
Abstract
Bacteria have evolved diverse immunity mechanisms to protect themselves against the constant onslaught of bacteriophages1,2,3. Similar to how eukaryotic innate immune systems sense foreign invaders through pathogen-associated molecular patterns4 (PAMPs), many bacterial immune systems that respond to bacteriophage infection require phage-specific triggers to be activated. However, the identities of such triggers and the sensing mechanisms remain largely unknown. Here we identify and investigate the anti-phage function of CapRelSJ46, a fused toxin–antitoxin system that protects Escherichia coli against diverse phages. Using genetic, biochemical and structural analyses, we demonstrate that the C-terminal domain of CapRelSJ46 regulates the toxic N-terminal region, serving as both antitoxin and phage infection sensor. Following infection by certain phages, newly synthesized major capsid protein binds directly to the C-terminal domain of CapRelSJ46 to relieve autoinhibition, enabling the toxin domain to pyrophosphorylate tRNAs, which blocks translation to restrict viral infection. Collectively, our results reveal the molecular mechanism by which a bacterial immune system directly senses a conserved, essential component of phages, suggesting a PAMP-like sensing model for toxin–antitoxin-mediated innate immunity in bacteria. We provide evidence that CapRels and their phage-encoded triggers are engaged in a ‘Red Queen conflict’5, revealing a new front in the intense coevolutionary battle between phages and bacteria. Given that capsid proteins of some eukaryotic viruses are known to stimulate innate immune signalling in mammalian hosts6,7,8,9,10, our results reveal a deeply conserved facet of immunity.
Similar content being viewed by others


Main
Innate immunity in eukaryotes relies on pattern recognition receptors that directly sense PAMPs, which are conserved molecules like bacterial lipopolysaccharide and flagellin, or viral RNA or DNA4. These innate immune signalling pathways must remain silent before infection, but be poised for rapid activation to defend against foreign invaders. Bacteria also encode innate immune systems to protect themselves against diverse invading bacteriophages, but how they sense infection is poorly understood. One exception is restriction-modification systems, but these distinguish self from non-self using DNA methylation patterns and thus do not need the nuclease effector to be specifically activated during phage infection. Similarly, for CRISPR–Cas systems, the adaptive immune system of some bacteria, guide RNAs enable a cell to specifically target foreign DNA without a need for infection-triggered activation. Dozens of bacterial defence systems have been identified in recent years11,12,13,14,15, but unlike restriction modification and CRISPR–Cas, many of them must be specifically activated upon phage infection. This is particularly critical for abortive infection systems, in which a defence system uses a lethal effector to kill an infected cell and prevent propagation of the virus through a population16. The phage-encoded triggers for such bacterial immunity mechanisms are largely unknown.
Toxin–antitoxin systems are prevalent genetic elements in bacteria that are emerging as key components of anti-phage innate immunity13,14,17,18, often serving as abortive infection modules that kill infected cells to prevent spread of phages through a population. How toxin–antitoxin systems sense and respond to phage infection remains poorly understood. For the toxIN system, toxin (ToxN) activation relies on efficient, phage-induced shutoff of host transcription coupled to the intrinsically fast turnover of the antitoxin toxI19,20,21. However, toxI is an RNA, whereas most toxin–antitoxin systems feature a protein antitoxin. For systems with a protein antitoxin, the mechanism of activation is often assumed to arise through antitoxin degradation. Although protein antitoxins are often more proteolytically unstable than their cognate toxins, their turnover may not be fast enough to enable toxin activation on the timescale of a phage infection22, suggesting the existence of alternative mechanisms for toxin–antitoxin system activation. Bacterial retrons function as tripartite toxin–antitoxin systems and can be activated by overexpressing various prophage genes23, but whether these activators function as such during phage infection is unknown.
