
Abstract
The use of saliva as a diagnostic fluid has always been appealing due to the ability for rapid and non-invasive sampling for monitoring health status and the onset and progression of disease and treatment progress. Saliva is rich in protein biomarkers and provides a wealth of information for diagnosis and prognosis of various disease conditions. Portable electronic tools which rapidly monitor protein biomarkers would facilitate point-of-care diagnosis and monitoring of various health conditions. For example, the detection of antibodies in saliva can enable rapid diagnosis and tracking disease pathogenesis of various auto-immune diseases like sepsis. Here, we present a novel method involving immuno-capture of proteins on antibody coated beads and electrical detection of dielectric properties of the beads. The changes in electrical properties of a bead when capturing proteins are extremely complex and difficult to model physically in an accurate manner. The ability to measure impedance of thousands of beads at multiple frequencies, however, allows for a data-driven approach for protein quantification. By moving from a physics driven approach to a data driven approach, we have developed, for the first time ever to the best of our knowledge, an electronic assay using a reusable microfluidic impedance cytometer chip in conjunction with supervised machine learning to quantifying immunoglobulins G (IgG) and immunoglobulins A (IgA) in saliva within two minutes.
Similar content being viewed by others

1 Introduction
Even though blood is the most well-studied bodily fluid in terms of validated biomarkers, the use of saliva as a diagnostic fluid is more appealing due to the ability to sample it in a noninvasive way (Castagnola et al. 2011; Greabu et al. 2009; Kaczor-Urbanowicz et al. 2017; Malamud 2011; Streckfus and Bigler 2002). The states of numerous diseases can be assessed by monitoring the levels of various protein biomarkers in saliva. The presence of antibodies in saliva is particularly important for diagnosing various infectious diseases, autoimmune diseases, and also chronic inflammatory conditions. For example, hepatitis B virus (HBV) and hepatitis C virus (HCV) antibodies exist in saliva sample and correlate well with their levels in blood (Chen et al. 2009; Gonçalves et al. 2005; González et al. 2008; Yoshizawa et al. 2013). Patients with auto-immune diseases, like sepsis, can also benefit from monitoring their biomarker levels in saliva (Cho and Choi 2014). Biomarkers such as MMP-2, MMP9, and TNFα were found in oral cancer patients at higher levels compared to control subjects (Rhodus et al. 2005; Shpitzer et al. 2009). Immunoglobulin G (IgG) and immunoglobulin A (IgA) are two types of antibodies that play crucial roles in the immune system (Burton 1985; Fagarasan and Honjo 2003; Vidarsson et al. 2014; Woof and Keer 2006). The measurement of IgA and IgG in saliva or serum can be a diagnostic tool for certain conditions such as autoimmune hepatitis and celiac disease (Lakos et al. 2008; Dane and Gürbüz 2016). One of the disadvantages of using saliva as diagnostic sample in last few decades was the lack of inexpensive test tools (Giannobile 2011). The challenge with rapid protein biomarker monitoring at the point-of-need, however, lies in the fact that current technologies to perform biomarker assays usually involve bulky instrumentation using optical technologies, like sandwich ELISA or Luminex, which requires sending a sample out to the lab and waiting for analysis. Light-weight, highly sensitive, and inexpensive ultra-compact platforms can greatly improve the standard of care for patients that can benefit from frequent monitoring of biomarker levels whether in an acute or primary care setting. While we focused on detection of IgG and IgA in saliva, this technology is broadly applicable to detection of a wide array of protein biomarkers in various matrices including blood. This technique can even be applied to detection of IgG and IgM antibodies in serum to for diagnosis of viral infections such as Sars-Cov-2 (Li et al. 2020).
