
Abstract
The long-known presence of a sharp OH absorption band in the tetragonal fluoride mineral sellaite, MgF2, inspired us to conduct a detailed study of the OH incorporation modes into this IR-transparent (where IR stands for Infrared) material as well as to search for hydrogen traces in two other IR-translucent halides—villiaumite (NaF) and fluorite (CaF2). Among these three phases, sellaite is the only one to incorporate ‘intrinsic’ OH groups, most commonly as O–H∙∙∙F defects oriented nearly perpendicular to the c-axis along the shortest edge of the constituent MgF6 polyhedra, in analogy with the isostructural mineral rutile, TiO2. Another defect type, seen only scarcely in untreated natural material, develops when subjecting sellaite to temperatures above 900 °C. It involves an O–H∙∙∙O cluster along the 2.802 Å edge of the original MgF6 dipyramid, as fluorine atoms are progressively expelled from the structure, being replaced by O2- anions. This is corroborated by the appearance of spectral absorption features typical for brucite (Mg(OH)2) and ultimately periclase (MgO), the presence of which could be proven via powder diffraction of the heat-treated material. Except for a ‘dubious’ peak most probably caused by included phases, neither villiaumite (NaF) nor fluorite (CaF2) showed any presence of ‘intrinsic’ OH defects. They do however decompose along a similar route into the respective oxide and hydroxide phases at high temperature. This thermal decomposition of the studied halide phases is accompanied by the emission of gaseous (HF)n species at temperatures well below their established melting point - a subject which seems to be quite overlooked.
Similar content being viewed by others
Introduction
While the presence of trace amounts of hydrogen in nominally anhydrous silicate minerals was firmly established by numerous studies, comparatively little attention regarding this particular subject has been dedicated to anhydrous non-silicate phases. Based on IR spectroscopy, the presence of H2O molecules was established in barite (Beran et al. 2010), while hydrogen incorporated in the form of OH defects occurs in the phosphate minerals xenotime and triphylite (Talla et al. 2011; Libowitzky et al. 2012). Hydrogen-related defects have also been detected in wulfenite and in the ore mineral wolframite, (Talla et al. 2013, 2017). Oxide minerals, such as rutile and cassiterite, also contain considerable amounts of structural OH groups (rutile: Hammer and Beran 1991; Vlassopoulos et al. 1993; Swope et al. 1995; cassiterite: Losos and Beran 2004). Surprisingly, in a short note by Beran and Zemann (1985), OH groups have even been reported in the fluoride mineral sellaite.
IR spectroscopy is a sensitive method for the detection of trace hydrogen bound to oxygen atoms, thus forming OH groups or H2O molecules in the structure of nominally anhydrous minerals (Johnson 2006; Libowitzky and Beran 2006; Rossman 2006). By using polarized IR radiation, the pleochroic scheme of the absorption bands provides information on the orientation of the O–H vector; the band position suggests a certain O–H∙∙∙A bond length (with A representing different acceptor ions—N, O, F, Cl, Br, S, Se). Finally, the band amplitude and area relate to the analytical water content.
Based on this method and by considering the preliminary results of Beran and Zemann (1985), it is the principal aim of this paper to characterize the OH incorporation modes into the relatively rare fluoride mineral sellaite, MgF2, as well as changes occurring during its treatment at high temperature. Inspired by the observed phenomena, we opted to verify whether hydrogen or deuterium in the form of ‘intrinsic’ defects could be artificially ‘implanted’ into fluorite, CaF2, as well as villiaumite, NaF, by exposing them to elevated temperature in the presence of saturated H2O/D2O vapor.
It should be noted that all three studied compounds are manufactured artificially to serve as optically transparent elements for infrared beam-lines. Furthermore, MgF2 has gained great technical importance as coating material in the optical industry.
Experimental
Sample description and sample preparation
Four samples of sellaite, summarized in Table 1, four samples of fluorite (from four Austrian localities—Weißeck-Riedingsee, Vorderkrimml, Gnadenalm, Obernberg) and five samples of villiaumite from the Koashva quarry (Kola peninsula), Khomas region (Namibia), Mt. St. Hilaire (Québec), Illimaussaq (Greenland), as well as synthetic NaF, prepared by slow evaporation of an aqueous NaF solution at room temperature (RT), were available for the present study. Sample preparation of sellaite consisted of orienting selected single crystals by morphology and optical methods, followed by preparing double-polished crystal plates parallel to the c-axis, allowing polarized measurements along both principal optical directions of the tetragonal mineral. For general comparison as well as additional heating and deuteration experiments, synthetic flux-grown MgF2 single crystals supplied by the companies Leitz and Merck, prepared in the same manner, were used. All samples were inspected under the optical microscope prior to FTIR measurements, using an actual total magnification of 400x.
Preliminary FTIR measurements of the fluorite and villiaumite samples hinted at the presence of hydrogen only in villiaumite from the Koashva quarry, Kola peninsula. Therefore, crude slabs of both fluorite and villiaumite along their natural (111) and (100) cleavage direction, respectively, were used without any cutting or polishing. This included colorless cubes of villiaumite up to 8 mm in size, synthesized by slow evaporation.
IR spectroscopy, heating experiments and deuteration
Polarized single-crystal spectra in the infrared (IR) region were measured by means of the Bruker Hyperion 1000 microscope (LN2 cooled MCT detector) attached to the Bruker FTIR spectrometer Tensor 27 (Globar light source), equipped with a KRS-5 grid polarizer. The instrumental spectral resolution was 4 cm− 1, the lateral spectral resolution was 50 μm; spectra were averaged over 120 scans. The spectra are presented in their original form and are not background-corrected. The excellent quality and size of sellaite slabs from the Brumado mine (Bahia, Brazil) and the Signols quarry (Piemonte, Italy), enabled us to place point measurements along a line profile perpendicular to the c-axis, with the (x,y)-coordinates of each point defined as the distance in µm in respect to a particular corner of the slab serving as the origin point. This allowed to subsequently perform chemical analyses using the electron probe micro-analyser (EPMA) in the very same spots to check for correlations between sample chemistry and OH content.
To avoid damage of the crystals, low heating and cooling rates were established. The samples were thermally treated in air at heating rates of 1 °C min− 1. Peak temperatures progressing in 100 °C steps from 200 to 1100 °C were used for sellaite and 100 °C heating steps from 200 to 500 °C for villiaumite. The holding time was 25 h. Following each heating step, the samples were measured immediately after cooling down to room temperature.
For deuteration experiments at variable temperature, the samples were placed into an elongated suitably sized Pt-container, adjacent to a small glass vial containing 2.5 cm3 of ‘deuterating fluid’, consisting either of pure D2O or equal parts of H2O and D2O. Both vessels were transferred into a large near-to-horizontally mounted quartz glass test tube with the sample container closer to its slightly lowered terminal section. That particular region was subsequently inserted into the appropriately oriented cylindrical heating chamber of a programmable furnace, with the liquid-containing glass vessel farther away from the heat source. During the experiments, the quartz glass tube remained sealed by a cork lid to prevent the escape of the superheated H2O/D2O vapor, which instead condensed in its colder sections, the liquid eventually flowing back to the heated zone to re-evaporate. In these experiments, the sample heating rate amounted to 100 °C/hour, followed by a holding time of 16 h. The deuterated product was extracted and measured immediately after spontaneous cooldown of the experimental assembly.
