
Abstract
The clinical efficacy and safety of a drug is determined by its molecular properties and targets in humans. However, proteome-wide evaluation of all compounds in humans, or even animal models, is challenging. In this study, we present an unsupervised pretraining deep learning framework, named ImageMol, pretrained on 10 million unlabelled drug-like, bioactive molecules, to predict molecular targets of candidate compounds. The ImageMol framework is designed to pretrain chemical representations from unlabelled molecular images on the basis of local and global structural characteristics of molecules from pixels. We demonstrate high performance of ImageMol in evaluation of molecular properties (that is, the drug’s metabolism, brain penetration and toxicity) and molecular target profiles (that is, beta-secretase enzyme and kinases) across 51 benchmark datasets. ImageMol shows high accuracy in identifying anti-SARS-CoV-2 molecules across 13 high-throughput experimental datasets from the National Center for Advancing Translational Sciences. Via ImageMol, we identified candidate clinical 3C-like protease inhibitors for potential treatment of COVID-19.
Similar content being viewed by others
Main
Despite recent advances in biomedical research and technologies, drug discovery and development remains a challenging multidimensional task requiring optimization of vital properties of candidate compounds, including pharmacokinetics, efficacy and safety1,2. It was estimated that pharmaceutical companies spent $2.6 billion in 2015, up from $802 million in 2003, on drug approval by the US Food and Drug Administration3. The increasing cost of drug development resulted from lack of efficacy of the randomized controlled trials, and the unknown pharmacokinetics and safety profiles of candidate compounds4,5,6. Traditional experimental approaches are unfeasible in proteome-wide evaluation of molecular targets for all candidate compounds in humans, or even animal models. Computational approaches and technologies have been considered a promising solution7,8, which can substantially reduce costs and time during the complete pipeline of drug discovery and development.
The rise of advanced artificial intelligence technologies9,10 motivated their application to drug design11,12,13 and target identification14,15,16. One of the fundamental challenges is how to learn molecular representation from chemical structures17. Previous molecular representations were based on hand-crafted features, such as fingerprint-based features16,18, physiochemical descriptors and pharmacophore-based features19,20. However, these traditional molecular representation methods rely on a large amount of domain knowledge to extract molecular features, such as functional-connectivity fingerprints21 and extended-connectivity fingerprints22. Compared with traditional representation methods, automatic molecular representation learning models perform better on most drug discovery tasks23,24,25. With the rise of unsupervised learning in natural language processing26,27, recent approaches that incorporate unsupervised learning with one-dimensional sequential strings, such as the simplified molecular-input line-entry system (SMILES)28,29,30,31 and International Chemical Identifier (InChI)32,33,34, or two-dimensional (2D) graphs35,36,37,38,39, have also been developed for various computational drug discovery tasks. Yet, their accuracy in extracting informative vectors for description of molecular identities and biological characteristics of the molecules is limited. Recent advances of unsupervised learning in computer vision40,41 suggest that it is possible to apply unsupervised image-based pretraining models for computational drug discovery.
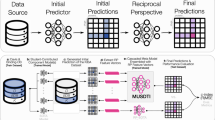
In this study, we developed an unsupervised molecular image pretraining framework (named ImageMol) with chemical awareness for learning the molecular structures from large-scale molecular images. ImageMol combines an image processing framework with comprehensive molecular chemistry knowledge for extracting fine pixel-level molecular features in a visual computing way. Compared with state-of-the-art methods, ImageMol has two important improvements: (1) it utilizes molecular images as the feature representation of compounds with high accuracy and low computing cost; (2) it exploits an unsupervised pretrained learning framework to capture the structural information of molecular images from 10 million drug-like compounds with diverse biological activities at the human proteome (Fig. 1). We demonstrated the high accuracy of ImageMol in a variety of drug discovery tasks. Via ImageMol, we identified anti-SARS-CoV-2 molecules across 13 high-throughput experimental datasets from the National Center for Advancing Translational Sciences. In summary, ImageMol provides a powerful pretraining deep learning framework for computational drug discovery.

a, A molecular encoder (light blue throughout) is used to extract the latent features of the molecular images. b, Five strategies are used to pretrain the molecular encoder. The structural classifier (dark blue) in MG3C is used to predict chemical structural information in molecular images. The rationality classifier (green) in MRD is used to distinguish rational and irrational molecules. The jigsaw classifier (light grey) in jigsaw puzzle prediction is used to predict rational permutations. The contrastive classifier (dark grey) in mask-based contrastive learning is used to maximize the similarity between the original image and the masked image. The generator (yellow) in molecular image reconstruction is used to restore latent features to the molecular image and the discriminator (purple) is used to discriminate between real and fake molecular images. c, ImageMol for discovery of anti-SARS-CoV-2 inhibitors. A fully connected layer is appended to the pretrained molecular encoder for fine-tuning on the COVID-19 dataset. Subsequently, the fine-tuned model is used for virtual screening from approved drugs in DrugBank. The 75% success rate of the top 20 drugs as potential inhibitors of COVID-19 has been validated by experimental and clinical evidence.
Results
Description of ImageMol
Here, we developed a pretraining deep learning framework, ImageMol, for accurate prediction of molecular targets. ImageMol pretrained 9,999,918 images of drug-like, bioactive molecules from PubChem databases42. We assembled five pretext tasks to extract biologically relevant structural information: (1) a molecular encoder is designed to extract latent features from ~10 million molecular images (Fig. 1a); (2) five pretraining strategies (Supplementary Figs. 1–5) are utilized to optimize the latent representation of the molecular encoder by considering the chemical knowledge and structural information from molecular images (Fig. 1b); (3) a pretrained molecular encoder is fine-tuned on downstream tasks to further improve model performance (Fig. 1c).
Benchmark evaluation of ImageMol
We first evaluated the performance of ImageMol using eight types of benchmark dataset for drug discovery (Supplementary Tables 1–4): (1) molecular targets—human immunodeficiency virus (HIV), beta-secretase (BACE, a key target in Alzheimer’s disease) and maximum unbiased validation (17 targets for virtual screening); (2) blood–brain barrier penetration (BBBP); (3) the drug’s metabolism and side effect resource; (4) molecular toxicities—toxicity using the Toxicology in the 21st Century (Tox21) and clinical trial toxicity (ClinTox) databases and Toxicity Forecaster (ToxCast); (5) solubility—Free Solvation (FreeSolv) and Estimated Solubility (ESOL)—and lipophilicity; (6) quantum—Quantum Machine 7 (QM7) and Quantum Machine 9; (7) ligand–GPCR (G protein-coupled receptor) binding activity; (8) compound–kinase binding activity (Methods).
We used the three popular split strategies (scaffold split36,43,44 balanced scaffold split35, and random scaffold split37,45) to evaluate the performance of ImageMol on all benchmark datasets. In scaffold, random scaffold split and balanced scaffold split, the datasets are divided according to the molecular substructures: the substructures in the training set, validation set and test set are disjoint, making them ideal to test robustness and generalizability of models. In a classification task, using the area under the receiver operating characteristic (ROC) curve (AUC), ImageMol achieves high AUC values (Fig. 2a) with random scaffold split across BBBP (AUC = 0.952), Tox21 (AUC = 0.847), ClinTox (AUC = 0.975), BACE (AUC = 0.939), Side Effect Resource (AUC = 0.708) and ToxCast (AUC = 0.752). We found similar results on scaffold split as well (Fig. 2a). In a regression task, ImageMol achieves low error values with scaffold split (Supplementary Table 5) across FreeSolv (root-mean-square error, RMSE = 2.02), ESOL (RMSE = 0.97), lipophilicity (RMSE = 0.72) and Quantum Machine 9 (mean absolute error = 3.724) and with random scaffold split (Supplementary Table 6) across FreeSolv (RMSE = 1.149), ESOL (RMSE = 0.690), lipophilicity (RMSE = 0.625) and QM7 (mean absolute error = 65.9). In addition, the probability distributions of ImageMol on BBBP and BACE datasets have similarity greater than 95%, revealing that ImageMol has high consistency and stability during training (Supplementary Fig. 6). On the basis of more comprehensive evaluation metrics (including accuracy, AUC, AUPR—area under the precision–recall curve, F1 score, precision, recall, kappa and confusion matrix), we found that ImageMol is able to achieve high performance across these metrics (Supplementary Tables 7 and 8 and Supplementary Figs. 7 and 8). For a fair comparison in Fig. 2b and Supplementary Fig. 9, we used the same experimental set-up as Chemception46, a state-of-the-art convolutional neural network (CNN) framework. ImageMol achieves elevated AUC values on HIV (AUC = 0.814) and Tox21 (AUC = 0.826) compared with Chemception, suggesting that ImageMol can capture more biologically relevant information from molecular images than Chemception. We further evaluated the performance of ImageMol in prediction of drug metabolism across five major metabolic enzymes: CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 (Methods). Figure 2c shows that ImageMol achieves higher AUC values (ranging from 0.799 to 0.893) in the prediction of inhibitors versus non-inhibitors across five major drug metabolism enzymes as well, compared with three state-of-the-art molecular image-based representation models: Chemception46, ADMET-CNN12 and QSAR-CNN47. Additional results of the detailed comparison are provided in Supplementary Fig. 10.