Among various sensing modalities, electrical impedance based technologies(Xie et al. 2017a, 2017b; Emaminejad et al. 2016; Javanmard et al. 2010; Gholizadeh et al. 2016) have great potential for building miniaturized ultra-compact platforms. Previously, Javanmard et al. demonstrated detection of protein biomarkers in bioactivated microchannels (Javanmard and Talasaz 2009) and used antibody coated beads (for capture of target analyte) and impedance cytometry for detection of proteins with low abundance (Mok et al. 2014). Lin et al. (2015) also demonstrated another method for detection of proteins based on bead aggregate sizing in single frequency. This method takes more than 3.5 h to have beads aggregated together in sample preparation, and the biomarker was in pure PBS. Previous work requires finding high affinity antibody pairs that can allow bead aggregate. Carbonaro et al. demonstrated a method for label free biomarker detection based on size changes of beads resulting from protein binding (Carbonaro and Sohn 2005). Chang et al. (2007) described a detection method for proteins based on nanogap electrodes. Lasseter et al. (2004) also demonstrated use of frequency dependent electrochemical impedance spectroscopy to detect protein biomarkers. For the first time, to the best of our knowledge, we have developed an electronic assay using impedance cytometry in conjunction with supervised machine learning, capable of quantifying immunoglobulin G and immunoglobulin A in saliva within two minutes.
2 Theory
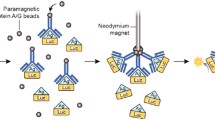
Figures 1 and 2 illustrates the assay steps and device operation. The assay works by using magnetic anti-IgG/anti-IgA (primary antibody) coated beads to capture soluble IgG/IgA (target antigen) molecules in saliva. Anti-IgG/anti-IgA (detector antibody) is then added to the magnetic beads to further increase the effective radius of the bead-protein complex. The beads flow through an impedance cytometer that probes the impedance of the beads at eight frequencies. The use of multi-frequency electrical impedance cytometry and supervised machine learning allows for electronically determining the level to which beads have captured target IgG/IgA molecules in saliva. On the other hand, we independently confirmed that binding properly occurred by fluorescently tagging (FITC) the IgG/IgA bound beads and imaged optically.

A Schematic of biochip. Presence of IgG/IgA in saliva sample results in beads binding to IgG/IgA which enables the labeling of FITC markers to the beads. Multi-frequency impedance based sizing in conjunction with machine learning classification were used for differentiating between FITC coated beads and unbound beads; B Raw data of peaks due to beads passing. The frequencies shown are 600 K, 5 M, 21 M. The amplitude of the input AC voltage was 400 mV

A Image of the device in comparison to a one cent coin B Micrograph of microfabricated electrodes, C Entrance of micropore, and D Schematic of sample preparation steps
A particle inside the channel with the electrode/electrolyte interface can be modeled into a circuit as shown in Fig. 3. We assumed an ideal polarizable electrode model without charge transfer resistance at the interface as we used gold as our electrode material, which is inert. Previous works from Sun and Morgan (2010), Gawad et al. (2001) and Ahuja et al. (2019) demonstrated that a cell can be modeled as a cell resistance in parallel with cell capacitance. The beads were coated with antibody and protein on its surface, so we also modelled it as capacitor in parallel with a resistor. The complete model consists of two double layer capacitances (Cdl) at each electrode interface in series with resistors due to solution resistance and electrodes resistance (Rs), and also the impedance components representing a bead. Our lumped model of the bead consists of a capacitance (Cbead) at its surface in parallel to a resistance (Rbead) along the surface. The impedance of the analyte attached to the surface of the bead can be modeled as a resistor (ΔR) in series and a capacitor (ΔC) in parallel.

The equivalent circuit model for a two electrode system in a microfluidic channel with a bead suspended in buffer
The impedance of the beads depends on bead size and conductivity and dielectric permittivity at the bead surface. The binding of IgG or IgA modulates these properties in a manner, which is difficult to fully model physically, thus we use a data-driven approach leveraging the power of multi-frequency lock-in-amplification measurements (to capture the frequency dependent dielectric changes) in conjunction with supervised machine learning to quantify the amount of protein captured (and thus present in saliva).
