals. Chem. Eng. J. 357, 140–149 (2019).
Kinghorn, R., Milner, C. & Zweigenbaum, J. Analysis of trace residues of explosive materials by time-of-flight LC/MS. Agilent Technologies publication 230, U359–U360 (2005).
Carlos, L. et al. Intermediate distributions and primary yields of phenolic products in nitrobenzene degradation by Fenton’s reagent. Chemosphere 72, 952–958 (2008).
Zhao, L., Ma, J. & Sun, Z.-Z. Oxidation products and pathway of ceramic honeycomb-catalyzed ozonation for the degradation of nitrobenzene in aqueous solution. Appl. Catal. B-Environ. 79, 244–253 (2008).
Zhu, J., Zhu, Y. & Zhou, W. Cu-doped Ni-LDH with abundant oxygen vacancies for enhanced methyl 4-hydroxybenzoate degradation via peroxymonosulfate activation: key role of superoxide radicals. J. Colloid Interface Sci. 610, 504–517 (2021).
Zhu, M. et al. Efficient wastewater remediation enabled by self-assembled perovskite oxide heterostructures with multiple reaction pathways. ACS Sustain. Chem. Eng. 8, 6033–6042 (2020).
Zhao, G. et al. Activation of peroxymonosulfate by Fe3O4–Cs x WO3/NiAl layered double hydroxide composites for the degradation of 2, 4-dichlorophenoxyacetic acid. Ind. Eng. Chem. Res. 57, 16308–16317 (2018).
Tan, C. et al. Degradation of antipyrine by UV, UV/H2O2 and UV/PS. J. Hazard. Mater. 260, 1008–1016 (2013).
Khan, J. A. et al. Kinetic and mechanism investigation on the photochemical degradation of atrazine with activated H2O2, S2O82− and HSO5−. Chem. Eng. J. 252, 393–403 (2014).
Zhou, S. et al. Heterogeneous co-activation of peroxymonosulfate by CuCoFe calcined layered double hydroxides and ultraviolet irradiation for the efficient removal of p-nitrophenol. J. Mater. Sci.: Mater. Electron. 30, 19009–19019 (2019).
Gao, Q., Cui, Y., Wang, S., Liu, B. & Liu, C. Enhanced photocatalytic activation of peroxymonosulfate by CeO2 incorporated ZnCo–layered double hydroxide toward organic pollutants removal. Sep. Purif. Technol. 263, 118413 (2021).
Cai, C., Kang, S., Xie, X. & Liao, C. Ultrasound-assisted heterogeneous peroxymonosulfate activation with Co/SBA-15 for the efficient degradation of organic contaminant in water. J. Hazard. Mater. 385, 121519 (2020).
Ye, C. et al. Multifunctional capacity of CoMnFe-LDH/LDO activated peroxymonosulfate for p-arsanilic acid removal and inorganic arsenic immobilization: Performance and surface-bound radical mechanism. Sci. Total Environ. 806, 150379 (2022).
Miao, W. et al. The role of Fe-Nx single-atom catalytic sites in peroxymonosulfate activation: Formation of surface-activated complex and non-radical pathways. Chem. Eng. J. 423, 130250 (2021).
Cao, J. et al. Efficient charge transfer in aluminum-cobalt layered double hydroxide derived from Co-ZIF for enhanced catalytic degradation of tetracycline through peroxymonosulfate activation. Chem. Eng. J. 382, 122802 (2020).
Naderi, M. & Soltani, R. D. C. Hybrid of ZnFe layered double hydroxide/nano-scale carbon for activation of peroxymonosulfate to decompose ibuprofen: Thermodynamic and reaction pathways investigation. Environ. Technol. Innov. 24, 101951 (2021).
He, S. et al. Consolidated 3D Co3Mn-layered double hydroxide aerogel for photo-assisted peroxymonosulfate activation in metronidazole degradation. Chem. Eng. J. 423, 130172 (2021).
Wang, H. et al. Ultra-adsorption enhancing peroxymonosulfate activation by ultrathin NiAl-layered double hydroxides for efficient degradation of sulfonamide antibiotics. J. Clean. Prod. 369, 133277 (2022).
Wang, Y. et al. Fabricating iron-cobalt layered double hydroxide derived from metal-organic framework for the activation of peroxymonosulfate towards tetracycline degradation. J. Solid State Chem. 294, 121857 (2021).