The performance was evaluated in a variety of drug discovery tasks, including molecular properties (that is, drug metabolism, toxicity, brain penetration) and molecular target profiles (that is, HIV and BACE). a–c, FPR, false positive rate; TPR, true positive rate The AUC values are given in each panel. a, ROC curves of ImageMol across eight datasets (BBBP, Tox21, HIV, ClinTox, BACE, Side Effect Resource (SIDER), maximum unbiased validation (MUV) and ToxCast) with scaffold split and random scaffold split. b, ROC curves of Chemception46 and ImageMol on HIV and Tox21 datasets with the same experimental set-up as for Chemception, which is a classical CNN for predicting molecular images. c, ROC curves of Chemception, ADMET-CNN12, QSAR-CNN47 and ImageMol on five CYP isoform validation sets (PubChem data set II). ADMET-CNN and QSAR-CNN are the latest molecular image-based drug discovery models. d, The ROC-AUC performance of sequence-based, graph-based and fingerprint-based models and ImageMol across six classification datasets (BBBP, Tox21, BACE, ClinTox, SIDER and ToxCast) with random scaffold split. For each type of method the maximum value is selected for display. e, The ROC-AUC performance across four regression datasets (FreeSolv, ESOL, lipophilicity (Lipo) and QM7) with random scaffold split. For each type of method the maximum value is selected for highlighting. For aesthetic presentation, the results of FreeSolv and QM7 are scaled down by a factor of 10 and 100, respectively. MAE, mean absolute error. f, The ROC-AUC performance of fingerprint-based (MACCS-based and FP4-based) methods and ImageMol across five major CYP isoform validation sets (PubChem data set II). g, The ROC-AUC performance of sequence-based and graph-based models across CYP450 datasets with balanced scaffold split.
We further compared the performance of ImageMol with three types of state-of-the-art molecular representation model: (1) fingerprint-based, (2) sequence-based and (3) graph-based models. As shown in Fig. 2d,e, ImageMol has better performance compared with fingerprint-based (for example, AttentiveFP11), sequence-based (for example, TF_Robust48) and graph-based models (for example, N-GRAM45, GROVER35 and MPG37) using a random scaffold split. In addition, ImageMol achieved higher AUC values (Fig. 2f) on CYP1A2 (AUC = 0.852), CYP2C9 (AUC = 0.870), CYP2C19 (AUC = 0.871), CYP2D6 (AUC = 0.893) and CYP3A4 (AUC = 0.799) compared with traditional MACCS-based methods and FP4-based methods49 across multiple machine learning algorithms, including support vector machine, decision tree, k-nearest neighbours, naive Bayes and their ensemble models49, across all five cytochrome P450 (CYP) isoform datasets (Supplementary Table 9). Compared with sequence-based (including RNN_LR, TRFM_LR, RNN_MLP, TRFM_MLP, RNN_RF, TRFM_RF50 and CHEM-BERT51) and graph-based models (including MolCLRGIN, MolCLRGCN39 and GROVER35), ImageMol achieves better AUC performance than other methods (Fig. 2g) across CYP1A2 (AUC = 0.912), CYP2C9 (AUC = 0.858), CYP2C19 (AUC = 0.873), CYP2D6 (AUC = 0.827) and CYP3A4 (AUC = 0.903) (the AUCs and confusion matrix are provided in Supplementary Figs. 11 and 12 respectively) and achieves elevated performance using other performance metrics (Supplementary Table 10) as well. We extended CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 to multilabelled classification, and ImageMol achieved state-of-the-art performance (AUCs and confusion matrix in Supplementary Figs. 13 and 14 respectively) on multiple evaluation metrics (Supplementary Table 11). In compound–protein binding prediction tasks, ImageMol achieved better performance (AUCs and confusion matrix in Supplementary Figs. 15 and 16) on ten GPCRs (regression task) and ten kinases (classification task) compared with existing approaches as well (Supplementary Tables 12 and 13).
We further used the McNemar test to assess the statistical significance of performance differences among state-of-the-art models and ImageMol. ImageMol shows statistically elevated performance compared with existing methods on multiple datasets (Supplementary Tables 11 and 13–17). The detailed comparisons of ImageMol with each model/method are provided in Supplementary Results. Altogether, ImageMol achieves improved performance in various drug discovery tasks, outperforming state-of-the-art methods (Fig. 2a–g, Supplementary Figs. 9–16 and Supplementary Tables 5–17).
Prediction of antiviral activities across 13 SARS-CoV-2 targets
The ongoing global COVID-19 pandemic caused by the SARS-CoV-2 virus has led to more than 1.1 billion confirmed cases and over 6 million deaths worldwide as of 15 March 2022. There is a critical, time-sensitive need to develop effective antiviral treatment strategies for the COVID-19 pandemic6,52. We therefore test ImageMol to identify potential anti-SARS-CoV-2 treatments across a variety of SARS-CoV-2 biological assays, including viral replication, viral entry, counterscreen, in vitro infectivity and live virus infectivity20. In total, we evaluated ImageMol across 13 SARS-CoV-2 bioassay datasets (Supplementary Table 18).
Across 13 SARS-CoV-2 bioassay datasets, ImageMol achieves high AUC values ranging from 72.6% to 83.7% (Fig. 3a). To test whether ImageMol capture biologically relevant features, we used the global average pooling layer of ImageMol to extract latent features and used t-distributed stochastic neighbour embedding (t-SNE) to visualize latent features. Figure 3a reveals that the latent features identified by ImageMol are well clustered according to whether they are active or inactive anti-SARS-CoV-2 agents across all 13 targets or endpoints. These observations show that ImageMol can accurately extract discriminative, antiviral features from molecular images for downstream tasks.

a, ROC curves and t-SNE visualizations across 13 high-throughput experimental SARS-CoV-2-related datasets. The ROC curves include Jure’s GNN and ImageMol. The t-SNE visualizations are produced by using the latent features of the global average pooling layer of our ImageMol. b, t-SNE visualization of the 3CL dataset from the antiviral activity prediction task. The molecules with activity concentration 50% of less than 10 were treated as inhibitors and those with greater than 10 were treated as non-inhibitors. From blue to red indicates the feature embedding of 3CL non-inhibitors and inhibitors. The grey dots indicate drugs from DrugBank. c, Drug discovery of 3CL potential inhibitors on the DrugBank dataset. The black dots represent the probability distribution of drug molecules from DrugBank, and the white dots represent the known 3CL inhibitors found by ImageMol. d, Molecular structure of the 3CL inhibitors discovered by ImageMol.