3 Experimental
3.1 Methods
3.1.1 Bead preparation
To demonstrate proof-of-concept and benchmark performance of the platform, we characterized the ability to detect presence of IgG and mouse IgA spiked in saliva. For IgG detection, we used 2.8 \(\mu\)m paramagnetic Dynal beads (Life Technologies, Carlsbad, CA) pre-coated with anti-mouse IgG antibodies to capture mouse IgG spiked in the saliva sample. Sheep anti-mouse IgG beads were washed three times in PBS 0.1% BSA. For IgA detection, beads coated with anti-IgA antibody were not readily commercially available, so we used the following protocol to couple anti-IgA antibodies directly to the beads. 2.8 \(\mu\)m Tosyl activated beads (Life Technologies, Carlsbad, CA) coated with anti IgA antibodies were used to capture human IgA in the saliva sample. The protocol for the 2 min IgA assay went as follows: To couple ligands to the beads, Tosyl activated beads were washed 3 times with 1 ml of buffer B (0.1 M Na-phosphate buffer, pH 7.4). The beads were then mixed with anti IgA antibodies in a mixing buffer consisting of 150 \(\mu\)L of buffer B and 100 \(\mu\)L of buffer C (3 M ammonium sulphate in Buffer B). The beads were incubated on a roller at 37 degrees Celsius for 18 h first, and then incubated in buffer D (PBS pH 7.4 with 0.5% (w/v) BSA) for another 1 h. Finally, the beads were washed with buffer E (PBS pH 7.4 with 0.1% (w/v) BSA) three times and ready for coupling to human IgA.
3.1.2 Sample preparation
For IgG characterization, saliva samples were spiked with mouse IgG at concentrations of 1667 nM, 167 nM and 17 nM. The protocol for performing the 2 min assay went as follows: The beads were mixed with mouse IgG spiked into the saliva sample off-chip in an epindorf tube and rotated for 2 min at 25 degrees Celsius. In order to confirm that binding properly occurred, we washed the beads three times with PBS 0.1% BSA and mixed with FITC tagged anti-IgG. Beads were washed with PBS to ensure that all the unbound antibodies were washed away before injecting into the microfluidic impedance cytometry chip. For IgA experiments, saliva samples were spiked with human IgA at a concentration of 8.4 \(\mu\)M. To couple IgA, the pretreated beads were mixed with human IgA spiked saliva sample in the epindorf tube and rotated for 2 min at 37 degrees Celsius. For validation purposes, in parallel with electrical measurements, the beads were washed three times and mixed with FITC tagged with anti-IgA. The beads as well were washed three times with PBS to ensure that all unbound antibodies were washed away before the electrical testing.
3.1.3 Biochip fabrication
The microfabricated biochip (Fig. 2A–C) consists of two gold microelectrodes on a glass substrate with a PDMS (Polydimethyl Siloxane) channel above it. To fabricate the gold electrodes, standard photolithography, electron beam evaporation of metal, and liftoff processing were performed on a 3″ fused silica wafer. The photo-pattering process consists of wafer cleaning, spin coating of photoresist, soft bake, ultra-violet exposure through a 4″ chromium mask, resist development, and a hard bake. 5 nm of chromium was deposited on the wafer via electron beam evaporation for enhancing adhesion of gold. A 100 nm layer of gold was then deposited on top of the chromium. Finally, lift off was performed using an ultrasonic cleaner in acetone. The spacing between the two electrodes is 20 µm, and the width of each electrode is 15 µm.
3.1.4 Microfluidic channel fabrication
The micro-channel consistes of a wider channel at the width of 300 µm and height of 20 µm, and then tapers down to a 30 µm wide and 20 µm high aperture for sensing. The smaller cross sectional area of the sensing pore improves the focusing of particles above the sensor, and increases electrical sensitivity, to the extent that biomarker can be differentiated. The micro-channel was made in PDMS using a master mold fabricated using soft lithography. The channel fabrication process consists of wafer cleaning, spin coating, soft bake, photo-pattering an inverse feature onto a 3″ silica wafer, UV light exposure, development, and finally a hard bake. To make the PDMS channels, pre-polymer and curing agent were mixed at a ratio of 10 to 1 and poured onto the master mold. After curing the PDMS for about 1 h, we peeled it off, cut the channels out and punched holes to make inlets and outlets. At last, the microfluidic channel was aligned to the electrode substrate and bonded onto the electrode wafer by treating both substrates with oxygen plasma.