Quantitative IR spectroscopic water determination
For an evaluation of the water content in sellaite, the integrated absorbance values Ai (cm− 1) measured with polarized radiation on sections cut parallel to the c-axis were summed to get the total integrated absorbance value Ai tot (Ai tot = 2 Ai ∟c + Ai //c), which was used for the calculation of the integrated absorption coefficient αi (cm− 2; αi = Ai tot/t; t is the thickness of the crystal slabs measured in cm). As expressed by Beer’s law, αi is directly related to the concentration c (mol L− 1) via the integrated molar absorption coefficient εi (L mol− 1 cm− 2). The water content in wt∙% is cH2O = (1.8/D) (αi/εi), where D is the density of the corresponding mineral (using 3.15 g cm− 3 for sellaite). The εi value can be derived from the linear calibration curve of Libowitzky and Rossman (1997) correlating the integrated molar absorption coefficient of the OH stretching vibration of hydrous mineral phases versus the wavenumber (Paterson’s 1982 method). Although this approach is generally applied to nominally anhydrous minerals, a correlation of εi with the molar volume and the density has to be considered for nominally anhydrous high-pressure phases (Koch-Müller and Rhede 2010). The calculated εi value of 37,730 L mol− 1 cm− 2 was used to infer the H2O content in sellaite associated with the main sharp OH absorption band at 3615 cm− 1 as well as its weaker ‘satellites’, all regarded as a singular type of OH defect. The εi value of 71,021 L mol− 1 cm− 2 was derived for the rather weak absorption band at 3462 cm− 1 observed exclusively in the Brumado sample. It should be noted that the εi value associated with H2O trace contents determined by an independent calibration for the particular mineral may substantially differ from εi calculated by the aforementioned method (Libowitzky and Rossman 1997; Rossman 2006).
Electron probe micro-analyser measurements
The sellaite sample material was inspected with a Cameca SX 100 electron probe micro-analyser (EPMA) at 15 kV operating voltage, 20 nA sample current, 3 μm beam size and counting times of 20 s for all elements. The measured X-ray line for all elements with the exception of Sr (Lα-line measured) was their particular Kα-line. The standard materials used were albite for Na, pyrope for Mg, synthetic SrSO4 for Sr, almandine for Al and Fe, wollastonite for Ca, vanadinite for Cl, topaz for F and titanite for Si. In case of the samples from Brazil and Italy, the analytical points were placed on the coordinates of previous FTIR micro-spectroscopic measurements. Raw X-ray intensities were corrected for matrix effects with a φ(ρz) algorithm of the X-PHI routine (Merlet 1994).
Since no significant presence of OH defects could be determined neither in fluorite nor villiaumite samples (apart from a very weak ‘dubiously behaved’ band observed in sample material from Koashva, Kola peninsula), chemical analyses were omitted for both types of phases. In addition, information on the chemical composition of the fluorite samples handled can be found in the work of Koss et al. (2022) and citations therein.
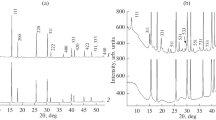
Powder X-ray diffraction
To verify the kind and presence of newly formed compounds after thermal decomposition ensuing the treatment of the samples at highest temperatures, part of the material was ground by hand in an agate mortar, pressed against a Si-plate mounted on the appropriate sample holder and measured on the Philips PW 3710 powder diffractometer (CuKα radiation, 2ʘ range from 5 to 90° in steps of 0.02°, counting time per step 5 s). Subsequent phase identification was accomplished using the Bruker EVA 2013 software with its inbuilt database.
Results
Chemical composition of sellaite
Analytical EPMA data of the natural sellaite samples are summarized in Table 1. Apart from the occasional presence of Ca, Fe, Al and Cl contents for the most part below the detection limit of the instrument, small yet relevant contents of Si (0.03–0.11 wt %) can be seen in measured points across all sellaite samples regardless of their provenance, including the ‘spectroscopic-grade’ synthetic material. Elevated Ca-contents in sample material from Grube Clara most probably originate from the interference of the fluorite matrix, in which sellaite occurs as scarce very fine needles (thickness ~ 30 μm).
IR spectra of sellaite
Polarized OH absorption spectra in the OH fundamental vibrational region of the examined sellaite crystals from the various localities always show a similar spectral pattern (Fig. 1). Taking as an example the spectrum of sellaite from the Brumado mine, Brazil, measured perpendicular and parallel to the c-axis, an extremely strong sharp band at 3615 cm− 1 is immediately evident, accompanied by a weaker band at 3591 cm− 1. An additional weak, somewhat broader peak can be observed at 3462 cm− 1. With the polarization direction of the IR beam oriented parallel to the c-axis, only very weak absorption components are visible for the 3615/3591 cm− 1 doublet band, thus indicating an extreme pleochroism of the OH dipole. For the broad weak 3462 cm− 1 band measured perpendicular to the c-axis, a less pronounced but still significant absorption can also be observed parallel to c.

Polarized IR absorption spectra of sellaite from the Brumado mine, Piemonte, Grube Clara and Mt. Bischoff, measured perpendicular and parallel to the c-axis at different absorption scales; (a) overall appearance of the spectral envelope in the 3800 − 3000 cm− 1 wavenumber region, (b) detail of weaker ‘satellite’ bands
Microscopic measurements within one single-crystal slab show slight differences in the band amplitudes, pointing to a somewhat uneven distribution of the OH groups. As clearly seen in Fig. 1a, a sellaite crystal from Piemonte, Italy, shows a very similar OH absorption pattern to the Brumado sample, dominated by the sharp band at 3615 cm− 1. However, the band at 3591 cm− 1 is barely visible and the 3462 cm− 1 peak is missing completely.
Besides the ‘classic’ dominant band at 3615 cm− 1, two weaker OH absorption peaks occur at distinct wavenumbers in the sellaite sample from Mt. Bischoff (Tasmania), which deviate from the situation in the other natural samples (Fig. 1a,b). While the weak 3600 cm− 1 band could roughly be associated with the 3591 cm− 1 peak of the Brumado sample, the additional absorption at 3577 cm− 1 in the spectrum measured perpendicular to the c-axis has no comparable feature in the other samples. The band at 3462 cm− 1 is missing.