We further compared ImageMol with both deep learning and machine learning frameworks: (1) a graph neural network (GNN) with a series of pretraining strategies (termed Jure’s GNN53) and (2) REDIAL-202020, a suite of machine learning models for estimating small-molecule activities in a range of SARS-CoV-2-related assays. We found that ImageMol notably outperforms Jure’s GNN models across all 13 SARS-CoV-2 targets (Fig. 3a and Supplementary Table 19). For instance, the AUC values of ImageMol (AUC = 0.837) compared with Jure’s GNN model (AUC = 0.704) in prediction of 3-chymotrypsin-like (3CL) protease inhibitors are elevated by over 12%. We further evaluated the AUPR, which is highly sensitive to the imbalance issues of positive versus negative labelled data. Compared with Jure’s GNN models, the elevated AUPR of ImageMol ranges from 1.9% to 25.6% with a performance advantage of 6.4% on average across 13 SARS-CoV-2 bioassay datasets, in particular for 3CL protease inhibitors (12.3% AUPR improvement) and ACE2 enzymatic activities (25.6% AUPR improvement). To compare with REDIAL-202020, we used the same experimental settings and performance evaluation metrics, including accuracy, sensitivity, precision, F1 (the harmonic mean between sensitivity and precision) and AUC. We found that ImageMol outperformed REDIAL-2020 as well (Supplementary Table 20). To test the generalization of ImageMol across 13 anti-SARS-CoV-2 bioassay datasets, we split the datasets using a balanced scaffold split. Compared with both sequence-based and graph-based models, ImageMol achieves better performance (including accuracy, AUC, AUPR, F1 score, kappa, confusion matrix) than other methods (Supplementary Table 21 and Supplementary Figs. 17 and 18). In addition, a McNemar test showed that ImageMol achieves statistically higher performance compared with existing methods on multiple anti-SARS-CoV-2 bioassay datasets.
In summary, these comprehensive evaluations reveal a high accuracy of ImageMol in identifying anti-SARS-CoV-2 molecules across diverse viral targets and phenotypic assays. Furthermore, ImageMol is more capable on datasets with extreme imbalance of positive and negative samples compared with traditional deep learning pretrained models53 or machine learning approaches20 and has strong generalization compared with sequence-based50,51 and graph-based models35,39.
Identifying anti-SARS-CoV-2 inhibitors via ImageMol
We next turned to identification of potential anti-SARS-CoV-2 inhibitors using 3CL protease as a prototypical example, as it has been shown to be a promising target for therapeutic development in treating COVID-1954,55. We focused on 2,501 US Food and Drug Administration-approved drugs from DrugBank56 to identify ImageMol-predicted 3CL protease inhibitors as drugs repurposable for COVID-19 using a drug repurposing strategy52.
Via molecular image representation of the 3CL protease inhibitor versus non-inhibitor dataset under the ImageMol framework, we found that 3CL inhibitors and non-inhibitors are well separated in a t-SNE plot (Fig. 3b). Molecules with activity concentration 50% less than 10 μM were defined as inhibitors; otherwise they were non-inhibitors. We showed the probability of each drug in DrugBank being inferred as a 3CL protease inhibitor (Supplementary Table 22) and visualized their overall probability distribution (Supplementary Fig. 19). We found that 11 of the top 20 drugs (55%) have been validated (including cell assay, clinical trial or other evidence) as potential SARS-CoV-2 inhibitors (Supplementary Table 22), among which two drugs are further verified as potential 3CL protease inhibitors by biological experiments. To test the generalization ability of ImageMol, we used 16 experimentally reported 3CL protease inhibitors as an external validation set (Supplementary Table 23). ImageMol identified 10 out of 16 known 3CL protease inhibitors and visualized these 10 drugs to embedding space in Fig. 3c (62.5% success rate, Fig. 3d), suggesting a high generalization ability in anti-SARS-CoV-2 drug discovery.
We further used the HEY293 assay to predict anti-SARS-CoV-2 repurposable drugs. We collected experimental evidence for the top 20 drugs as potential SARS-CoV-2 inhibitors (Supplementary Table 24). We found that 15 out of 20 drugs (75%) have been validated by different experimental assays as potential inhibitors for the treatment of SARS-CoV-2 (such as in vitro cellular assays and clinical trials) as shown in Supplementary Table 24. Meanwhile, 122 drugs have been identified to block SARS-CoV-2 infection57. From these drugs, we selected a total of 70 small molecules overlapping in DrugBank to evaluate the performance of the KEY293 model. We found that ImageMol successfully predicted 42 out of 70 (60% success rate, Supplementary Table 25), suggesting a high generalizability of ImageMol for inferring potential candidate drugs in the HEY293 assay as well.
Biological interpretation of ImageMol
We next turned to using t-SNE to visualize molecular representations from different models to test the biological interpretation of ImageMol. We used the clusters identified by the multigranularity chemical cluster classification (MG3C) task (Methods) to split the molecular structures. We randomly selected 10% of clusters obtained from MG3C and sampled 1,000 molecules for each cluster. We performed three comparisons for each molecule: (1) MACCS fingerprints with 166-dimensional features, (2) ImageMol without pretrained models with 512-dimensional features and (3) ImageMol pretrained 512-dimensional features. We found that ImageMol distinguishes molecular structures very well (Fig. 4e and Supplementary Fig. 20c), outperforming MACCS fingerprints (Supplementary Fig. 20a) and non-pretrained models (Supplementary Fig. 20b). ImageMol can capture prior knowledge of chemical information from the molecular image representations, including the =O bond, –OH bond, –NH3 bond and benzene ring (Fig. 4a). We further used the Davies–Bouldin (DB) index58 to quantitatively evaluate the clustering results: smaller DB index represents better performance. We found that ImageMol (DB index 1.92) was better than MACCS fingerprint (DB index 2.93); furthermore, pretrained models can greatly improve the molecular representation as well (DB index of ImageMol without pretraining is 19.40).

a, Examples of ImageMol’s feature maps. Hotter colour area indicates higher ImageMol attention. b,c, ImageMol’s heatmaps (b, global; c, local) of several molecular images whose structures are highlighted by Grad-CAM. The warmer the colour, the higher the attention of the area, and the colder the colour, the lower the attention of the area. In particular, the red area indicates that the model pays the highest attention to it, while the light blue indicates that the model does not pay any attention to it. d, The average heat map of all molecular images on each dataset, using Grad-CAM to obtain the heat map of each molecular image in the dataset and calculate the average of these heat maps in each dimension. e, The variable probability distribution figures (principal diagonal) and the kernel density estimate figures (subdiagonal) of representations learned by ImageMol. The representations extracted by ImageMol are dimensionally reduced by t-SNE. The different colours indicate different clusters.
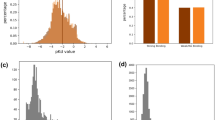
Gradient-weighted Class Activation Mapping (Grad-CAM)59 is a commonly used CNN visualization method60,61. Figure 4b,c illustrates 12 example molecules of the Grad-CAM visualization of ImageMol (Supplementary Figs. 21 and 22). ImageMol accurately captures attention to the global (Fig. 4b) and the local (Fig. 4c) structural information simultaneously. Since the warmer colour in Grad-CAM is sometimes centred in the blank areas of the image and covers more than 10 atoms, we found that setting a high attention value to capture the highlighted areas was more informative for chemists (Supplementary Table 26). Specifically, it captures key substructures related to a certain biological function by observing a pair of molecules with similar structures but different biological activities (Supplementary Fig. 23). In addition, we counted the proportion of blank areas in the images to the entire molecular image across 11 SARS-CoV-2 datasets (Supplementary Table 27). We found an average sparsity (sparsity refers to the proportion of blank areas in an image) of 94.9% across the entire dataset, suggesting that ImageMol models are easily inclined to use blank areas of the image for meaningless inferences47. Figure 4d shows that ImageMol primarily pays attention to the middle area of the image during predictions. Thus, ImageMol indeed predicts on the basis of the molecular structures rather than using meaningless blank areas. We further calculated the coarse-grained and fine-grained hit rates (Supplementary Fig. 24). The coarse-grained hit rate illustrates that ImageMol can utilize molecular structures of all images for inference, with a ratio of 100%, compared with the QSAR-CNN models47 with 90.7%. The fine-grained hit rate shows that ImageMol can leverage almost all structural information in molecular images to inference, with a ratio of over 99%, reflecting its ability to capture global information of molecules.
In summary, ImageMol captures the biologically relevant chemical information of molecular images with both local and global levels of structural information, outperforming existing state-of-the-art deep learning approaches (Fig. 4).
Ablation analysis of ImageMol
The robustness of the model to hyperparameters is important because different hyperparameters can affect the performance of the model62. As shown in Supplementary Fig. 25, we found that ImageMol has lower s.d. than ImageMol_NonPretrained (average s.d. of 0.5% versus 8.9% on the classification task and 0.654 versus 1.68 on the regression task), which demonstrates that pretraining strategies improve the robustness of ImageMol to hyperparameters.