3.2 Measurements
Impedance measurement of beads with different concentrations of IgG/IgA were performed three times each. In order to make the beads flow without the help of a syringe pump (to minimize external noise and interference), we first oxygen plasma treated the channel to make it hydrophilic and filled it with PBS to retain its hydrophilicity. Due to the pressure difference between the inlet and outlet, which was generated by the different volumes of buffer at the inlet and outlet, the beads were then injected into the inlet of the biosensor and injected through the multi-frequency impedance sensor, which was connected to a lock-in amplifier (Zurich Instrument HF2 series, Zurich, SI). The input AC voltage was 400 mV peak-to-peak and the transimpedance gain was 1 kV/amp. As particles passed over the electrodes, impedance responses at 8 discrete frequencies from 600 kHz to 25 MHz were captured. A metal box was used to cover the sensor to further decrease the external electrical interference during testing. The recorded data was then processed using a custom-written Matlab code which performed detrending, denoising, peak finding and differentiating the beads bound with IgG/IgA from the normal unbound beads using supervised machine learning. At the same time, in order to confirm the binding both electrically and optically, we observed the beads under a fluorescent microscope immediately after the electronic assay.
3.3 Machine learning algorithm
To improve classification accuracy and be able to detect the differences between IgG positive and IgG negative beads, we made use of a supervised machine learning classifier, the support vector machine (SVM). SVMs are an important class of supervised learning algorithms in classification and regression given that they have the ability to capture much more complex relationships between data points without having to perform difficult transformations. Peak amplitude at 8 frequencies is used as features in the feature vector. The benefit of using machine learning is that classification of different particles can be improved in hyper dimensional space. We used SVM with a Gaussian Kernel in MATLAB (MathWorks, Natick, MA, USA) to improve the classification accuracy where the box constraint level is 1 and manual kernel scale is 11.
The SVM classifier gets trained using a portion of the data, and the remainder of the data is used for testing. The training data consists of features collected from control sample where bead samples were incubated with 0 nM IgG/IgA and test sample where bead samples were incubated with IgG/IgA at different concentrations. The other features like antibody, bead types, incubation time remain all the same. In data training process, we labeled the features from beads with no IgG/IgA as 0, and features from beads with IgG/IgA as 1. The beads that are recorded by our sensors are all over 1500. Accuracy is calculated by comparing the accurately predicted number of target biomarker (IgG or IgA positive beads) and control beads with the true number of beads.
TP (true positive) is the number of times the classifier accurately predicted the bead has IgG/IgA, TN (true negative) is the number of times the machine accurately predicted the bead has no IgG/IgA. N is the total number of beads passing through the sensor. If the both control sample and testing samples have 0 nM IgG/IgA, the classifier would not be able to tell the difference between two samples, since they are exactly the same, and the accuracy would be 50% since the classifier can only guess between group 1 and 0. However, if testing sample has certain amount of IgG/IgA, the classifier would be able to detect the differences between the two particle types and thus the classification accuracy would be higher than 50%. The classification accuracy increases as concentration increases.
To quantify the amount of biomarker bound to the beads, we formulated a biomarker quantification score, which is defined as:
The utilization of the biomarker quantification score helps to provide a self-calibrated method of quantifying biomarker levels scaled from 0 to 100.