Given that sellaite from Grube Clara (Germany) occurs as scarce fine needles randomly dispersed in fluorite, no polarized IR spectra could be recorded. The dominant broad strong hump in the 3400 cm− 1 stretching vibrational region corresponds to the microscopically verified presence of countless hydrous fluid inclusions. Nevertheless, the sharp OH band at 3615 cm− 1 is clearly visible, corroborating the existence of OH defects analogical to those present in the aforementioned samples. As was to be expected in the case of synthetic MgF2 crystals grown at high temperature from the melt in a vacuum, no bands occur in the OH stretching vibrational region of untreated samples.
‘Water’ contents in sellaite
‘Water’ contents associated with the major sharp absorption feature at 3615 cm− 1 as well as adjacent weaker ‘satellite bands’ showing the same pleochroic scheme range from 88 to 156 wt∙ppm across the measured profile with more H2O enrichment in the central region of the crystal from Brumado (Table 1). H2O contents associated with the weak 3462 cm− 1 peak only visible in this sample are rather stable, slightly varying around the value of a mere 3 wt∙ppm. The point profile in the slab from Piemonte shows the highest H2O content with values ranging from 382 to 588 wt∙ppm, all associated with the 3615 cm− 1 absorption feature. While the nature of the material from Grube Clara with sellaite finely dispersed in a fluorite matrix prevented us from extracting any meaningful values as to its ‘water’ content, we were able to do so in the case of the sample from Mt. Bischoff, Tasmania, albeit achieving only one measurement due to the very small size of the crystals at hand. The overall H2O content is the lowest recorded, amounting to a mere 7 wt∙ppm (Table 1).
No correlation between the H2O content and sample chemistry could be determined. While trace amounts of both homovalent as well as heterovalent substituting cations can be presumed, the measured concentrations of Na, Sr, Ca, Al, Fe and Cl are for the most part lower than the detection limit of the instrument. Silicon, being the only element unambiguously present in quantities well above the EPMA detection limit, does not show any relation to the spectroscopically determined quantity of H2O (Table 1).
Heating and deuteration of sellaite
Heating experiments performed on crystal slabs from the Brumado mine reveal a relatively constant amplitude of the sharp 3615 cm− 1 band up to a temperature of 700 °C, followed by its sudden decrease at 800 °C and the appearance of a new band at 3734 cm− 1 (Fig. 2a). At 900 °C, both bands disappear and a weak sharp peak occurs instead at yet a higher wavenumber of 3767 cm− 1 together with a relatively broad absorption with two maxima centered around 3690 and 3465 cm− 1. The 3465 cm− 1 band, which becomes clearly visible above 800 °C, apparently corresponds to the weak 3462 cm− 1 absorption feature observed during measurement in some regions of the original Brumado sample at room temperature. After further heating to 1000 and 1100 °C, only the bands at 3690 and 3465 cm− 1 remain.

Unpolarized IR absorption spectra of sellaite from the Brumado mine, heat-treated from room temperature up to 1100 °C in intervals of 200 °C and 100 °C, respectively; (a) OH stretching vibrational region; (b) region of combination modes of fundamental lattice vibrations
As seen in Fig. 2a, major changes in the spectra occur above 700 °C. Starting from this temperature, the colorless transparent samples start to become turbid, eventually turning white and somewhat ‘porous’, while progressively losing their anisotropy. X-ray powder diffraction on sample material after its heating to 1100 °C indicates the predominant presence of periclase, MgO. Unpolarized FTIR spectra in the 1800 − 1200 cm− 1 IR spectral region are presented in Fig. 2b. The weak band at 1640 cm− 1 is assigned to the bending vibration of H2O molecules. The amplitude of this band clearly correlates with that of the broad absorption centered around 3400 cm− 1, which is due to the stretching vibration of adsorbed molecular H2O (Fig. 2a). Figure 3 shows in detail the IR spectrum of the Brumado sample heated to 800 °C. The IR absorption pattern of brucite, Mg(OH)2, is shown for comparison (RRUFF database ID R050455). A prominent asymmetric OH absorption band at 3687 cm− 1 dominates the OH absorption region of brucite (Fig. 3a), while a characteristic combination mode occurs at 1415 cm− 1 (Fig. 3b). Despite the greater part of hydrogen still being in the form of ‘intrinsic defects’ as evidenced by the sharp peaks at 3615 and 3734 cm− 1, both features indicative of brucite formation (OH band at 3690 cm− 1—Fig. 3a, arrow) and its combination mode at 1415 cm− 1 (Fig. 3b), are also present, confirming the conversion of sellaite to brucite above 800°C as well as the onset of periclase (MgO) formation (Fig. 3b), indicated by the presence of its combination mode at 1495 cm− 1 (Nyquist and Kagel 1973).

Detailed presentation of the polarized IR spectra of sellaite from the Brumado mine, heat-treated at 800 °C in comparison with the IR spectrum of brucite from the RRUFF database; (a) OH stretching vibrational region, with the developing brucite-related 3690 cm− 1 absorption marked by an arrow; (b) region of combination modes of fundamental lattice vibrations. In addition to the lattice modes observed in sellaite, the 1415 cm− 1 lattice vibration of brucite (unpolarized measurement) from the RRUFF database is shown for comparison
Deuteration experiments (Ryskin 1974) performed at 700 °C on oriented single crystals of initially OH-free synthetic MgF2 as well as natural sellaite from the Brumado mine (Fig. 4a) lead to the same absorption pattern in both sample types with bands at 2754, 2720, 2663 and 2630 cm− 1, which are due to stretching vibrations of OD (deuteroxyl) groups, offset from their respective OH (hydroxyl) analogues by a certain wavenumber factor (Fig. 4b). The OD analogue of the characteristic narrow OH band at 3615 cm− 1 is represented by the 2663 cm− 1 peak, equally showing strongest absorption with the polarization direction perpendicular to the c-axis (Fig. 4b). The derived OH/OD wavenumber factor of 1.36 agrees perfectly with the theoretical value of 1.37 (Ryskin 1974). Using the calculated figure, the broader OD peak at 2720 cm− 1 perfectly matches the OH band at 3690 cm− 1 by its broader shape, wavenumber position as well as the pleochroic scheme with a stronger absorption component parallel to the c-axis. This particular OH absorption band associated with brucite formation appears in sellaite heated to sufficiently high temperature (Fig. 2a). In the same manner, the narrow OD band at 2754 cm− 1 (Fig. 4a) represents the analogue of the 3734 cm− 1 OH absorption present in heated natural sample material between 700 and 800 °C (Fig. 2a).

Polarized IR absorption spectra of untreated and deuterated sellaite from the Brumado mine, measured perpendicular and parallel to the c-axis; (a) overall shape of the spectral envelope; (b) close up of the OD absorption pattern and the OH bands. The weak doublet band marked by an asterisk in both (a) and (b) is caused by the presence of atmospheric CO2
Only a very weak OH analogue, probably obscured by the broad H2O stretching band, does apparently exist for the 2630 cm− 1 OD band (the corresponding O-H stretching vibration should occur at 3570 cm− 1). While it would seem tempting to associate this OD signal with the absorption peak at 3577 cm− 1 observed in the sample from Mt. Bischoff (Fig. 1b), its inverted pleochroic scheme (strongest absorption parallel to the c-axis, weak pleochroism) renders such an assignment implausible.