We explored the impact of pretraining with different data scales and found that the average ROC-AUC performance increased from 1.2% to 10.2% as the pretrained data scale increased (Supplementary Fig. 26). Thus, ImageMol can be further improved as more drug-like molecules can be pretrained. We further investigated the impact of different pretext tasks (Methods) and found that each pretext task improves the mean AUC value of ImageMol from 0.7% to 4.9% (Supplementary Fig. 27). More details of ablation studies (including data augmentation) can be found in Supplementary Note C.5 and Supplementary Table 28. In summary, each task integrated implemented the ImageMol framework to synergistically improve performance, and models can be improved further by pretraining from larger drug-like chemical datasets in the future.
Discussion
We have presented a self-supervised image-processing-based pretraining deep learning framework that combines molecular images and unsupervised learning to learn molecular representations. We demonstrated the high accuracy of ImageMol across multiple benchmark biomedical datasets with a variety of drug discovery tasks (Figs. 2 and 3). In particular, we identified candidate anti-SARS-CoV-2 agents, which were validated by ongoing clinical and experimental data across 13 biological anti-SARS-CoV-2 assays. If broadly applied, our pretraining deep learning framework will offer a powerful tool for rapid drug discovery and development for various emerging diseases, including the COVID-19 pandemic and future pandemics as well.
We highlighted several improvements of ImageMol compared with existing state-of-the-art methods. First, ImageMol achieved high performance across diverse tasks of drug discovery, including drug-like property assessment (brain permeability, drug metabolism and toxicity) and molecular target prediction across diverse targets, such as Alzheimer’s disease (that is, BACE) and emerging infectious diseases caused by HIV and SARS-CoV-2 virus. Furthermore, ImageMol outperforms state-of-the-art methods, including sequence-based, fingerprint-based and graph-based representation methods (Fig. 2). Finally, ImageMol has better interpretability and is more intuitive in identifying biologically relevant chemical structures or substructures for molecular properties and target binding (Fig. 4a–c).
We acknowledge several limitations. Although we mitigated the effects of different representations of molecular images through data augmentation, perturbed views (that is, rotation and scaling) of the input images may still affect the prediction results of ImageMol. We did not optimize for the sparsity of molecular images, which may affect the latent features extracted by the model. Our current ImageMol framework cannot capture three-dimensional (3D) structural information on ligands, receptors and ligand–receptor interactions. It is challenging to explicitly define the chemical properties of atoms and bonds compared with graph-based methods45,53, which will inevitably lead to insufficient chemical information. Several potential directions may improve ImageMol further: (1) integration of larger-scale biomedical data and larger-capacity models (such as ViT63) in molecular images will inevitably be the focus of future work; (2) multiview learning of joint images and other representations (for example SMILES and graph) is an important research direction; (3) incorporating more chemical knowledge (such as atomic properties, chemical properties and 3D structural information) into each image or pixel area is also a promising future direction. Specifically, we can visualize atoms or atomic fragments of different chemical properties as different colours in molecular images. For example, high hydrophobicity versus low hydrophobicity (for example, on the basis of Ghose–Crippen atom types) and different charge properties (for example, Gasteiger–Marsili partial charges) can be visualized as differently coloured atomic fragments. Meanwhile, integration of 3D structural information on ligands, receptors and ligand–receptor interactions is also crucial for determining molecular properties and drug effects, which is promising for further improving the performance of ImageMol. Therefore, we will integrate more atomic properties and 3D information into molecular images (for example, ligand–receptor complex) to further develop ImageMol version 2.0. In summary, ImageMol is an active self-supervised image processing-based strategy that offers a powerful toolbox for computational drug discovery in a variety of human diseases, including COVID-19.
Methods
Strategies for pretraining ImageMol
Pretraining aims to make the model learn how to extract expressive representations by training on large-scale unlabelled datasets and then applying the well pretrained model to related downstream tasks and fine-tuning to improve performance. Definition of several effective and task-related pretext tasks is required to pretrain the model. In this Article, the core of our pretraining strategy is the visual representation of molecules by considering three principles: consistency, relevance and rationality. These principles lead ImageMol to capture meaningful chemical knowledge and structural information from molecular images. Specifically, consistency means that the semantic information of the same chemical structure in different images is consistent, such as –OH, =O, benzene. Relevance means that different augmentations of the same image (such as mask, shuffle) are related in the feature space. For example, the distribution of the image after the mask should be close to that of the original image. Rationality means that the molecular structure must conform to chemical common sense. The model needs to recognize the rationality of the molecule to promote the understanding of the molecular structure. Unlike graph-based and SMILES-based pretraining methods (they consider either only consistency or only correlation), ImageMol is a molecular image-based pretraining framework and considers multiple principles comprehensively by five defined effective pretext tasks.
Consistency for pretraining
Considering that the semantic information of the same chemical structure in different images is consistent, the MG3C task (Supplementary Fig. 1), which discovers semantic consistency by predicting the chemical structure of the molecule, is proposed. Briefly, multigranularity clustering is first used to assign multiple clusters of different granularities to each chemical structural fingerprint. Then, each cluster is assigned as a pseudolabel to the corresponding molecule and each molecule has multiple pseudolabels with different granularities. Finally, a molecular encoder is employed to extract the latent features of the molecular images and a structural classifier is used to classify the pseudolabels.
Specifically, we employed the MACCS key, which is a 166-length sequence composed of 0 and 1 values, as the descriptor of molecular fingerprints. These molecular fingerprint sequences can be used as a basis for clustering: the shorter the distance between molecular fingerprints the more likely they are to belong to a cluster. Finally, we use the K means64 with different K = 100, 1,000, 10,000 (see Supplementary Note A.2 and Supplementary Fig. 28 concerning selection of K) to cluster molecules to obtain clusters with different granularities from coarse grained to fine grained. According to the clustering results, we assigned three pseudolabels to each molecular image and then applied ResNet1865 as a molecular encoder to extract a latent feature and a structural classifier to predict the pseudolabels of the latent feature. The structural classifier is multitask, consisting of three parallel fully connected layers corresponding to three different clustering granularities. The numbers of neurons of each fully connected layer are 100, 1,000 and 10,000, respectively. Formally, the molecular image and the three corresponding pseudolabels are represented by \(x_n \in {\Bbb R}^{224 \times 224 \times 3}\), \({{{\mathcal{Y}}}}_n^{100} \in \left\{ {0,1,...,99} \right\}^{100}\), \({{{\mathcal{Y}}}}_n^{1{,}000} \in \left\{ {0,1,...,999} \right\}^{1{,}000}\) and \({{{\mathcal{Y}}}}_n^{10{,}000} \in \left\{ {0,1,...,9{,}999} \right\}^{10{,}000}\), respectively, and the cost function \({{{\mathcal{L}}}}_{\mathrm{MG3C}}\) of the MG3C task is as follows:
where fθ and θ refer to the mapping function and corresponding parameters of the molecular encoder, respectively. w100, w1,000 and w10,000 represent the parameters of three fully connected classification layers in a structural classifier with 100, 1,000 and 10,000 neurons, respectively. W represents all parameters of w100, w1,000 and w10,000. \(\ell\) is the multinomial logistic loss or the negative log-softmax function.
Relevance for pretraining
On the basis of the assumption that different augmentations (such as mask, shuffle) of the same image are related in the feature space, we use a pixel-level task to reconstruct molecular images from latent features and use an image-level task to maximize the correlation between the original sample and the mask sample in that space.