4 Results and discussion
The experiments were performed both optically and electrically at multiple IgG/IgA concentrations to verify the viability and repeatability of the two-minute assay. Figure 4A shows the fluorescent image of the positive assay where beads were incubated for two minutes in an IgG positive saliva sample at 1.67 μM and Fig. 4B for the assay where beads incubated for the same duration in an IgG positive saliva sample at 167 nM. Figure 4C shows the fluorescent image of the IgG negative assay. Similarly, we performed a 2-min assay for IgA in saliva sample both optically and electrically. The optical images of the IgA positive test and negative control assay were similar to that of the IgG sample, confirming that binding had occurred, which we do not show here. For both IgG and IgA, the IgG/IgA positive tests show stronger florescent signals at higher concentration. The IgG/IgA negative assay shows a black background, indicating that binding has not occurred. The images confirm that the IgG/IgA bind to the beads. As for the electrical test, when the sample was injected into the biosensor, pressure driven particles pass over the electrodes, which were connected to an AC voltage source and a multi-frequency lock-in amplifier, a frequency dependent change in current is exhibited as shown in the raw data (Fig. 1B). The amplitude of the current peak in different frequencies depends on bead size and conductivity and dielectric permittivity on the bead surface. Single and even dual frequency analysis alone makes it difficult to distinguish different bead types from each other as shown in the scatter plot for voltage peak intensity at 600 kHz and 14 or 21 MHz (Figs. 5A, B and 6A, B). When looking at average intensity of peak current response in 8 frequencies independently, a drop in average peak intensity is observed from high to low concentrations as a result of the amount of protein that bind to the beads (Figs. 5C and 6C). Nevertheless, when the concentration is lower than 150 nM, differences in average amplitude are too small to observe. It is also shown in Fig. 5C that the blue curve and purple curve which represent 0 nM and 16.7 nM samples are hard to differentiate by its impedance changing in a single frequency. The use of eight frequencies, in conjunction with machine learning, however, makes feasible of bead differentiation and thus target IgG/IgA quantification at tens of nanomolars. To achieve this, we first trained a machine learning classifier, the SVM (Support Vector Machine) model with peak intensity data from IgG/IgA positive and IgG/IgA negative samples at eight frequencies, and then used this model to test the accuracy of its ability to discriminate between IgG/IgA positive and IgG/IgA negative beads. As the concentration of target IgG/IgA in saliva decreases, SVM classification accuracy decreases. We used this classification accuracy as a metric to quantify IgG/IgA levels. We formulated a biomarker quantification score, defined as in (Eq. 3), to provide a self-calibrated method of quantifying biomarker levels. Figure 7A, B shows that the biomarker quantification score dynamic range is 3 orders of magnitude and has a repeatable detection limit (performed in triplicate) of 16.67 nM. Figure 7C shows that the score increases as more frequencies are measured, which reflects the improvement in detection limit.

(A) IgG labeled beads (high concentration) under fluorescent microscope (B) Fluorescent microscope image of IgG labeled beads (low concentration) (C) Fluorescent image of non-IgG labeled unbound beads

A Scatter plot for IgG sample peak voltage intensity at 600 kHz and peak voltage intensity at 21 MHz. B Scatter plot for IgG sample peak voltage intensity at 600 kHz and voltage intensity at 14 MHz. C Average amplitude changes for IgG when beads pass over electrodes at 8 different frequencies

A Scatter plot for IgA sample peak voltage intensity at 600 kHz and peak voltage intensity at 21 MHz. B Scatter plot for IgA sample peak voltage intensity at 600 kHz and peak voltage intensity at 14 MHz. C Average amplitude changes for IgA when beads pass over electrodes at 8 different frequencies

A Bargraph of Biomarker Quantification Score with respect to IgG concentrations ranging from 0 to 1667 nmol/L; B Bargraph of Biomarker Quantification Score with respect to IgA concentrations ranging from 0 to 1667 nmol/L; C Bargraph of Biomarker Quantification Score with respect to the number of frequencies used as features by the classifier. The increase in features improves classification accuracy
5 Conclusions
In conclusion, our experimental results show that we are able to qualify immunoglobulin G and immunoglobulin A in saliva within two minutes, through the combination of multi-frequency microfluidic impedance cytometry and supervised machine learning. Although in this work, IgG/IgA was only used as the biomarker for testing in saliva sample, the method can be applied to a wide variety of proteins as long as a comparable high affinity antibody pair is available. The work shown here has potential to be developed into an integrated biochip to rapidly quantify certain biomarker levels in complex samples like blood, saliva, and urine. Our future work will be focused on increasing the sensitivity for this assay from nanoMolar to picoMolar levels, applying this assay to diagnose specific diseases and integration of sample preparation onto the microfluidic chip.
Data availability
All study data are included in the article and/or SI Appendix.