It is important to note that the deuteration of OH-free synthetic MgF2 crystals at the same conditions yielded to some extent also the brucite-related OH band at 3690 cm− 1 besides the OD absorptions. The most probable source of hydrogen was moisture entering the large quartz glass test tube due to some degree of leakiness between it and the cork lid, as well as during the necessary refills of D2O into the particular glass container within.
IR spectra of villiaumite
The unpolarized OH absorption spectrum in the OH fundamental region of villiaumite from Koashva, Kola peninsula, is characterized by a sharp band at 3676 cm− 1 (Fig. 5). Following the storage of the sample for three days in air at room-temperature conditions after cleaving the small fragment out of the matrix and its immediate measurement, the band completely disappeared. Inspection of villiaumite samples from the other localities did not confirm the presence of this suspicious peak anywhere else. Stepwise heating of another much larger freshly cleaved piece off the Koashva sample in 100 °C increments lead to the disappearance of the band at 400 °C. Interestingly, the pinkish to dark red color of natural villiaumite samples also vanishes at this particular temperature.

Detail of the 3800 − 2500 cm− 1 IR spectral region of villiaumite from Koashva, Kola peninsula, featuring the peculiar narrow 3676 cm− 1 absorption band. The broad hump centered at 3400 cm− 1 is indicative for the presence of molecular H2O within submicroscopic fluid inclusions. The weak C–H triplet band marked by two asterisks is caused by the presence of contaminating hydrocarbons
Deuteration experiments performed on villiaumite from Koashva at 550 °C in a D2O atmosphere also lead to the disappearance of the 3676 cm− 1 band without producing an OD band analogue. However, as shown in Fig. 6, an OH band at 3575 cm− 1 appeared together with its OD equivalent at 2636 cm− 1. The broad D2O absorption hump centered at 2560 cm− 1 is equivalent to the 3400 cm− 1 stretching band of adsorbed H2O molecules. The use of higher deuteration temperatures causes the NaF samples to ‘melt’, leaving behind a whitish ‘stain’ of residue, which was determined to consist of poorly crystalline Na2CO3 by subsequent powder diffraction measurement.

IR absorption spectra of villiaumite from Koashva, untreated and after deuteration at 550 °C. The respective OD absorption is marked by an arrow. The weak triplet band marked by an asterisk is due to the presence of contaminating hydrocarbons, the broad hump centered at around 3400 cm− 1 is due to adsorbed moisture
IR spectra of deuterated fluorite
We found no indication of the presence of intrinsic OH defects in any of the measured fluorite crystals—a result well in accord with the study of Koss et al. (2022) on a much larger pool of CaF2 samples. Yet, the observed similarity as to the decomposition mechanism of both sellaite and villiaumite at elevated temperature (with both of them apparently exchanging F− ligands for OH− or other oxygenated function groups) inspired us to attempt at artificially ‘implanting’ OH groups into initially OH-free fluorite via its deuteration, either with pure D2O or a 1:1 D2O/H2O mixture in use.
Deuteration at temperatures of up to 650 °C does not invoke any changes of the spectral envelope. At 800 °C, however, an OD band starts to appear at 2684 cm− 1 (Fig. 7). With the D2O/H2O mixture in use, the OH band corresponding to the aforementioned OD absorption is clearly discernible at 3640 cm− 1. As in the case of sellaite, the spectral changes are accompanied by a progressive increase in sample turbidity.

Spectral envelope of untreated and deuterated fluorite material from Weißeck-Riedingsee (showing but the broad 3406 cm− 1 hump due to H2O in hydrous inclusions), Austria, in the 4000 − 2500 cm− 1 spectral region. Both pure D2O as well as a 1:1 H2O/D2O mixture were used, the latter contributing to the formation of the respective OH absorption peak, but no sooner than at temperatures above 700 °C (red curve). A spectrum of portlandite (RRUFF ID R070210) is shown for comparison. All spectra were recorded after cooling down of the samples to room temperature
Fluorite material deuterated at 1000 °C becomes white. The powder diffraction measurement still reveals a substantial content of CaF2 in the sample, yet Bragg peaks matching the diffraction pattern of portlandite and lime (Ca(OH)2 and CaO, respectively), are evident. Indeed, the wavenumber position of the new OH absorption band at 3640 cm− 1 (Fig. 7) perfectly corresponds to that of portlandite (RRUFF ID R070210.1).
Discussion
Sellaite
The tetragonal crystal structure of sellaite, MgF2, is the same as that of rutile, TiO2. Comparably to the TiO6 polyhedra in rutile, edge-sharing MgF6 ‘octahedra’ form straight chains parallel to the c-axis in sellaite; these chains are cross-linked via the remaining two corner F− anions. Considering the detailed structural topology of sellaite (Baur 1956), the six fluorine ligands coordinating each Mg2+ cation actually form a rhombic dipyramid, with the two F–F edges shared with neighboring MgF6 polyhedra being the shortest (2.578 Å) and oriented perpendicular to the c-axis. These constitute in part the rectangular base of the rhombic dipyramid oriented parallel to the (100) plane, together with two other edges perpendicular to the c-axis, which are the longest (3.016Å). The remaining eight F–F edges have the same length (2.802 Å) and are oriented at an angle of about 30° in respect to the (001) plane.
Correlation trends between the OD∙∙∙FA donor-acceptor distance of an oxygen-fluorine anion pair hosting a hydrogen atom and the wavenumber of the associated absorption feature, established by Mikenda and Steinböck (1994, 1996), allow to determine ligand-ligand vectors of corresponding length in the mineral structure at hand, which could host the particular O–H∙∙∙F defect. The pleochroic scheme of the given peak enables a rough estimate of the required orientation of the O∙∙∙F vector in respect to the crystal axes, further reducing the pool of plausible OH incorporation modes.
With this in mind, the omnipresent major absorption at 3615 cm− 1 would require a donor-acceptor distance of 2.46 Å, which is well in accord with the shortest F–F edge shared by adjacent MgF6 polyhedra within their chains. In addition, these shared edges are oriented strictly perpendicular to the c-axis, which perfectly corresponds to the observed very strong pleochroism of the discussed band, showing maximum absorption with the electric vector of the incident polarized IR radiation (E) oriented perpendicular to the c-axis, while exhibiting minimal absorption with E parallel to it. The derived OH incorporation mode, labelled as ‘A’ in Fig. 8, represents (rather unsurprisingly) the analogy to the hydrogen incorporation mode into the isostructural mineral rutile, TiO2, as established by Beran and Zemann (1971). Correspondingly to the 3300 cm− 1 peak observed in TiO2, a weak absorption component of the 3615 cm− 1 band also exists in the direction parallel to the c-axis of sellaite, indicating a slight out-of-plane orientation of the O–H dipole in respect to (001), as was also described for rutile by the aforementioned authors. The weaker ‘satellite’ bands at 3591, 3600 and 3577 cm− 1 (with the presence of the latter two limited to the Tasmania sample) exhibit the same strong pleochroism and band shape as the ‘main’ 3615 cm− 1 absorption. They are thus interpreted to originate via its splitting, either due to minor presence of foreign cations (such as Fe2+ replacing Mg2+—Burns and Strens 1966) and, perhaps, also in part by Cl− anions substituting for the more electronegative F acceptor atoms (Mikenda and Steinböck 1994). Unfortunately, the huge analytical error associated with the concentration of these substituents, being under the detection limit of the electron probe micro-analyser, hinders us at determining any correlation between their content and the variation of spectroscopically determined absorbance of said minor bands. No correlation between the analytically significant content of Si and H2O content could be established either.