Molecular image reconstruction
Molecular image reconstruction reconstructs the latent features to the molecular images. We input the original molecular image xn into the molecular encoder to obtain the latent feature fθ(xn). To make the model learn the correlation between the molecular structures in the image, we shuffle and rearrange the input image xn (as in Rationality for pretraining) in the hope that the correct image can be reconstructed. After this, we define a generator G and a discriminator D to reconstruct the latent features. G is composed of four 2D deconvolution layers with a batch normalization 2D layer and ReLU (rectified linear unit) activation function, and one deconvolution layer with a tanh activation function. The discriminator is also composed of four 2D convolutional layers with a batch normalization 2D layer, a LeakyReLU activation function and one 2D convolutional layer with a sigmoid activation function. For further details of the generative adversarial network model see Supplementary Fig. 5. Since it is difficult for the generator to reconstruct the latent features to 224 × 224 molecular images, we simplify the task to reconstruct the latent features to 64 × 64 molecular images. The discriminator accepts 64 × 64 molecular images and distinguishes real or fake images. In detail, first the generator is used to reconstruct the latent feature fθ(xn) to a 64 × 64 molecular image \({\tilde{x}}_{n}^{64 \times 64} = G\left( f_{\theta} \left( {x_{n}} \right) \right)\). Then, we resize the original molecular image xn of 224 × 224 to the molecular image \(x_{n}^{64 \times 64}\) of 64 × 64 and input it into D together with the molecular image generated by G at the same time to obtain \(D\left(x_{n}^{64 \times 64}\right)\) and \(D\left(G\left(f_{\theta} \left(x_n\right)\right)\right)\). Finally, we update the parameters of the generator and the discriminator through their cost functions \({{{\mathcal{L}}}}_G\) and \({{{\mathcal{L}}}}_D\) respectively, which are defined as
For \({{{\mathcal{L}}}}_G\), the first term represents Wasserstein loss, and the second term represents the Euclidean distance between the generated image G(fθ(xn)) and the corresponding real image \(x_n^{64 \times 64}\). For \({{{\mathcal{L}}}}_D\), we use this loss to approximate the Wasserstein distance of the distribution of real images \(x_n^{64 \times 64}\) and fake images \(G(f_{\theta}(x_n))\). Finally, the molecular encoder model is updated by using the cost function \({{{\mathcal{L}}}}_{\mathrm{MIR}}\), which was formalized as
Mask-based contrastive learning
Recently, the performance gap between unsupervised pretraining and supervised learning in computer vision has narrowed, notably owing to the achievements of contrastive learning methods40,41. However, these methods typically rely on a large number of explicit pairwise feature comparisons, which is computationally challenging66. Furthermore, to maximize the feature extraction ability of the pretraining model, contrastive learning must select good feature pairs, which obviously increases the huge cost in computing resources. Therefore, to save computing resources and mine the fine-grained information in the molecule images, we introduce a simple contrastive learning method in molecular images, namely mask-based contrastive learning (Supplementary Fig. 4). We first use a 16 × 16 square area to randomly mask the molecular images (Supplementary Fig. 29), denoted by \(\tilde x_n\). Then, the masked molecular images \(\tilde x_n\) and the unmasked molecular images xn are simultaneously input into the molecular encoder to extract latent features \(f_\theta \left( {\tilde x_n} \right)\), fθ(xn). Finally, the cost function \({{{\mathcal{L}}}}_{\mathrm{MCL}}\) is introduced to ensure consistency between the latent feature extracted by the molecular image before and after the mask, formalized as
where \(||\,f_\theta \left( {\tilde x_n} \right),f_\theta \left( {x_n} \right)||_2\) means the Euclidean distance between \(f_\theta \left( {\tilde x_n} \right)\) and fθ(xn).
Rationality for pretraining
Inspired by human understanding of the world, we proposed the rationality principle, which means that the structural information described by molecular images must conform to chemical common sense. We rearranged the original images to construct irrational molecular images and designed two pretraining tasks to predict them (Supplementary Figs. 2 and 3), which can effectively improve the model’s understanding of molecular images.
Molecular rationality discrimination (MRD)
The reason why people can easily judge whether things in the image are reasonable on the basis of the knowledge they have learned is because people are very good at summarizing the spatial structure information in the image scene. For example, with an image of a blue sky under the grass and an image of a blue sky above the grass we can easily distinguish that the former is unreasonable and the latter is reasonable. However, it is difficult for an artificial intelligence model to pay attention to this global-level spatial structure information spontaneously during the learning process. Motivated by these phenomena, we construct a rational and an irrational molecular image pair for each molecular image to guide the model to learn the structural information. Specifically, as shown in Supplementary Fig. 2, we use a 3 × 3 grid to decompose each molecular image xn into nine patches and number these 1 to 9. Then, these patch numbers are randomly shuffled and respliced according to the shuffled patch to form an image with the same dimensions as the original image. Finally, these disordered images are viewed as irrational samples \(\hat x_n\). Subsequently, the original ordered image xn and the shuffled image \(\hat x_n\) are forward propagated to the molecular encoder to extract latent features fθ(xn) and \(f_\theta \left( {\hat x_n} \right)\), and these features are further input into a rationality classifier to obtain the probability value wMRDfθ(xn) for whether the sample is reasonable. Here, we define the cost function of the MRD task \({{{\mathcal{L}}}}_{\mathrm{MRD}}\) to update ResNet18, formalized as
where the first term and the second term represent the binary classification loss of the rational image and the irrational image respectively. wMRD represents the parameters of the rationality classifier. \({{{\mathcal{Y}}}}_n^{\mathrm{MRD}}\) represents the real label, which consists of 0 (irrational) and 1 (rational).
Jigsaw puzzle prediction
Compared with MRD, jigsaw puzzle prediction provides a more fine-grained prediction to discover the invariance and regularity of molecular images (Supplementary Fig. 3), and is widely used in computer vision67. Solving a jigsaw puzzle on the same molecular images can help the model pay attention to the more global structural information and learn the concepts of spatial rationality to improve the generalization of the pretraining model. In this task, by using the maximal Hamming distance algorithm in ref. 68, we assign an index (ranging from 0 to 100, where 0 represents original permutation) to each permutation of patch numbers, which will be used as the classification label \({{{\mathcal{Y}}}}_n^{\mathrm{Jig}}\) of the molecular image. Similar to the MRD task, the original ordered image xn and the shuffled image \(\hat x_n\) are forward propagated to the molecular encoder to extract latent features fθ(xn) and \(f_\theta \left( {\hat x_n} \right)\). Then, an additional jigsaw classifier is introduced to classify the permutation to which the image belongs. The molecular encoder is updated by using cost function \({{{\mathcal{L}}}}_{\mathrm{JPP}}\), which is formalized as
where the first term and the second term represent the classification losses of the original ordered image and the shuffled image respectively. wJig represents the parameters of the jigsaw classifier.
Pretraining process
In pretraining, we used ~10 million unlabelled molecules from PubChem42 for unsupervised pretraining. The pretraining of ImageMol consists of two steps, which are data augmentations and the training process. A detailed pretraining data flow can be found in Supplementary Fig. 30 and Supplementary Note B.2.
Data augmentations
Data augmentation is a simple way to effectively augment a limited number of samples and improve the generalization ability and robustness of the model, and has been widely used in supervised and unsupervised representation learning. However, compared with ordinary images, the molecular images are sparser as they are filled mostly (>90%) by zeros, resulting in ‘usable’ data being limited to a very small fraction of the image46. In view of the above limitation, ‘random cropping’ is not applied in our model. Finally, three augmentations are selected in the pretraining stage, RandomHorizontalFlip, RandomGrayscale and RandomRotation, which do not change the original structure of the molecular image and allow the model to learn invariance to data augmentation (Supplementary Table 29). Hence, before the original images are input into our pretraining model, each image has a 50% probability of being horizontally flipped, 20% probability of being converted to greyscale and 100% probability of being rotated between 0° and 360°. The augmentations are provided by PyTorch (https://pytorch.org/).
Training process
Here, we used ResNet18 as our molecular encoder. After using data augmentations to obtain molecular images xn, we forward these molecular images xn to the ResNet18 model to extract latent features fθ(xn). Then, these latent features are used by five pretext tasks to calculate the total cost function \({{{\mathcal{L}}}}_{\mathrm{ALL}}\), which is defined as
Finally, the total loss function \({{{\mathcal{L}}}}_{\mathrm{ALL}}\) is used for backpropagation to update ResNet18. Specially, the cost function \({{{\mathcal{L}}}}_{\mathrm{ALL}}\) is minimized using mini-batch stochastic gradient descent. See Supplementary Note A.3 and Supplementary Table 30 for more detailed hyperparameter settings and Supplementary Note C.1 and Supplementary Fig. 31 for the loss record during pretraining.