CapRelSJ46 is an anti-phage fused toxin–antitoxin system
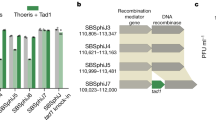
To investigate the molecular basis of phage-induced activation of bacterial immunity, we focused on toxSAS toxin–antitoxin systems, which feature toxins homologous to bacterial small alarmone synthetases (SAS) that pyrophosphorylate purine nucleotides24. Whereas most housekeeping alarmone synthetases produce the growth regulator (p)ppGpp25,26, toxSAS toxins can synthesize (p)ppApp to deplete ATP24,27 or pyrophosphorylate tRNAs to inhibit translation24,28. Their cognate antitoxins can either bind and neutralize the toxin or act as hydrolases to reverse toxin-catalysed pyrophosphorylation24,28. One subfamily of translation-inhibiting toxSAS is called CapRel, on the basis of their prevalence in Cyanobacteria, Actinobacteria, and Proteobacteria and sequencesimilarity to the (p)ppGpp synthetase/hydrolase Rel. The CapRel subfamily is the most broadly distributed subfamily of toxSAS, being found across multiple phyla of Gram-positive and Gram-negative bacteria. In addition to its prevalence in Cyanobacteria, Actinobacteria, and Proteobacteria, CapRel representatives are found in Spirochetes, Bacteroidetes, and Firmicutes24. This subfamily often features systems that are—in contrast to canonical bicistronic toxin–antitoxin systems—encoded by a single open reading frame, with an N-terminal domain homologous to toxSAS toxins and a C-terminal domain homologous to the corresponding antitoxins29 (Fig. 1a and Extended Data Figs. 1 and 2a). This fused architecture is the predominant form of CapRel, except in Actinobacteria.

a, Domain organization of long RelA-SpoT homologues (RSH), SAS, toxSAS and the fused subclass of toxSAS toxin–antitoxin systems including CapRelSJ46. b, Cell viability assessed by serial dilutions for strains expressing the N-terminal toxin domain of CapRelSJ46 alone or with the C-terminal antitoxin domain. Ara, arabinose; glu, glucose; Para, arabinose-inducible, glucose-repressible promoter; Plac, IPTG-inducible promoter. c, EOP data for the indicated phages when infecting cells producing CapRelSJ46, CapRelEbc or CapRelKp. d, Serial, tenfold dilutions of the indicated phages spotted on lawns of cells harbouring plasmid expressing CapRelSJ46 or an empty vector (EV). Relative phage concentration is indicated by the height of the wedge. e,f, One-step growth curves measuring plaque-forming units (PFU) during the first round of infection by T4 (e) or SECΦ27 (f) in cells harbouring plasmid expressing CapRelSJ46 or an empty vector. g, Serial dilutions of T7 phage spotted on lawns of cells harbouring plasmids expressing CapRelEbc, CapRelEbc(Y153A) or an empty vector. h, Growth of cells producing CapRelSJ46 or CapRelSJ46(Y155A) or harbouring an empty vector, following infection with T4 at a MOI of 10 or 0.001. Data are mean ± s.d. of eight plate replicates and representative of three independent experiments.
Prophages in bacterial genomes often encode anti-phage systems, helping both the temperate phage and its host to defend against other phages11,30,31. Here, we selected a fused CapRel encoded by the Salmonella temperate phage SJ46 and also encoded (with 100% amino acid sequence identity) in prophages of several E. coli strains (Extended Data Fig. 2b). The toxin and antitoxin-like regions of CapRelSJ46 are related to the PhRel toxSAS toxin and its antitoxin ATphRel, respectively, from the mycobacterial temperate phage Phrann30 (Extended Data Fig. 1 and 2a). This Phrann-encoded system can inhibit superinfection by other temperate mycophages30, although the molecular basis of PhRel activation is not known. To test whether CapRelSJ46 is a fused toxin–antitoxin system, we cloned the N-terminal region containing the conserved alarmone synthetase domain and the C-terminal region containing the putative antitoxin domain under the control of separate inducible promoters. Expression of the N-terminal fragment alone was toxic, and its toxicity was rescued in trans by co-expression with the C-terminal fragment (Fig. 1b and Extended Data Fig. 2c), suggesting that CapRelSJ46 is a fused toxin–antitoxin system.
To determine whether fused CapRels can defend against phages, we transformed E. coli MG1655 with three different systems expressed from their native promoters on low copy number plasmids, and then tested whether each conferred protection against a panel of 12 diverse coliphages. In addition to CapRelSJ46, we also tested CapRelEbc from Enterobacter chengduensis and CapRelKp from Klebsiella pneumoniae (Fig. 1c and Extended Data Fig. 2b,d). CapRelSJ46 expressed from a low copy number plasmid or the E. coli genome decreased the efficiency of plaquing (EOP) for T2, T4, T6, RB69 and SECΦ27 by 10- to 1,000-fold (Fig. 1c,d and Extended Data Fig. 2e,f), indicating that this system provides strong protection against phages. T4 phage formed smaller plaques when plated onto CapRelSJ46-containing cells, and one-step growth curves confirmed that CapRelSJ46 reduced the burst size of T4 by around 70% (Fig. 1e) and prevented bursting of SECΦ27 (Fig. 1f). CapRelEbc protected strongly against T7 and CapRelKp protected, albeit less efficiently, against SECΦ18 (Fig. 1g and Extended Data Fig. 2d,g).