Part of the rutile-type sellaite structure viewed along the the a-axis, demonstrating the proposed incorporation mode of the O–H∙∙∙F defect corresponding to the 3615 cm− 1 band and weaker satellite peaks (‘A’) and of the O–H∙∙∙O defect related to the weak band at 3462 cm− 1, pointing along an ‘octahedral’ edge with an original F–F distance of 2.80 Å (‘B’). In the case of both depicted H-related defects, the O atom replaces original F ions. The blue dotted line connects two F atoms at a distance of 3.34 Å, for explanation see text
Linking the provenance of these absorption bands to small contents of phyllosilicate impurities, mainly mica, is not feasible despite the close wavenumber value between the sellaite OH absorption feature and that of, for example, muscovite at ~ 3630 cm− 1 (Beran 2002). Already the extreme pleochroism of all respective bands relative to the structure of sellaite rules out the role of submicroscopic inclusions, the absorption bands of which never show such extreme amplitude ratios. Moreover, the very same feature (the 3615 cm− 1 band) is the major OH-related absorption phenomenon in all studied sellaite samples and can even be induced via deuteration of originally OH-free synthetic MgF2 material, again ruling out included phases.
The standard routine to quantitatively assess the ‘water’ content could in itself be another source of error in this case. The value of the molar absorption coefficient (εi) has been derived from the calibration curve of Libowitzky and Rossman (1997) for O–H∙∙∙O bonds. Taking the position of the OH band at 3615 cm− 1 into account, the respective calculated εi value is 37,730 L mol− 1 cm− 2 with that for the 3462 cm− 1 absorption amounting to 71,021 L mol− 1 cm− 2. As discussed above, it is most probable that we are dealing with an O–H∙∙∙F bonding scheme. Nevertheless, we apply the existing calibration—having to put up with a certain degree of error in the determined water content (Table 1).
A different scenario must be considered for the hydrogen incorporation mode associated with the presence of the 3462 cm− 1 band detected exclusively in the sample from Brumado (Brazil). Presuming a second type of an OD–H∙∙∙FA defect, the required length of the associated F–F vector, under consideration of the aforementioned trends derived by Mikenda and Steinböck (1994, 1996), would amount to a mere 2.20 Å, a value well below the absolute minimum anion-anion distance in sellaite (2.578 Å). Furthermore, this shortest F–F vector (as discussed above) is oriented strictly perpendicular to the c-axis. This is in sharp contrast with the observed pleochroic scheme of the 3462 cm− 1 band, showing significant absorption components both parallel and perpendicular to the c-axis (the latter being stronger) and relatively weak pleochroism.
As of untreated material, the given peak was observed only in the Brazilian sample, and is associated with a rather constant and minuscule quantity of ‘water’—a mere 3 wt∙ppm H2O. However, upon heating of another crystal from the Brumado mine, where this absorption feature was initially absent, it appeared after having exposed the material to a temperature of 900 °C (Fig. 2a), well above the threshold at which both macroscopic as well as spectral changes start to occur in MgF2, with the samples becoming turbid and showing new absorption features, respectively (Fig. 2a,b). As indicated in the ‘Experimental’ section, new combination modes simultaneously appear in the 1400–1650 cm− 1 region (Fig. 2b), which can be associated with the formation of brucite (furthermore evidenced by the progressive appearance of a broad OH absorption feature at 3690 cm− 1—Fig. 2a) and periclase. The presence of both could be confirmed via powder diffraction measurement of part of the heat-treated material.
In all, it can be asserted that the progressive decomposition of MgF2 into brucite (Mg(OH)2) and ultimately periclase (MgO) at sufficiently high temperature is associated with continuous replacement of the F− anions by O2− atoms. As the number of oxygen atoms increases, so does the probability of the formation of O–O anion pairs along some of the original MgF6 polyhedral edges, potentially hosting a hydrogen atom, aiding in partly mitigating the accompanying charge imbalance. Considering the correlation trends for such OD–OA donor-acceptor distances versus the wavenumber of the associated absorption band (Libowitzky 1999), the required OD–OA distance related to the band observed at 3462 cm− 1 would amount to 2.85 Å, a value surprisingly close to the 2.80 Å edge length of the original MgF6 polyhedron. Furthermore, the given edges are tilted by about 60° in respect to the c-axis, implying a somewhat stronger absorption perpendicular to the [001] direction, nevertheless with a significant measurable component parallel to it as well—exactly matching the observed behavior of the 3462 cm− 1 peak (Fig. 1b). We therefore assign this particular absorption feature to such an O–H∙∙∙O cluster in sellaite (Fig. 8, case ‘B’).
In spite of having covered the nature of the ‘intrinsic’ defects in sellaite, further aspects of the decomposition mechanism of MgF2 deserve to be addressed. No pleochroism should be expected for the broad 3690 cm− 1 band, appearing above 800 °C (Fig. 2a), which can unambiguously be associated with brucite formation (Mg(OH)2), as evidenced by the simultaneous development of the corresponding combination modes in the 1400–1600 cm− 1 spectral region (Fig. 3b). However, the 3690 cm− 1 peak displays a somewhat stronger absorption component parallel to the c-axis of the original sellaite, implying a certain degree of preferential orientation of the layered brucite structure in respect to the MgF2 matrix. The trigonal structure of brucite (Ryskin 1974; Catti et al. 1995) consists of layers of Mg atoms arranged in a hexagonal motif along its (001) plane. Each triplet of Mg atoms forming an equilateral triangle within the said pattern is bound to one OH group, with the donor oxygen atom lying out-of-plane in respect to the Mg sheet. The O–H vector is oriented strictly parallel to the c-axis of brucite. Assuming preferential formation of brucite layers along a suitable template represented by appropriately arranged Mg atoms in the host structure, the most advantageous orientation of these would be along the (102) planes in sellaite, which contain solely Mg atoms in the appropriate distribution pattern. With the given scheme in mind, the O–H vector perpendicular to the brucite layers would be oriented at an angle of about 60° in respect to the c-axis of sellaite, implying absorption both perpendicular and parallel to it, with the latter component being stronger. This matches the behavior of the 3690 cm− 1 band observed both during the heating as well as deuteration experiments, with the situation well visible in Fig. 4.