References
K. Ahuja, G.M. Rather, Z. Lin, J. Sui, P. Xie, T. Le, J.R. Bertino, M. Javanmard, Toward point-of-care assessment of patient response: a portable tool for rapidly assessing cancer drug efficacy using multifrequency impedance cytometry and supervised machine learning. Microsyst. Nanoeng. 5(1), 1–11 (2019)
D.R. Burton, Immunoglobulin G: functional sites. Mol. Immunol. 22(3), 161–206 (1985)
A. Carbonaro, L. Sohn, A resistive-pulse sensor chip for multianalyte immunoassays. Lab Chip 5(10), 1155–1160 (2005)
M. Castagnola, P.M. Picciotti, I. Messana, C. Fanali, A. Fiorita, T. Cabras, L. Calo, E. Pisano, G.C. Passali, F. Iavarone, Potential applications of human saliva as diagnostic fluid. Acta Otorhinolaryngol. Ital. 31(6), 347 (2011)
L. Chen, F. Liu, X. Fan, J. Gao, N. Chen, T. Wong, J. Wu, S.W. Wen, Detection of hepatitis B surface antigen, hepatitis B core antigen, and hepatitis B virus DNA in parotid tissues. Int. J. Infect. Dis. 13(1), 20–23 (2009)
T.L. Chang, C.-Y. Tsai, C.-C. Sun, C.-C. Chen, L.-S. Kuo, P.-H. Chen, Ultrasensitive electrical detection of protein using nanogap electrodes and nanoparticle-based DNA amplification. Biosens. Bioelectron. 22(12), 3139–3145 (2007)
S.-Y. Cho, J.-H. Choi, Biomarkers of sepsis. Infect. Chemother. 46(1), 1–12 (2014)
A. Dane, T. Gürbüz, Clinical evaluation of specific oral and salivary findings of coeliac disease in eastern Turkish paediatric patients. Eur. J. Paediatr. Dent. 17(1), 53–56 (2016)
S. Fagarasan, T. Honjo, Intestinal IgA synthesis: regulation of front-line body defences. Nat. Rev. Immunol. 3(1), 63–72 (2003)
S. Gawad, L. Schild, P. Renaud, Micromachined impedance spectroscopy flow cytometer for cell analysis and particle sizing. Lab Chip 1(1), 76–82 (2001)
W. Giannobile, J. McDevitt, R. Niedbala, D. Malamud, Translational and clinical applications of salivary diagnostics. Adv. Dent. Res. 23(4), 375–380 (2011)
P.L. Gonçalves, C.B. Cunha, S.C. Busek, G.C. Oliveira, R. Ribeiro-Rodrigues, F.E. Pereira, Detection of hepatitis C virus RNA in saliva samples from patients with seric anti-HCV antibodies. Braz. J. Infect. Dis. 9(1), 28–34 (2005)
V. González, E. Martró, C. Folch, A. Esteve, L. Matas, A. Montoliu, J. Grifols, F. Bolao, C. Tural, R. Muga, Detection of hepatitis C virus antibodies in oral fluid specimens for prevalence studies. Eur. J. Clin. Microbiol. Infect. Dis. 27(2), 121–126 (2008)
M. Greabu, M. Battino, M. Mohora, A. Totan, A. Didilescu, T. Spinu, C. Totan, D. Miricescu, R. Radulescu, Saliva–a diagnostic window to the body, both in health and in disease. J. Med. Life 2(2), 124–132 (2009)
M. Javanmard, A.H. Talasaz, M. Nemat-Gorgani, F. Pease, M. Ronaghi, R.W. Davis, Electrical detection of protein biomarkers using bioactivated microfluidic channels. Lab Chip 9(10), 1429–1434 (2009)
K.E. Kaczor-Urbanowicz, C. Martin Carreras-Presas, K. Aro, M. Tu, F. Garcia-Godoy, D.T. Wong, Saliva diagnostics–Current views and directions. Exp. Biol. Med. 242(5), 459–472 (2017)
T.L. Lasseter, W. Cai, R.J. Hamers, Frequency-dependent electrical detection of protein binding events. Analyst 129(1), 3–8 (2004)
G. Lakos, L. Soos, A. Fekete, Z. Szabo, M. Zeher, I.F. Horvath, K. Dankó, A. Kapitány, A. Gyetvai, G. Szegedi, Anti-cyclic citrullinated peptide antibody isotypes in rheumatoid arthritis: association with disease duration, rheumatoid factor production and the presence of shared epitope. Clin. Exp. Rheumatol. 26(2), 253 (2008)
Z. Li, Y. Yi, X. Luo, N. Xiong, Y. Liu, S. Li, R. Sun, Y. Wang, B. Hu, W. Chen, Development and clinical application of a rapid IgM‐IgG combined antibody test for SARS‐CoV‐2 infection diagnosis. J. Med. Virol. (2020)