Fine-tuning
After completing the pretraining, we fine-tune the pretrained ResNet18 in the downstream task. Clearly, the performance of the model can be further improved by establishing a complex fine-tuning task for the pretrained model. However, fine-tuning is not the research focus of this Article, so we only use a simple and common fine-tuning method to adapt the model to different downstream tasks. In detail, we only add an additional full connection layer gω (ω is the parameter of the full connection layer) after ResNet18, and the output dimension of the full connection layer is equal to the number of classifications of downstream tasks. In fine-tuning, we first input the molecular image \(x_n^{\mathrm{ft}}\) (ft is an abbreviation for fine-tuning) from the downstream task into ResNet18 to obtain the latent feature representation \(f_\theta \left( {x_n^{\mathrm{ft}}} \right)\). Then, we forward the latent feature representation to the full connection layer gω to obtain the logical value \(g_\omega \left( {f_\theta \left( {x_n^{\mathrm{ft}}} \right)} \right)\) related to the category and use the softmax activation function to normalize these logical values to obtain the predicted category probability \(\tilde {{{\mathcal{Y}}}}_n^{\mathrm{ft}} = {\mathrm{softmax}}\left( {g_\omega \left( {f_\theta \left( {x_n^{\mathrm{ft}}} \right)} \right)} \right)\). Finally, our model will be fine-tuned by calculating the cross-entropy loss between the category probability \(\tilde {{{\mathcal{Y}}}}_n^{\mathrm{ft}}\) and the true label \({{{\mathcal{Y}}}}_n^{\mathrm{ft}}\). Specifically, since the data in the downstream task have the problem of category imbalance, we also added the category weight in the cross-entropy loss, formalized as
where N and K respectively represent the number of samples and the number of categories in downstream tasks. λk represents the category weight, which is calculated as \(1 - \frac{{N_k}}{N}\) (Nk is the number of samples of category k). \({{{\mathcal{Y}}}}_{i,k}^{\mathrm{ft}}\) and \(\tilde {{{\mathcal{Y}}}}_{i,k}^{\mathrm{ft}}\) represent the true label and predicted probability on the kth category of the ith sample, respectively. Finally, the loss function \({{{\mathcal{L}}}}_{\mathrm{CE}}\) is used for backpropagation to update the parameters of the model. The more detailed hyperparameter settings can be found in Supplementary Note A.3 and Supplementary Table 30.
Downstream task details
To evaluate our proposed pretraining model, we designed four types of downstream task related to molecular representation learning for testing: molecular property prediction, drug metabolism prediction, drug–protein binding prediction and antiviral activity prediction. More experimental settings for downstream tasks can also be found in Supplementary Note A.1.
Datasets and splitting methods
In molecular property prediction, we used multiple benchmark datasets from MoleculeNet13, including eight classification datasets and five regression datasets (Supplementary Table 1). In drug metabolism prediction, we use PubChem data set I (training set) and PubChem data set II (validation set) from Cheng et al.49, which includes five human CYP isoforms (Supplementary Table 2). In drug–protein binding prediction, we used the top ten GPCR datasets (Supplementary Table 3) from the ChEMBL database and KINOMEscan datasets (Supplementary Table 4). In antiviral activity prediction, we used 13 high-throughput experimental datasets from the COVID-19 portal20 of the National Center for Advancing Translational Sciences (Supplementary Table 18) and 11 existing COVID-19 datasets from REDIAL-202020 (Supplementary Table 27). To comprehensively evaluate the performance of ImageMol, we used several popular splitting methods, including cross-validation, stratified split, scaffold split, random scaffold split and balanced scaffold split. See Supplementary Note A.1 for more details of the dataset and splitting method.
Baselines
We summarized four different types of popular and competitive baseline to compare with ImageMol: fingerprint-based methods (AttentiveFP11, MACCS-based and FP4-based models across multiple machine learning algorithms—support vector machine, decision tree, k-nearest neighbours, naive Bayes and their ensemble models49—and REDIAL-202020), sequence-based methods (TF_Robust48, X-MOL30, RNN_LR, TRFM_LR, RNN_MLP, TRFM_MLP, RNN_RF, TRFM_RF50 and CHEM-BERT51), graph-based methods (GraphConv69, Weave70, SchNet71, MPNN72, DMPNN73, MGCN23, Hu et al. (Jure’s GNN)53, N-GRAM45, MolCLR39, GCC74, GPT-GNN75, Grover35, MGSSL36, 3D InfoMax44, G-Motif35, GraphLoG76, GraphCL77, GraphMVP43 and MPG37) and molecular image-based methods (Chemception46, ADMET-CNN12 and QSAR-CNN47). See Supplementary Note A.1 for more details of the comparison baselines. For our reproduced models (such as CHEM-BERT, MolCLR and Chemception), the details of hyperparameter optimization can be found in Supplementary Table 31.
Evaluation metrics
For comprehensive evaluation, we used various evaluation metrics, including accuracy, ROC-AUC, AUPR, F1, precision, recall and kappa. Furthermore, we also used the one-sided McNemar significance test with a significance threshold of 0.05 to demonstrate the significant performance difference between ImageMol and compared methods. We reported the mean and the s.d. of the metrics by executing three independent runs with different random seeds for each method.
Data availability
The datasets used in this project can be found at the following links: 10 million-molecule pretraining dataset, https://deepchemdata.s3-us-west-1.amazonaws.com/datasets/pubchem_10m.txt.zip; 13 molecular property prediction datasets (Supplementary Table 1), https://deepchemdata.s3-us-west-1.amazonaws.com/datasets/BBBP.csv (replace the BBBP in the hyperlink with another dataset name to download other datasets); 13 SARS-CoV-2 targets, https://opendata.ncats.nih.gov/covid19/assays (Supplementary Table 18); five drug metabolism enzymes, https://drive.google.com/file/d/1mBsgGWXYqej5McsLwy1_fs_-VGGQnCro/view?usp=sharing (Supplementary Table 2); ten GPCR datasets, https://drive.google.com/file/d/1HVHrxJfW16-5uxQ-7DxgQTxroXxeFDcQ/view?usp=sharing (Supplementary Table 3); ten KINOMEscan datasets, https://lincs.hms.harvard.edu/kinomescan/ (Supplementary Table 4); US Food and Drug Administration-approved drugs in DrugBank, https://go.drugbank.com/releases/5-1-9/downloads/approved-drug-links; 122 drugs that block SARS-CoV-2, https://static-content.springer.com/esm/art%3A10.1038/s41586-022-04482-x/MediaObjects/41586_2022_4482_MOESM1_ESM.pdf.
Code availability
All of the codes are freely available at GitHub (https://github.com/ChengF-Lab/ImageMol). The version used in this publication is available at https://doi.org/10.5281/zenodo.7088986.
References
Schneider, G. Automating drug discovery. Nat. Rev. Drug Discov. 17, 97–113 (2018).
De Rycker, M., Baragaña, B., Duce, S. L. & Gilbert, I. H. Challenges and recent progress in drug discovery for tropical diseases. Nature 559, 498–506 (2018).
Avorn, J. The $2.6 billion pill—methodologic and policy considerations. N. Engl. J. Med. 372, 1877–1879 (2015).
Galson, S. et al. The failure to fail smartly. Nat. Rev. Drug Discov. 20, 259–260 (2021).
Dowden, H. & Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 18, 495–496 (2019).
Zhou, Y., Wang, F., Tang, J., Nussinov, R. & Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2, e667–e676 (2020).
Falivene, L. et al. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 11, 872–879 (2019).
Swain, S. S. et al. Computer-aided synthesis of dapsone–phytochemical conjugates against dapsone-resistant Mycobacterium leprae. Sci. Rep. 10, 6839 (2020).
Pathak, D., Krahenbuhl, P., Donahue, J., Darrell, T. & Efros, A. A. Context encoders: feature learning by inpainting. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 2536–2544 (IEEE, 2016).
Wang, G., Ye, J. C. & De Man, B. Deep learning for tomographic image reconstruction. Nat. Mach. Intell. 2, 737–748 (2020).
Xiong, Z. et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 63, 8749–8760 (2020).
Shi, T. et al. Molecular image-based convolutional neural network for the prediction of ADMET properties. Chemom. Intell. Lab. Syst. 194, 103853 (2019).
Wu, Z. et al. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
Tsubaki, M., Tomii, K. & Sese, J. J. B. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 35, 309–318 (2019).
Zheng, S., Li, Y., Chen, S., Xu, J. & Yang, Y. Predicting drug–protein interaction using quasi-visual question answering system. Nat. Mach. Intell. 2, 134–140 (2020).