Two new OH absorption bands appear following the onset of sellaite disintegration at about 700 °C prior to the formation of large amounts of brucite and ultimately periclase. A sharp peak appears at 3734 cm− 1 at 800 °C (Figs. 2a and 3a). Its OD analogue has successfully been reproduced during the deuteration experiments with the corresponding band appearing at 2754 cm− 1. Bearing in mind the severe distortion of the original structure at the prevailing conditions, it should not come as a surprise that the band assignment to a particular type of OH defect becomes problematic. Both the correlation trend of Mikenda and Steinböck (1994) for OD∙∙∙FA donor-acceptor distances as well as the one for OD∙∙∙OA oxygen pairs (Libowitzky 1999) indicate no more H-bridging. The only viable anion-anion distance in the original sellaite structure coming close to the required value well above 3 Å is present between two corner atoms of adjacent MgF6 polyhedra within a particular ‘octahedral’ chain, amounting to 3.34 Å (blue dotted line in Fig. 8), oriented strictly parallel to the c-axis. However, the pleochroic scheme of the band in consideration shows a strong absorption component also with E vibrating parallel to the c-axis (Fig. 3a). Alas, it must be assumed that while certainly caused by the presence of an OH group as evidenced by the appearance of its OD analogue band upon deuteration, the given hydrous defect is already consequential to the severe distortion of the original sellaite structure and cannot be characterized in further detail.
This applies more so to the absorption band detected at 3767 cm− 1 after exposing natural sellaite to 900 °C. In this case, its position effectively lies out of the wavenumber range associated with OH-related stretching vibration modes. One may thus speculate whether it could be assigned to the F-H stretching mode of a weakly bound H–F monomer, as F–H∙∙∙F bonding is realized. According to Andrews and Johnson (1984), various (HF)n species in solid argon produce abundant sharp absorptions in the high-energetic 4000 − 3600 cm− 1 region.
Villiaumite
The cubic crystal structure of villiaumite, NaF, corresponds to that of halite, NaCl, with a single F–F anion distance of 3.2767 Å. The only hint pointing at the presence of ‘intrinsic’ OH defects in untreated natural material was a single sharp band at 3676 cm− 1, which has only been observed in villiaumite crystals from the Koashva quarry (Kola peninsula). Moreover, its peculiar behavior, involving spontaneous disappearance from a small crystal slab while persisting in a more massive piece until its thermal treatment at 400 °C, strongly hints at an included foreign component. This assessment is corroborated by the absence of its OD analogue upon deuteration, as will be discussed further below. Despite that the pinkish to dark red color observed in most natural samples vanishes at the same temperature as the 3676 cm− 1 band, no relation is to be expected, with the original sample color most probably linked to a certain type of color center (artificially grown NaF crystals are colorless).
Regardless, given its abundance, we opted to subject the Koashva material to deuteration at various temperatures, with the aim of artificially ‘implanting’ OH defects into its structure. The only success occurred while deuterating the sample at 550 °C. Experimental runs at higher temperatures always resulted in the same outcome—the crystal ‘liquefied’, leaving behind a whitish stain inside the Pt-vessel, subsequently proven to consist of poorly crystalline Na2CO3. In the sample deuterated at 550 °C, a new sharp band at 3575 cm− 1 appeared, which is due to an OH defect, as evidenced by the appearance of the corresponding OD band at 2636 cm− 1.
With respect to the singular F–F edge length of NaF6 octahedra constituting the structure of villiaumite (3.2767 Å), an OD–H∙∙∙FA defect is unlikely, since the appropriate OD∙∙∙FA distance would have to amount to a mere 2.35 Å (Mikenda and Steinböck 1994, 1996), an unrealistically low value with regard to the structure of the host phase. In comparison, a spectral absorption feature related to an OD–H∙∙∙OA cluster would imply an OD∙∙∙OA distance of 3.11 Å (Libowitzky 1999), due to the much lower electronegativity of the acceptor atom. This value comes much closer to the actual F–F distance in villiaumite. In this scenario, a pair of fluorine ligands constituting the edge of the NaF6 octahedron is replaced by two O2− anions, one acting as the donor and the other as acceptor of a rather weak H-bridge from the incorporated hydrogen atom. Of course, the charge imbalance imposed by the second bivalent acceptor O2− ion necessitates compensation via one Schottky-type F− vacancy on a further anion site. In any case, it is apparent that villiaumite also tends towards replacement of its F− anions by O2− atoms or oxygenated function groups (OH−, CO32−) in the presence of moisture (or D2O vapor) at elevated temperature, in the same fashion as sellaite.
In the prevailing hot environment saturated with D2O and H2O vapor, it is possible to envisage at least a partial decomposition of NaF to NaOD or NaOH, (corresponding to bands at 2636 and 3575 cm− 1, respectively, as seen in Fig. 6), while producing volatile (HF)n species. The wavenumber position of the OH absorption peak in NaOH is 3636 cm− 1 (Busing 1955). Despite the notable wavenumber difference in respect to the observed OH band in villiaumite at 3575 cm− 1, a comparison can nevertheless be drawn. Taking into account the influence of the local NaF environment rich in F− anions with higher electronegativity in respect to the donor-acceptor oxygen pair, a decrease in wavenumber position of the said band is to be expected, as valence electrons from both O2− atoms are partly ‘dislocated’ towards the adjacent F anions. This in turn causes stronger attractive Coulomb forces within the O–H∙∙∙OA ensemble, thus shortening the OD–OA distance.
The ‘liquefaction’ of NaF starting at temperatures much lower than its declared melting point of 993 °C is a further hint for its decomposition into NaOH (or rather NaOD in this case) at temperatures above 600 °C, since the NaOH melting point amounts to a mere 318 °C (with an expected similar value for solid NaOD absent in literature). Thus, a NaF/NaO(D, H) mixture also melts at a much lower temperature than expected, subsequently reacting with atmospheric CO2, ultimately leading to its conversion into Na2CO3, as confirmed by the results of powder diffraction analyses of the whitish powdery product. This swift carbonation process is well-known to occur even in aqueous NaO(D, H) solutions at room temperature, rendering them useful for the purpose of CO2 sequestration (Tavan and Hosseini 2017).
Fluorite
While deuteration experiments conducted on initially hydrogen-free natural samples of fluorite, CaF2, could be regarded by some as a futile (or ‘excessive’) effort, the obtained results allowed us to draw parallels as to the high-temperature behavior of all three studied fluoride phases (and possibly many others). Regardless of sample origin or variations in chemical composition, fluorite always undergoes the same type of changes at a deuteration temperature above 650 °C, with an OD absorption band progressively developing at 2684 cm− 1, together with its OH analogue at 3640 cm− 1 with a 1:1 H2O/D2O mixture in use (Fig. 7). As can be seen in the respective figure, the newly developed OH absorption phenomenon perfectly corresponds to the OH absorption band of portlandite, Ca(OH)2, as illustrated in Fig. 7 by a comparison with its FTIR spectrum (RRUFF ID R070210). Powder diffraction on fluorite samples treated at 1100 °C shows visible Bragg peaks related to the aforementioned portlandite as well as lime, despite the majority of CaF2 still being preserved.