Next, we tested whether CapRelSJ46 provides direct immunity or functions through abortive infection, in which an infected cell dies but prevents the production of mature virions, thereby sparing uninfected cells in a population. To this end, we infected cells containing CapRelSJ46 with T4 at a multiplicity of infection (MOI) of either 10 or 0.001, and found that defence only manifested at the low MOI, indicating that CapRelSJ46 probably functions through abortive infection (Fig. 1h). Phage protection by CapRelSJ46 depended on the predicted enzymatic activity of the N-terminal synthetase domain, as substituting the conserved tyrosine (Y155A) in the G-loop that is critical for substrate binding abolished phage protection32 (Fig. 1h). A similar catalysis-compromising substitution, Y153A in CapRelEbc, also abolished phage protection (Fig. 1g). Collectively, our results established that fused CapRels can provide anti-phage defence, with variable phage specificity.
To understand what determines the specificity of phage protection by fused CapRels, we compared CapRelSJ46 and CapRelEbc. These two proteins share 70% amino acid identity overall, but differ substantially in their C-terminal regions, which are only 47% identical (Fig. 2a). In addition, this region is the least conserved when we compared a more diverse set of fused CapRel homologues (Extended Data Fig. 3a). Because CapRelSJ46 and CapRelEbc protected against different phages, we made a chimera in which the C-terminal region of CapRelSJ46 was replaced by the corresponding region of CapRelEbc. This chimeric CapRel no longer protected against SECΦ27 and gained protection against T7, manifesting as a tenfold decrease in EOP and smaller plaques (Fig. 2b and Extended Data Fig. 3b). The chimera protected against T7 less efficiently than CapRelEbc, despite similar expression levels (Extended Data Fig. 3c), probably owing to a non-optimal interaction interface between the N and C termini. Nevertheless, this result indicates that the C-terminal region of CapRel is critical for phage specificity.

a, Sequence alignment of CapRelSJ46 and CapRelEbc, with the more variable pseudo-ZFD labelled. b, Serial dilutions of the indicated phages spotted on lawns of cells harbouring the indicated CapRel constructs. Left, schematic of the CapRel constructs. c, Cartoon representation of the crystal structure of CapRelSJ46 with active site G-loop Y155 and the ATP-coordination residues R79 and R116 highlighted in red. Structural elements (toxSYNTH, pseudo-ZFD and the anchors) are coloured as in a. Wavy lines and arrows indicate alpha helices and beta strands, respectively. d, The closed conformation of CapRelSJ46 predicted by AlphaFold and coloured as in c. e, Superposition of the active (open, light purple) and inactive (closed, dark purple) states of CapRelSJ46 as observed in the crystal structure and predicted by AlphaFold. f, Details of the autoinhibited active site of CapRelSJ46 in the closed state. In this conformation, the YXXY neutralization motif blocks the adenine coordination site, preventing catalysis.
Structural analysis of CapRelSJ46
To further understand the mechanistic basis of anti-phage defence by CapRelSJ46, we solved a crystal structure to 2.3 Å resolution (Fig. 2c and Extended Data Table 1). CapRelSJ46 contains a conserved, N-terminal nucleotide pyrophosphokinase domain (toxSYNTH) that is also present in alarmone synthetases and tRNA-pyrophosphotransferase enzymes, which mediates toxicity. The smaller C-terminal antitoxin domain consists of a central antiparallel three-stranded β-sheet with an α-helix connecting β-strands β7 and β8 (Fig. 2c and Extended Data Fig. 3d,e). The antitoxin domain is topologically analogous to the classical zinc-finger domain (ZFD), but lacks the conserved cysteines (Extended Data Fig. 3f); we refer to this domain as a pseudo-ZFD. The pseudo-ZFD is connected to the toxSYNTH domain via α-helices α7, α8 and α9, and has a C-terminal α-helical extension that anchors the domain to α8 and α9 (Extended Data Fig. 3d). In this structure, the toxSYNTH and pseudo-ZFD are distant from each other, revealing the ATP donor nucleotide binding pocket and the conserved G-loop Y155 of toxSYNTH (Fig. 2c), indicating that this state may represent the active, toxic conformation of CapRelSJ46. This open state captured by crystallography is probably stabilized by crystal lattice contacts that are incompatible with a closing of the active site owing to clashes with symmetry-related partners (Extended Data Fig. 3g).