Quan, Z., Guo, Y., Lin, X., Wang, Z.-J. & Zeng, X. GraphCPI: graph neural representation learning for compound–protein interaction. In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 717–722 (IEEE, 2019).
Li et al. An effective self-supervised framework for learning expressive molecular global representations to drug discovery. Brief. Bioinform. 22, bbab109 (2021).
Lee, I., Keum, J. & Nam, H. J. DeepConv-DTI: prediction of drug–target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 15, e1007129 (2019).
Pradeepkiran, J. A., Reddy, A. P. & Reddy, P. H. Pharmacophore-based models for therapeutic drugs against phosphorylated tau in Alzheimer’s disease. Drug Discov. Today 24, 616–623 (2019).
Bocci, G. et al. A machine learning platform to estimate anti-SARS-CoV-2 activities. Nat. Mach. Intell. 3, 527–535 (2021).
Gobbi, A. & Poppinger, D. Genetic optimization of combinatorial libraries. Biotechnol. Bioeng. 61, 47–54 (1998).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Informat. Model. 50, 742–754 (2010).
Lu, C. et al. Molecular property prediction: a multilevel quantum interactions modeling perspective. Proc. AAAI Conf. Artif. Intell. 33, 1052–1060 (2019).
Li, C., Wang, J., Niu, Z., Yao, J. & Zeng, X. A spatial–temporal gated attention module for molecular property prediction based on molecular geometry. Brief. Bioinform. 22, bbab078 (2021).
Wang, Z. et al. Advanced graph and sequence neural networks for molecular property prediction and drug discovery. Bioinformatics 38, 2579–2586 (2022).
Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst. 30, 5998–6008 (2017).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies Vol. 1 (eds Burstein, J. et al.) 4171–4186 (Association for Computational Linguistics, 2019).
Zhang, X.-C. et al. MG-BERT: leveraging unsupervised atomic representation learning for molecular property prediction. Brief. Bioinform. 22, bbab152 (2021).
Chen, D. et al. Algebraic graph-assisted bidirectional transformers for molecular property prediction. Nat. Commun. 12, 3521 (2021).
Xue, D. et al. X-MOL: large-scale pre-training for molecular understanding and diverse molecular analysis. Sci. Bull. 67, 899–902 (2022).
Shrivastava, A. D. & Kell, D. B. FragNet, a contrastive learning-based transformer model for clustering, interpreting, visualizing, and navigating chemical space. Molecules 26, 2065 (2021).
Winter, R., Montanari, F., Noé, F. & Clevert, D.-A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 10, 1692–1701 (2019).
Handsel, J., Matthews, B., Knight, N. J. & Coles, S. J. Translating the InChI: adapting neural machine translation to predict IUPAC names from a chemical identifier. J. Cheminform. 13, 79 (2021).
Yang, Q., Ji, H., Lu, H. & Zhang, Z. Prediction of liquid chromatographic retention time with graph neural networks to assist in small molecule identification. Anal. Chem. 93, 2200–2206 (2021).
Rong, Y. et al. Self-supervised graph transformer on large-scale molecular data. Adv. Neural Inf. Process. Syst. 33, 12559–12571 (2020).
Zhang, Z., Liu, Q., Wang, H., Lu, C. & Lee, C.-K. Motif-based graph self-supervised learning for molecular property prediction. Adv Neural Inf. Process. Syst. 34, 15870–15882 (2021).
Li, P. et al. An effective self-supervised framework for learning expressive molecular global representations to drug discovery. Brief. Bioinform. 22, bbab109 (2021).
Ying, C. et al. Do transformers really perform badly for graph representation? Adv. Neural Inf. Process. Syst. 34, 28877–28888 (2021).
Wang, Y., Wang, J., Cao, Z. & Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 4, 279–287 (2022).
Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A simple framework for contrastive learning of visual representations. Proc. Mach. Learning Res. 119, 1597–1607 (2020).
He, K., Fan, H., Wu, Y., Xie, S. & Girshick, R. Momentum contrast for unsupervised visual representation learning. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 9729–9738 (IEEE, 2020).
Kim, S. et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109 (2019).
Liu, S. et al. Pre-training molecular graph representation with 3D geometry. In Proc. 10th International Conference on Learning Representations (ICLR) (eds Hofmann, K. et al.) 1–18 (OpenReview.net, 2022).
Stärk, H. et al. 3D Infomax improves GNNs for molecular property prediction. In Proc. 39th International Conference on Machine Learning (eds Kamalika, C. et al.) 20479–20502 (PMLR, 2022).
Liu, S., Demirel, M. F. & Liang, Y. N-gram graph: simple unsupervised representation for graphs, with applications to molecules. Adv. Neural Inf. Process. Syst. 32, 8466–8478 (2019).
Goh, G. B., Siegel, C., Vishnu, A., Hodas, N. O. & Baker, N. Chemception: a deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. Preprint at https://arxiv.org/abs/1706.06689 (2017).
Zhong, S., Hu, J., Yu, X. & Zhang, H. Molecular image-convolutional neural network (CNN) assisted QSAR models for predicting contaminant reactivity toward OH radicals: transfer learning, data augmentation and model interpretation. Chem. Eng. J. 408, 127998 (2021).
Ramsundar, B. et al. Massively multitask networks for drug discovery. Preprint at https://arxiv.org/abs/1502.02072 (2015).
Cheng, F. et al. Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J. Chem. Informat. Model. 51, 996–1011 (2011).
Honda, S., Shi, S. & Ueda, H. R. SMILES transformer: pre-trained molecular fingerprint for low data drug discovery. Preprint at https://arxiv.org/abs/1911.04738 (2019).
Kim, H., Lee, J., Ahn, S. & Lee, J. R. A merged molecular representation learning for molecular properties prediction with a web-based service. Sci. Rep. 11, 11028 (2021).
Pan, X. et al. Deep learning for drug repurposing: methods, databases, and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 12, e1597 (2022).
Hu, W. et al. Strategies for pre-training graph neural networks. In Proc. 8th International Conference on Learning Representations (ICLR) (eds Rush, A. et al.) 1–22 (OpenReview.net, 2020).
Zhu, W. et al. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci. 3, 1008–1016 (2020).
Boras, B. et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 12, 6055 (2021).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Schultz, D. C. et al. Pyrimidine inhibitors synergize with nucleoside analogues to block SARS-CoV-2. Nature 604, 134–140 (2022).
Davies, D. L. & Bouldin, D. W. A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. 2, 224–227 (1979).
Selvaraju, R. R. et al. Grad-CAM: visual explanations from deep networks via gradient-based localization. Proc. IEEE International Conference on Computer Vision 618–626 (IEEE, 2017).
Ozturk, T. et al. Automated detection of COVID-19 cases using deep neural networks with X-ray images. Comput. Biol. Med. 121, 103792 (2020).
Wu, Y.-H. et al. JCS: an explainable COVID-19 diagnosis system by joint classification and segmentation. IEEE Trans. Image Process. 30, 3113–3126 (2021).
Sutskever, I., Martens, J., Dahl, G. & Hinton, G. On the importance of initialization and momentum in deep learning. Proc. Mach. Learning Res. 28, 1139–1147 (2013).
Dosovitskiy, A. et al. An image is worth 16 × 16 words: transformers for image recognition at scale. In Proc. 8th International Conference on Learning Representations (ICLR) (eds Mohamed, S. et al.) 1–21 (OpenReview.net, 2021).
Johnson, J. et al. Billion-scale similarity search with GPUs. IEEE Trans. Big Data 7, 535–547 (2019).
He, K., Zhang, X., Ren, S. & Sun, J. Deep residual learning for image recognition. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 770–778 (IEEE, 2016).
Caron, M. et al. Unsupervised learning of visual features by contrasting cluster assignments. Adv. Neural Inf. Process. Syst. 33, 9912–9924 (2020).
Carlucci, F. M., D’Innocente, A., Bucci, S., Caputo, B. & Tommasi, T. Domain generalization by solving jigsaw puzzles. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition 2229–2238 (IEEE, 2019).
Noroozi, M. & Favaro, P. Unsupervised learning of visual representations by solving jigsaw puzzles. In Computer Vision—ECCV 2016 (eds Leibe, B. et al.) 69–84 (Lecture Notes in Computer Science Vol. 9910, Springer, 2016).