This realization is by far nothing new—in fact, the reaction of molten CaF2 with moisture under an imperfect vacuum caused serious setbacks in the initial attempts at producing artificial fluorite crystals of optical quality, with all product being turbid due to Ca(OH)2 and CaO formation. The synthesis was, of course, accompanied by the emission of gaseous HF (Krivandina 1991). The reaction of CaF2 to portlandite also occurs during measurements of fluid inclusion homogenization temperature in fluorite (Götzinger 1990). An analogical process apparently takes place in nature, as evidenced, to give one example, by the presence of accessory portlandite in fluorite-rich carbonatite-related ores in the Latium region, Italy (Stoppa et al. 2019).
Similarities in the behavior of sellaite, villiaumite and fluorite at elevated temperature
While the tetragonal mineral sellaite (MgF2) is the only one of the three minerals studied, which unambiguously proves to incorporate ‘intrinsic’ hydrous defects in nature, it appears to undergo a comparable type of decomposition at elevated temperature as villiaumite and fluorite. In all three cases, the F− anions are progressively replaced by OH− groups and O2− anions, in combination with the liberation of gaseous (HF)n species. In the course of this process, mainly sellaite develops bands associated with new types of OH defects.
By discussing the incorporation mode of hydrogen traces in fluoride minerals on a general basis, four processes occur in succession, as the temperature is increased: (1) formation of O–H∙∙∙F defects, (2) formation of O–H∙∙∙O clusters, (3) formation of O–H groups without H-bridging, accompanied by (4) adsorption of part of the escaping (HF)n polymers onto the remaining substrate. In the first case, F− is replaced by an (OH)− group; no charge compensation is required, thus the local distortion of the host structure is presumably minimal. The very good match between the required OD∙∙∙FA distance related to the 3615 cm− 1 absorption band and associated ‘satellite’ peaks in sellaite with the F–F distance along the shortest edge shared by adjacent MgF6 polyhedra (Fig. 8, case ‘A’) strongly suggests such O–H∙∙∙F bonding. Besides, the given hydrogen incorporation mode is analogic to the situation described in rutile (Beran and Zemann 1971) and consistent with the ‘principle of least action’ (Gray 2009).
The second incorporation mode involving the formation of O–H∙∙∙O clusters within the halide matrix requires additional charge compensation due to the second bivalent O2− atom, be it by a trivalent cation, e.g. Fe3+ or Al3+ substituting Mg2+ in sellaite, or a Schottky-type vacancy on a nearby anion site. In all cases, the particular scheme is certainly less energetically favorable than the former one. This is the probable reason for the appearance of such a defect (Fig. 8, case ‘B’) related to the 3462 cm− 1 absorption band in sellaite at rather high temperature (Fig. 2a), being linked to the progressive degradation of the material (with the exception of some regions of the original Brumado sample, where it was present from the start—Fig. 1). Ongoing structural damage upon further increase in temperature leads to the presence of OH groups without any acceptor anion, as evidenced by the appearance of the sharp peak at 3734 cm− 1 in natural sellaite heated to 800 °C, the OD analogue of which formed at 2754 cm− 1 during sample deuteration at ~ 700 °C.
The fourth model involving F− and H+ ions forming a neutral F–H group with a second F− atom acting as the acceptor ion in analogy with the situation in F-topaz from Spitzkoppe, Namibia (Libowitzky and Beran 2006), is most certainly not involved. However, the formation of certain (HF)n polymers resulting in absorption bands at extremely high wavenumbers (Andrews and Johnson 1984) is highly probable, in relation to the fluorine escape accompanying the decomposition of the particular fluoride phase. Such weakly bonded (if not merely adsorbed) species may be responsible for the weak absorption band at 3767 cm− 1, observed in thermally treated sellaite at 900 °C (Fig. 2a). At these conditions, the sample already displays extensive transformation into brucite and periclase, as evidenced by the apparition of the broad 3690 cm− 1 band as well as combination modes of both MgO and Mg(OH)2 (Fig. 2b).
All studied fluoride phases (and possibly others as well) apparently tend towards decomposition into the respective Me-oxide or hydroxide (Me = Mg, Na, Ca), emitting gaseous (HF)n species at temperatures well below their melting point (provided that enough H2O or D2O is present). The fact that the said detail is not featured in the respective Material Safety Data Sheets (MSDS) is somewhat worrisome, since it must be anticipated, that thermal treatment of large quantities of MgF2, NaF or CaF2 above a certain temperature threshold in the presence of moisture will lead to such emission of significant amounts of toxic hydrogen fluoride.
Conclusions
Of all three fluoride minerals studied (sellaite, villiaumite and fluorite), only natural samples of sellaite, MgF2, contain intrinsic OH defects, which can even be ‘implanted’ into an initially ‘hydrogen-free’ structure by hydrating/deuterating synthetic MgF2 above 700 °C. However, all phases mentioned ultimately decompose upon their heating to sufficiently high temperatures. In the presence of moisture (albeit H2O or D2O vapor), all three compounds tend to transform into the respective Me-oxide or hydroxide (Me = Mg, Na, Ca for sellaite, villiaumite and fluorite, respectively).
While the corresponding Me-oxide and/or hydroxide is the final product in the case of sellaite and fluorite, the NaOH (or NaOD upon deuteration) formed in the case of villiaumite swiftly reacts to Na2CO3 with atmospheric CO2. Nevertheless, the temperature-induced decomposition process of all investigated fluoride phases can be considered similar and is always accompanied by the release of toxic gaseous (HF)n species, already well under their respective melting points in the presence of moisture—an important fact not mentioned in the respective MSDS (Material Safety Data Sheets).