Aziz, F. et al. Composites with alginate beads: A novel design of nano-adsorbents impregnation for large-scale continuous flow wastewater treatment pilots. Saudi J. Biol. Sci. 27, 2499–2508 (2020).
Zhang, L., Yang, X., Zhou, R., Lin, S. & Zhou, L. High-efficiency carbamazepine degradation using a Ni/Co-LDH as the peroxymonosulfate activator: Performance, mechanism and degradation pathway. Appl. Surf. Sci. 574, 151580 (2022).
Chen, M. et al. Immobilized Co2+ and Cu2+ induced structural change of layered double hydroxide for efficient heterogeneous degradation of antibiotic. J. Hazard. Mater. 403, 123554 (2021).
Zeng, H. et al. Degradation of sulfamethoxazole using peroxymonosulfate activated by self-sacrificed synthesized CoAl-LDH@ CoFe-PBA nanosheet: Reactive oxygen species generation routes at acidic and alkaline pH. Sep. Purif. Technol. 268, 118654 (2021).
Guo, R. et al. Efficient degradation of sulfamethoxazole by CoCu LDH composite membrane activating peroxymonosulfate with decreased metal ion leaching. Chem. Eng. J. 417, 127887 (2021).
Zeng, H., Deng, L., Shi, Z., Luo, J. & Crittenden, J. Heterogeneous degradation of carbamazepine by Prussian blue analogues in the interlayers of layered double hydroxides: performance, mechanism and toxicity evaluation. J. Mater. Chem. A 7, 342–352 (2019).
He, S. et al. Femtosecond time-resolved diffuse reflectance study on facet engineered charge‐carrier dynamics in Ag3PO4 for antibiotics photodegradation. Appl. Catal. B-Environ. 281, 119479 (2021).
Ma, R. et al. Enhanced catalytic degradation of aqueous doxycycline (DOX) in Mg-Fe-LDH@ biochar composite-activated peroxymonosulfate system: Performances, degradation pathways, mechanisms and environmental implications. Chem. Eng. J. 425, 131457 (2021).
Rahman, S. et al. High capacity aqueous phosphate reclamation using Fe/Mg-layered double hydroxide (LDH) dispersed on biochar. J. Colloid Interface Sci. 597, 182–195 (2021).
Guo, R., Zhu, Y., Cheng, X., Li, J. & Crittenden, J. C. Efficient degradation of lomefloxacin by Co-Cu-LDH activating peroxymonosulfate process: Optimization, dynamics, degradation pathway and mechanism. J. Hazard. Mater. 399, 122966 (2020).
Kohantorabi, M., Moussavi, G. & Giannakis, S. A review of the innovations in metal- and carbon-based catalysts explored for heterogeneous peroxymonosulfate (PMS) activation, with focus on radical vs. non-radical degradation pathways of organic contaminants. Chem. Eng. J. 411, https://doi.org/10.1016/j.cej.2020.127957 (2021).
Zhao, C. et al. Activation of peroxymonosulfate by biochar-based catalysts and applications in the degradation of organic contaminants: A review. Chem. Eng. J. 416, https://doi.org/10.1016/j.cej.2021.128829 (2021).
Zhu, J., Zhu, Z., Zhang, H., Lu, H. & Qiu, Y. Calcined CoAl-layered double hydroxide as a heterogeneous catalyst for the degradation of acetaminophen and rhodamine B: activity, stability, and mechanism. Environ. Sci. Pollut. Res. 26, 33329–33340 (2019).
Gong, C. et al. Heterogeneous activation of peroxymonosulfate by Fe-Co layered doubled hydroxide for efficient catalytic degradation of Rhoadmine B. Chem. Eng. J. 321, 222–232 (2017).
Jaafarzadeh, N., Ghanbari, F. & Ahmadi, M. Efficient degradation of 2,4-dichlorophenoxyacetic acid by peroxymonosulfate/magnetic copper ferrite nanoparticles/ozone: A novel combination of advanced oxidation processes. Chem. Eng. J. 320, 436–447 (2017).
Zhao, J. et al. CoMn-layered double hydroxide nanowalls supported on carbon fibers for high-performance flexible energy storage devices. J. Mater. Chem. A 1, 8836–8843 (2013).
Luo, S. et al. Manganese oxide octahedral molecular sieve (OMS-2) as an effective catalyst for degradation of organic dyes in aqueous solutions in the presence of peroxymonosulfate. Appl. Catal. B-Environ. 164, 92–99 (2015).