To explore the conformational dynamics of the enzyme, we used AlphaFold33 to predict possible alternative structures of CapRelSJ46. In addition to predicting the open conformation observed in the crystal structure (Extended Data Fig. 3h), AlphaFold also predicted a closed conformational state in which the C-terminal domain folds back 110° onto the toxSYNTH central β-sheet and blocks the ATP-binding site (Fig. 2d). Comparison of the two states suggested that a conserved YXXY motif (Extended Data Fig. 3a) located in the hinge connecting the two C-terminal α-helices in the open state morphs into a short 310-helix in the closed state (Fig. 2e,f). This 310-helix projects into the toxSYNTH active site and intercalates between β1 R79 and β2 R116 to block the adenine coordination site (Fig. 2e,f).
We hypothesized that this closed-to-open switch underlies the activation of CapRelSJ46, with the docking of the pseudo-ZFD onto toxSYNTH precluding substrate binding in the absence of phage infection (Fig. 2f). To test this hypothesis, we made single substitutions to the YXXY motif (Y352A and Y355A) and residues from the predicted interface that serves as a scaffold to orient and stabilize the 310-helix (A77K, R116A, V338A, L339A, A341K and A351K), which are highly conserved among diverse CapRel homologues (Extended Data Fig. 3a,i). Whereas wild-type CapRelSJ46 was not toxic when expressed in cells, each of the substitutions predicted to disrupt the intramolecular recognition interface, on either the N- or the C-terminal domain, rendered CapRelSJ46 toxic (Extended Data Fig. 3j,k). These substitutions probably lead to constitutive activation of CapRelSJ46 by disrupting an autoinhibited state. As a control, we showed that substitutions in different structural elements of the pseudo-ZFD but not pointing towards the interface did not lead to constitutive activation (Extended Data Fig. 3j,k). Collectively, our results indicate that the pseudo-ZFD docks onto the ATP-binding site of CapRelSJ46 to prevent switching to the open state captured in our crystal structure. Conservation of the YXXY motif and the interface residues suggests that this auto-inhibitory regulation is probably retained in other CapRels.
SECΦ27 capsid protein triggers CapRelSJ46
Because full-length, wild-type CapRelSJ46 was not toxic when expressed in the absence of phage infection, we inferred that it must somehow be activated by phages. The toxins of some toxin–antitoxin systems are activated by the degradation of the more labile antitoxin19,34,35. To test whether the C-terminal antitoxin of CapRelSJ46 is proteolytically cleaved off and degraded upon phage infection, we N-terminally tagged CapRelSJ46 and first verified that the tagged protein still defends against phage (Extended Data Fig. 4a). We then tracked the size of CapRelSJ46 by immunoblotting following infection with SECΦ27. The overall protein levels of CapRelSJ46 remained constant and we observed only the full-length product, suggesting that CapRelSJ46 was not proteolytically processed (Fig. 3a and Extended Data Fig. 4b,c). Thus, we hypothesized that a specific phage product regulates the C-terminal domain of CapRelSJ46 to relieve autoinhibition. To identify such a factor, we sought to identify SECΦ27 mutants that escape CapRelSJ46 defence. As no spontaneous escape mutants could be isolated, we used an experimental evolution approach (Fig. 3b). In brief, we infected cells containing an empty vector or CapRelSJ46 with serial dilutions of phage in microtitre plates. After overnight incubation, we collected and pooled the phages from cleared wells, which indicated successful infection, and used these to seed the next round of infections. Initially, cells with the empty vector were infected more efficiently, but after 13 rounds, each phage population had evolved to infect cells containing empty vector or CapRelSJ46 to a similar degree (Fig. 3c and Extended Data Fig. 4d). We isolated ten mutant SECΦ27 clones from five independently evolved populations and sequenced their genomes. Remarkably, all ten clones contained a point mutation in the same gene that encodes a hypothetical protein, Gp57, with nine clones producing the same L114P substitution and one clone yielding an I115F substitution (Fig. 3d and Supplementary Table 1).