References
Andrews L, Johnson GL (1984) Fourier-Transform Infrared spectra of (HF)n species in solid argon. J Phys Chem 88:425–432
Baur WH (1956) Über die Verfeinerung der Kristallstrukturbestimmung einiger Vertreter des Rutiltyps: TiO2, SnO2, GeO2 und MgF2. Acta Cryst 9:515–520
Beran A (2002) Infrared spectroscopy of micas. In: Mottana A (eds) Micas: crystal chemistry and metamorphic petrology. Rev Mineral Geochem 46:351–369
Beran A, Zemann J (1971) Messung des Ultrarot-Pleochroismus von Mineralen. XI. Der Pleochroismus der OH-Streckfrequenz in Rutil, Anatas, Brookit und Cassiterit. Tschermaks Min Petr Mitt 15:71–80
Beran A, Zemann J (1985) Polarized absorption spectra of sellaite from the Brumado mine, Brazil, in the near infrared. Bull Geol Soc Finland 57:113–118
Beran A, Talla D, Losos Z, Pinkas J (2010) Traces of structural H2O molecules in barite. Phys Chem Miner 37:159–166
Burns RG, Strens RGJ (1966) Infrared study of the hydroxyl bands in clinoamphiboles. Science 153:890–892
Busing WR (1955) Infrared Spectra and structure of NaOH and NaOD. J Chem Phys 23:933–936
Catti M, Ferraris G, Hull S, Pavese A (1995) Static compression and H disorder in brucite, mg(OH)2, to 11 GPa: a powder neutron diffraction study. Phys Chem Miner 22:200–206
Götzinger MA (1990) Determination of aqueous salt solutions in fluid inclusions by infrared investigations. N Jb Miner Mh Jg 1:1–12
Gray CG (2009) Principle of least action. Scholarpedia 4:8291
Hammer VMF, Beran A (1991) Variations in the OH concentration of rutiles from different geological environments. Min Petrol 45:1–9
Johnson EA (2006) Water in nominally anhydrous crustal minerals: speciation, concentration, and geologic significance. In: Keppler H, Smyth JR (eds) Water in nominally anhydrous minerals. Rev Miner Geochem 62:117–154
Koch-Müller M, Rhede D (2010) IR absorption coefficients for water in nominally anhydrous high-pressure minerals. Am Mineral 95:770–775
Koss S, Götzinger MA, Beran A (2022) Zur Mineralogie und Genese von Fluoritvorkommen in Karbonatgesteinen des ‘Metamorphen Mesozoikums‘ in Österreich [in german with English abstract].Mitt Österr Miner Ges168: In print
Krivandina EA (1991) Preparation of single crystals of multicomponent fluoride materials with the fluorite type structure. Bull Soc Cat Cièn 12:393–412
Libowitzky E (1999) Correlation of O-H stretching frequencies and O-H∙∙∙O hydrogen bond lengths in minerals. Mh Chemie 130:1047–1059
Libowitzky E, Beran A (2006) The structure of hydrous species in nominally anhydrous minerals: Information from polarized IR spectroscopy. In: Keppler H, Smyth JR (eds) Water in nominally anhydrous minerals. Rev Miner Geochem 62:29–52
Libowitzky E, Rossman GR (1997) An IR absorption calibration for water in minerals. Am Mineral 82:1111–1115
Libowitzky E, Beran A, Wieczorek AK, Wirth R (2012) On the presence of a hydrous component in a gemstone variety of intermediate olivine-type triphylite-lithiophilite, Li(Fe,Mn)PO4. Min Petrol 105:31–39
Losos Z, Beran A (2004) OH defects in cassiterite. Min Petrol 81:219–234
Merlet C (1994) An Accurate Computer correction program for quantitative Electron probe Microanalysis. Microchim Acta 114/115:363–376
Mikenda W, Steinböck S (1994) Stretching frequency vs. bond distance correlation of O– D(H)∙∙∙F hydrogen bonds in solid hydrates. J Mol Struct 326:123–130
Mikenda W, Steinböck S (1996) Stretching frequency vs. bond distance correlation of hydrogen bonds in solid hydrates: a generalized correlation function. J Mol Struct 384:159–163
Nyquist RA, Kagel RO (1973) Infrared Spectra of Inorganic Compounds. 2nd reprint, Academic Press, New York, pp 495
Paterson MS (1982) The determination of hydroxyl by infrared absorption in quartz, silicate glasses and similar materials. Bull Min 105:20–29
Rossman GR (2006) Analytical methods for measuring water in nominally anhydrous minerals. In: Keppler H, Smyth JR (eds) Water in nominally anhydrous minerals. Rev Miner Geochem 62:1–28
Ryskin YI (1974) The vibrations of protons in minerals: hydroxyl, water and ammonium. In: Farmer VC (ed) The infrared spectra of minerals. Miner Soc, London, pp 137–181
Stoppa F, Schiazza M, Rosatelli G, Castorina F, Sharygin VV, Ambrosio FA, Vicentini N (2019) Italian carbonatite deposit: from mantle to ore deposit. Ore Geol Rev 114:103041
Swope RJ, Smyth JR, Larson AC (1995) H in rutile-type compounds: I. single-crystal neutron and X-ray diffraction study of H in rutile. Am Mineral 80:448–453
Talla D, Beran A, Škoda R, Losos Z (2011) On the presence of OH defects in the zircon-type phosphate mineral xenotime, (Y,REE)PO4. Am Mineral 96:1799–1808
Talla D, Wildner M, Beran A, Škoda R, Losos Z (2013) On the presence of hydrous defects in differently coloured wulfenites (PbMoO4): an infrared and optical spectroscopic study. Phys Chem Miner 40:757–769
Talla D, Beran A, Škoda R, Losos Z (2017) Polarized FTIR spectroscopic examination on hydroxylation in the minerals of the wolframite group, (fe,mn,mg)[W,(Nb,Ta)][O(OH)]4. Am Mineral 102:867–875
Tavan Y, Hosseini SH (2017) A novel rate of the reaction between NaOH with CO2 at low temperature in spray dryer. Petroleum 3:51–55
Vlassopoulos D, Rossman GR, Haggerty SE (1993) Coupled substitution of H and minor elements in rutile and the implication of high OH contents in Nb- and Cr-rich rutile from the upper mantle. Am Mineral 78:1181–1191
Acknowledgements
Thanks are due to Andreas Wagner for careful preparation of the oriented double-polished sellaite sections. We thank Stuart Mills from the Museum Victoria for supplying us with single-crystal sellaite material from Mt. Bischoff, Tasmania. In a comparable manner, we are indebted to Vera Hammer from the National Museum of Natural History in Vienna for providing sample material of villiaumite from the Khomas region in Namibia and Mt. St. Hilaire, Québec (samples N4284, M2222 I-1989 and M5209 III-1992, respectively). Furthermore, we gratefully acknowledge the help of Friedrich Koller, who provided us with villiaumite material from Illimaussaq, Greenland. We appreciate the helpful support of Christian L. Lengauer during the powder diffraction measurements on heat-treated samples. We are indebted to the companies Leitz and Merck for making synthetic MgF2 material available to us. Our gratitude also belongs to the work of the two anonymous reviewers, which helped us to improve the quality of this paper, as well as to the Chief Editor, Lutz Nasdala, and the respective Guest Editor, Dr. Tonci Balić-Žunić, for their suggestions concerning final revisions and corrections of this work.
Funding
Open access funding provided by University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: T. Balic-Zunic.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Talla, D., Beran, A. & Škoda, R. Natural and artificial OH defect incorporation into fluoride minerals at elevated temperature—a case study of sellaite, villiaumite and fluorite. Miner Petrol 117, 359–372 (2023). https://doi.org/10.1007/s00710-023-00824-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00710-023-00824-3