Hou, L. et al. Heterogeneous activation of peroxymonosulfate using Mn-Fe layered double hydroxide: Performance and mechanism for organic pollutant degradation. Sci. Total Environ. 663, 453–464 (2019).
Sun, D., Li, C., Lu, S., Yang, Q. & He, C. Magnetic Fe3O4@CoFe-LDH nanocomposite heterogeneously activated peroxymonosulfate for degradation of azo-dye AO7. RSC Adv. 11, 20258–20267 (2021).
Li, B. et al. LDHs-based 3D modular foam with double metal-fluorine interaction for efficiently promoting peroxymonosulfate activation in water pollutant control. Chem. Eng. J. 425, 131541 (2021).
Wang, L., Wang, Y., Lv, W. & Yao, Y. Activation of peroxymonosulfate by MgCoAl layered double hydroxide: Potential enhancement effects of catalyst morphology and coexisting anions. Chemosphere 286, 131640 (2022).
Soltani, R., Pelalak, R., Pishnamazi, M., Marjani, A. & Shirazian, S. A water-stable functionalized NiCo-LDH/MOF nanocomposite: green synthesis, characterization, and its environmental application for heavy metals adsorption. Arab. J. Chem. 14, 103052 (2021).
Kiani, M., Bagherzadeh, M., Ghadiri, A. M., Makvandi, P. & Rabiee, N. Multifunctional green synthesized Cu–Al layered double hydroxide (LDH) nanoparticles: anti-cancer and antibacterial activities. Sci. Rep. 12, 1–14 (2022).
Seron, A. & Delorme, F. Synthesis of layered double hydroxides (LDHs) with varying pH: A valuable contribution to the study of Mg/Al LDH formation mechanism. J. Phys. Chem. Solids 69, 1088–1090 (2008).
Zhu, J., Zhu, Y. & Zhou, W. Cu-doped Ni-LDH with abundant oxygen vacancies for enhanced methyl 4-hydroxybenzoate degradation via peroxymonosulfate activation: key role of superoxide radicals. J. Colloid Interface Sci. 610, 504–517 (2022).
Yang, Q. et al. In situ stabilization of the adsorbed Co2+ and Ni2+ in rice straw biochar based on LDH and its reutilization in the activation of peroxymonosulfate. J. Hazard. Mater. 416, 126215 (2021).
Balasubramanian, S., Kala, S. M. J., Pushparaj, T. L. & Kumar, P. V. Biofabrication of gold nanoparticles using Cressa cretica leaf extract and evaluation of catalytic and antibacterial efficacy. Nano Biomedicine Eng. 11, 58–66 (2019).
Hou, X. et al. A novel magnetic CuFeAl-LDO catalyst for efficient degradation of tetrabromobisphenol a in water. Chem. Eng. J. 430, 133107 (2022).
Zhao, J. et al. Insight into in-situ chemical oxidation by Fe (II)-containing minerals: The role of inherent Fe (II)-OH in Fe (II)-Al LDHs. Chem. Eng. J. 433, 133835 (2022).
Wu, L. et al. Oxygen Vacancy-induced nonradical degradation of organics: critical trigger of oxygen (O2) in the Fe–Co LDH/Peroxymonosulfate System. Environ. Sci. Technol. 55, 15400–15411 (2021).
Zhu, J., Zhu, Z., Zhang, H., Lu, H. & Qiu, Y. Efficient degradation of organic pollutants by peroxymonosulfate activated with MgCuFe-layered double hydroxide. RSC Adv. 9, 2284–2291 (2019).
Asif, M. B., Kang, H. & Zhang, Z. Gravity-driven layered double hydroxide nanosheet membrane activated peroxymonosulfate system for micropollutant degradation. J. Hazard. Mater. 425, 127988 (2022).
Deng, J. et al. Activation of peroxymonosulfate by CoFeNi layered double hydroxide/graphene oxide (LDH/GO) for the degradation of gatifloxacin. Sep. Purif. Technol. 255, 117685 (2021).
Li, X. et al. Efficient degradation of tetracycline by CoFeLa-layered double hydroxides catalyzed peroxymonosulfate: Synergistic effect of radical and nonradical pathways. J. Hazard. Mater. 398, 122884 (2020).