Z. Lin, X. Cao, P. Xie, M. Liu, M. Javanmard, PicoMolar level detection of protein biomarkers based on electronic sizing of bead aggregates: theoretical and experimental considerations. Biomed. Microdevice 17(6), 119 (2015)
D. Malamud, Saliva as a diagnostic fluid. Dent. Clin. 55(1), 159–178 (2011)
J. Mok, M.N. Mindrinos, R.W. Davis, M. Javanmard, Digital microfluidic assay for protein detection. Proc. Natl. Acad. Sci. 111(6), 2110–2115 (2014)
N.L. Rhodus, B. Cheng, S. Myers, L. Miller, V. Ho, F. Ondrey, The feasibility of monitoring NF-κB associated cytokines: TNF-α, IL-1α, IL-6, and IL-8 in whole saliva for the malignant transformation of oral lichen planus. Molecular Carcinogenesis: Published in Cooperation with the University of Texas MD Anderson Cancer Center 44(2), 77–82 (2005)
C. Streckfus, L. Bigler, Saliva as a diagnostic fluid. Oral Dis. 8(2), 69–76 (2002)
T. Shpitzer, Y. Hamzany, G. Bahar, R. Feinmesser, D. Savulescu, I. Borovoi, M. Gavish, R. Nagler, Salivary analysis of oral cancer biomarkers. Br. J. Cancer 101(7), 1194–1198 (2009)
T. Sun, H. Morgan, Single-cell microfluidic impedance cytometry: a review. Microfluid. Nanofluid. 8(4), 423–443 (2010)
G. Vidarsson, G. Dekkers, T. Rispens, IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 5, 520 (2014). Up-to-date review of IgG subclasse structure and their relationship to antibody effector functions. PubMedCentral 2014
J.M. Woof, M.A. Kerr, The function of immunoglobulin A in immunity. J. Pathol. 208(2), 270–282 (2006)
J.M. Yoshizawa, C.A. Schafer, J.J. Schafer, J.J. Farrell, B.J. Paster, D.T. Wong, Salivary biomarkers: toward future clinical and diagnostic utilities. Clin. Microbiol. Rev. 26(4), 781–791 (2013)
P. Xie, X. Cao, Z. Lin, Javanmard M. Topdown fabrication meets bottom-up synthesis for nanoelectronic barcoding of microparticles. Lab on a Chip 17(11), 1939–1947 (2017a)
P. Xie, X. Cao, Z. Lin, N. Talukder, S. Emaminejad, M. Javanmard, Processing gain and noise in multi-electrode impedance cytometers: comprehensive electrical design methodology and characterization. Sensors and Actuators B: Chemical 241, 672–680 (2017b)
S. Emaminejad, KH. Paik, V. Tabard-Cossa, M. Javanmard, Portable cytometry using microscale electronic sensing. Sens. Actuators B: Chem. 224, 275–281 (2016)
M. Javanmard, F. Babrzadeh, RW. Davis, Microfluidic force spectroscopy for characterization of biomolecular interactions with piconewton resolution. Appl. Phys. Lett. 97(17), 173704 (2010)
A. Gholizadeh, M. Javanmard, Magnetically actuated microfluidic transistors: Miniaturized micro-valves using magnetorheological fluids integrated with elastomeric membranes. J. Microelectromech. Syst. 25(5), 922–928 (2016)
Acknowledgements
This project was primarily funded by an anonymous corporation and partially by the PhRMA Foundation Starter Grant for Early Faculty. The devices were fabricated in the Weeks Hall Micro-Nanofabrication and Characterization Facilty in the School of Engineering at Rutgers University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
The author declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lin, Z., Sui, J. & Javanmard, M. A two-minute assay for electronic quantification of antibodies in saliva enabled through a reusable microfluidic multi-frequency impedance cytometer and machine learning analysis. Biomed Microdevices 25, 13 (2023). https://doi.org/10.1007/s10544-023-00647-1
Accepted:
Published:
DOI: https://doi.org/10.1007/s10544-023-00647-1