Welling, M. & Kipf, T. N. Semi-supervised classification with graph convolutional networks. In Proc. 5th International Conference on Learning Representations (ICLR) (eds Bengio, Y. et al.) 1–14 (OpenReview.net, 2017).
Kearnes, S., McCloskey, K., Berndl, M., Pande, V. & Riley, P. Molecular graph convolutions: moving beyond fingerprints. J. Comput. Aided Mol. Des. 30, 595–608 (2016).
Schütt, K. et al. SchNet: a continuous-filter convolutional neural network for modeling quantum interactions. Adv. Neural Inf. Process. Syst. 30, 1–11 (2017).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. Proc. Mach. Learning Res. 70, 1263–1272 (2017).
Yang, K. et al. Analyzing learned molecular representations for property prediction. J. Chem. Informat. Model. 59, 3370–3388 (2019).
Qiu, J. et al. GCC: graph contrastive coding for graph neural network pre-training. In Proc. 26th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (eds Gupta, R. et al.) 1150–1160 (Association for Computing Machinery, 2020).
Hu, Z., Dong, Y., Wang, K., Chang, K.-W. & Sun, Y. GPT-GNN: generative pre-training of graph neural networks. In Proc. 26th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (eds Gupta, R. et al.) 1857–1867 (Association for Computing Machinery, 2020).
Xu, M., Wang, H., Ni, B., Guo, H. & Tang, J. Self-supervised graph-level representation learning with local and global structure. Proc. Mach. Learning Res. 139, 11548–11558 (2021).
You, Y. et al. Graph contrastive learning with augmentations. Adv Neural Inf. Process. Syst. 33, 5812–5823 (2020).
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Author information
Authors and Affiliations
Contributions
F.C. conceived the study. H.X. and X.Z. implemented the pipeline, constructed the databases, developed the codes and performed all experiments. H.X., X.Z., L.Y., J.W., K.L. and F.C. performed data analyses. H.X., X.Z., F.C. and R.N. discussed and interpreted all results. H.X., X.Z., F.C. and R.N. wrote and critically revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Leng Han, Tudor Oprea and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–31 and Tables 1–4, 18, 19, 23, 26, 27, 29, 30, footnotes for Tables 5–17, 20–22, 24, 25, 28 and 31 and Materials and Methods.
Supplementary Table 5
The ROC-AUC (%) performance of different methods on benchmark datasets with scaffold split. All results are reported as mean ± s.d. The yellow and blue backgrounds represent methods without and with pretraining, respectively. The light-green background represents the results of our method. Rank represents the ranking of ImageMol in the comparison.
Supplementary Table 6
The ROC-AUC performance of different methods on benchmark datasets with random scaffold split. All results are reported as mean ± s.d. The yellow and blue backgrounds represent methods without and with pretraining, respectively. The light-green background represents the results of our method. Rank represents the ranking of ImageMol in the comparison.
Supplementary Table7
The comprehensive performance evaluation (including accuracy, AUC, AUPR, F1, precision, recall and kappa) of ImageMol on a molecular property prediction task with scaffold split.
Supplementary Table 8
The comprehensive performance evaluation (including accuracy, AUC, AUPR, F1, precision, recall and kappa) of ImageMol on a molecular property prediction task with random scaffold split.
Supplementary Table 9
The accuracy and ROC-AUC value of five major CYP isoforms from PubChem data set II. ImageMol_NonPretrained and ImageMol indicate that ImageMol is not pretrained and ImageMol is pretrained on 10M molecular images. ‘MACCS-’ represents the method based on the MACCS molecular fingerprint; ‘FP4-’ represents the method based on the FP4 molecular fingerprint. ‘CC-’ represents a combined algorithm (ensemble learning). For details of other methods, see ref. 24. ImageMol_NonPretrained and ImageMol are fine-tuned on PubChem data set I and evaluated on PubChem data set II. The best and second-best results for each dataset are bold and underlined, respectively. The methods in the blue background represent image-based methods. ‘3 times’ represents the mean ± s.d. of three runs with different random seeds.
Supplementary Table 10
The performance of different methods on single-task CYP450 datasets with balanced scaffold split.
Supplementary Table 11
The performance of different methods on multilabelled CYP450 datasets with balanced scaffold split.
Supplementary Table 12
The performance of different methods on the drug–protein binding efficiency datasets with balanced scaffold split.
Supplementary Table 13
The performance of different methods on the drug–protein binding activity datasets from KINOMEscan with balanced scaffold split.
Supplementary Table 14
Results of statistical significance tests between ImageMol and GROVER-10M, MPG-10M, X-MOL, MolCLRGIN on the eight molecular property prediction datasets, where numbers represent the P values (one-sided significance level) of the McNemar test between the models in the Supplementary Table (GROVER-10M, MPG-10M, X-MOL and MolCLRGIN) and ImageMol. The numbers in the green background indicate statistically different models, using a significance threshold of 0.05.
Supplementary Table 15
Results of statistical significance tests between ImageMol and Image-based methods (Chemception, ADMET-CNN, QSAR-CNN) on five CYP450 isoform training sets (PubChem data set I) and validation sets (PubChem data set II), where numbers represent the P values (one-sided significance level) of the McNemar test. The numbers in the green background indicate statistically different models, using a significance threshold of 0.05. 0 indicates statistical significance less than 10−100.
Supplementary Table 16
Results of statistical significance tests on the five CYP450 datasets with balanced scaffold split, where numbers represent the P values (one-sided significance level) of the McNemar test between the models in the table (CHEM-BERT, GROVER, MolCLRGIN, MolCLRGCN, RNN_LR, RNN_MLP, RNN_RF, TRFM_LR, TRFM_MLP and TRFM_RF) and ImageMol. The numbers in the green background indicate statistically different models, using a significance threshold of 0.05.
Supplementary Table 17
Results of statistical significance tests on the 13 SARS-CoV-2 datasets with balanced scaffold split, where numbers represent the P values (one-sided significance level) of the McNemar test between the models in the table (CHEM-BERT, GROVER, MolCLRGIN, MolCLRGCN, RNN_LR, RNN_MLP, RNN_RF, TRFM_LR, TRFM_MLP and TRFM_RF) and ImageMol. The numbers in the green background indicate statistically different models, using a significance threshold of 0.05.
Supplementary Table 20
The experimental results of REDIAL-2020, ImageMol_NonPretrained and ImageMol for anti-SARS-CoV-2 activity estimation. ACC, accuracy; F1, F1 score; SEN, sensitivity; PREC, precision. ImageMol (3 times) represents the mean ± s.d. of the results of three runs with different random seeds.
Supplementary Table 21
The performance of different methods on 13 SARS-CoV-2 datasets with balanced scaffold split.
Supplementary Table 22
Screening results of approved drugs in DrugBank for 3CL inhibitors via ImageMol.
Supplementary Table 24
Screening results of approved drugs in DrugBank for SARS-CoV-2.
Supplementary Table 25
Virtual screening of 70 validated anti-SARS-CoV-2 small-molecule drugs. These drugs were validated in Calu-3 cells.
Supplementary Table 28
Ablation results of data augmentation on benchmark datasets with ROC-AUC metric and random scaffold split. All results are reported as mean ± s.d. Bold font indicates the best result.
Supplementary Table 31
Hyperparameter optimization for MPG, GROVER, X-MOL, MolCLR, CHEM-BERT, SMILES_Transformer, Chemception, ADMET-CNN and QSAR-CNN.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zeng, X., Xiang, H., Yu, L. et al. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nat Mach Intell 4, 1004–1016 (2022). https://doi.org/10.1038/s42256-022-00557-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-022-00557-6
This article is cited by
-
DisoFLAG: accurate prediction of protein intrinsic disorder and its functions using graph-based interaction protein language model
BMC Biology (2024)
-
Prediction of blood–brain barrier penetrating peptides based on data augmentation with Augur
BMC Biology (2024)
-
Enhancing property and activity prediction and interpretation using multiple molecular graph representations with MMGX
Communications Chemistry (2024)
-
StackER: a novel SMILES-based stacked approach for the accelerated and efficient discovery of ERα and ERβ antagonists
Scientific Reports (2023)