Wang, R., Su, S., Ren, X. & Guo, W. Polyoxometalate intercalated La-doped NiFe-LDH for efficient removal of tetracycline via peroxymonosulfate activation. Sep. Purif. Technol. 274, 119113 (2021).
He, S. et al. Structure–dependent degradation of nitroimidazoles by cobalt–manganese layered double hydroxide catalyzed peroxymonosulfate process. Chemosphere 266, 129006 (2021).
Zeng, H. et al. Degradation of sulfamethoxazole using peroxymonosulfate activated by self-sacrificed synthesized CoAl-LDH@CoFe-PBA nanosheet: Reactive oxygen species generation routes at acidic and alkaline pH. Sep. Purif. Technol. 268, 118654 (2021).
Guo, R. et al. Efficient degradation of sulfamethoxazole by CuCo LDH and LDH@ fibers composite membrane activating peroxymonosulfate. Chem. Eng. J. 398, 125676 (2020).
Zeng, H., Deng, L., Zhang, H., Zhou, C. & Shi, Z. Development of oxygen vacancies enriched CoAl hydroxide@hydroxysulfide hollow flowers for peroxymonosulfate activation: A highly efficient singlet oxygen-dominated oxidation process for sulfamethoxazole degradation. J. Hazard. Mater. 400, 123297 (2020).
Chen, G. et al. Octadecylamine degradation through catalytic activation of peroxymonosulfate by FeMn layered double hydroxide. Sci. Total Environ. 695, 133963 (2019).
Li, T. et al. Efficient degradation of Rhodamine B by magnetically recoverable Fe3O4-modified ternary CoFeCu-layered double hydroxides via activating peroxymonosulfate. J. Environ. Sci. 108, 188–200 (2021).
Zhou, S., Zhao, G., Wang, Y., Liu, L. & Jiao, F. Fabrication of 3d multi-layered znco-ldh as a heterogeneous photoactivator of peroxymonosulfate for efficient degradation of rhodamine b. Desalination Water Treat. 216, 315–325 (2021).
Han, X., Wang, S., Huang, H. & Zhang, Y. Hydroxyl radicals and sulfate radicals synergistically boosting the photocatalytic and mineralization ability of 1D-2D Bi5O7I/NiFe-LDH heterojunction. Appl. Surf. Sci. 540, 148237 (2021).
Luo, L. et al. Co–Cu–Al layered double oxides as heterogeneous catalyst for enhanced degradation of organic pollutants in wastewater by activating peroxymonosulfate: performance and synergistic effect. Ind. Eng. Chem. Res. 58, 8699–8711 (2019).
Acknowledgements
The researcher (Mohamed Hosny) is funded by a full scholarship (MM32/21) from the Egyptian Ministry of Higher Education & Scientific Research represented by the Egyptian Bureau for Cultural & Educational Affairs in London.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
E.A., A.E. contributed to the idea and concept, analyzed/interpreted the literature studies, wrote the original draft, reviewing, editing. H.E., S.B., H.E., M.H., wrote the original draft. G.A., G.E., A.O., reviewing, editing. All authors contributed to the interpretation and discussion of reviewed studies.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abd El-Monaem, E.M., Elshishini, H.M., Bakr, S.S. et al. A comprehensive review on LDH-based catalysts to activate persulfates for the degradation of organic pollutants. npj Clean Water 6, 34 (2023). https://doi.org/10.1038/s41545-023-00245-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41545-023-00245-x
This article is cited by
-
Enhanced degradation and removal of ciprofloxacin and ofloxacin through advanced oxidation and adsorption processes using environmentally friendly modified carbon nanotubes
Environmental Science and Pollution Research (2024)
-
MOF-derived C, N-In2O3 with GdFeO3 Z-scheme heterostructure for the photocatalytic removal of tetracycline
npj Clean Water (2023)
-
Performance assessment of the MOF adsorbent MIL-101 for removal of gaseous benzene and toluene: kinetic column modeling and simulation studies of fixed-bed adsorption
Environmental Science and Pollution Research (2023)
-
Capturing Cu2+ and recycling spent Cu-adsorbents as catalyst for eliminating Rhodamine B: reactivity and mechanism
Environmental Science and Pollution Research (2023)
-
N self‐doped multifunctional chitosan biochar-based microsphere with heterogeneous interfaces for self-powered supercapacitors to drive overall water splitting
Biochar (2023)