a, Immunoblot of His6–CapRelSJ46 following infection with SECΦ27 compared with an uninfected control. Representative of two biological replicates. b, Schematic of the experimental evolution approach to identify SECΦ27 escape mutants that can infect CapRelSJ46-containing cells. White wells indicate clearing by phages and brown wells indicate bacterial growth. c, Serial dilutions of five independently evolved populations of SECΦ27 and a control population spotted on cells harbouring an empty vector or a CapRelSJ46 expression vector. d, Summary of identified escape mutants, all of which map to a hypothetical protein encoded by gene 57 of SECΦ27. The fraction of each escape mutant is indicated. e, AlphaFold-predicted structure of Gp57 compared with the major capsid protein Gp5 from phage HK97. f, Mass spectrometry analysis of SECΦ27 phage lysates (wild type or a mutant producing Gp57(L114P)). Spectrum count normalized to molecular mass is shown for individual phage proteins. g, Serial dilutions of cells expressing CapRelSJ46 and the indicated variant of Gp57 from an arabinose-inducible promoter on media containing glucose or arabinose. h, Cells expressing CapRelSJ46 and Gp57 (wild type (WT) or L114P) from an arabinose-inducible promoter or harbouring empty vector were pulse-labelled with 35S-cysteine and 35S-methionine after addition of arabinose. i, As in h, but for cells harbouring a CapRelSJ46 expression vector or empty vector and after infection with SECΦ27 (top) or the escape mutant expressing Gp57(L114P) (bottom). Asterisks indicate P = 0.022 (45 min) or P = 0.004 (60 min) (unpaired two-tailed t-test). j, In vitro transcription–translation assays using DHFR production from a DNA template as the readout of expression activity. Purified CapRelSJ46 was added along with a template for producing Gp57. Representative of two biological replicates. k, Autoradiography of reactions in which purified CapRelSJ46 was incubated with [γ-32P]ATP, bulk E. coli tRNAs and Gp57. SYBR Gold staining of bulk tRNAs serves as a loading control. Representative of two biological replicates.
The structure of the hypothetical protein Gp57 predicted by AlphaFold33 is highly similar (DALI z-score of approximately 17) to the HK97 fold commonly adopted by major capsid proteins of dsDNA viruses including bacteriophages and herpesviruses36 (Fig. 3e). By performing mass spectrometry on wild-type and escape-mutant SECΦ27 phages, we identified this hypothetical protein as the most abundant protein in mature virions, consistent with it being the major capsid protein of SECΦ27 (Fig. 3f and Extended Data Table 2).
Our results suggested that wild-type Gp57 from SECΦ27 activates CapRelSJ46, with the escape mutants preventing activation while retaining the ability to form a capsid. To test this hypothesis, we first examined whether Gp57 alone is sufficient to activate CapRelSJ46. Indeed, co-producing wild-type Gp57 with wild-type CapRelSJ46 was highly toxic to cells in the absence of phage infection, whereas neither evolved variant (L114P or I115F) of Gp57 had a measurable effect on growth when co-produced with CapRelSJ46 (Fig. 3g and Extended Data Fig. 4e,f). As controls, we confirmed that expressing the wild-type or either Gp57 variant was not toxic on its own or if co-produced with a catalytically compromised CapRelSJ46 (Extended Data Fig. 4g).
To examine the basis of CapRelSJ46 toxicity, we first co-produced it with wild-type or the L114P variant of Gp57 and then measured the effects on bulk transcription or translation by pulse-labelling with 3H-uridine or with 35S-methionine and 35S-cysteine, respectively. Active CapRelSJ46 produced with wild-type Gp57 robustly inhibited translation but not transcription (Fig. 3h and Extended Data Fig. 4h), whereas no effect was seen with Gp57(L114P). Similar effects were seen when overexpressing just the N-terminal domain of CapRelSJ46 (Extended Data Fig. 4i). We also measured bulk translation and transcription following SECΦ27 infection of CapRelSJ46-containing cells and observed a decrease in translation but not transcription with wild-type SECΦ27 (Fig. 3i and Extended Data Fig. 4j), with a less pronounced decrease in translation compared to overproducing Gp57 likely because poor adsorption of SECΦ27 phage leads to substantial asynchrony of infection dynamics (Extended Data Fig. 4k). No effect on translation was seen with the evolved mutant phage producing Gp57(L114P) (Fig. 3i). To compare the timing of CapRelSJ46 activation during infection to the timing of Gp57 production, we expressed a haemagglutinin (HA)-tagged version of Gp57 from its native promoter in the bacterial genome and infected cells with SECΦ27. Immunoblotting indicated substantial accumulation of Gp57 by around 30 min after infection (Extended Data Fig. 4l), which coincides with the detected inhibition of translation (Fig. 3i).
Next, we measured the ability of full-length CapRelSJ46 to affect translation in vitro using the reconstituted in vitro transcription–translation system. Purified CapRelSJ46 inhibited synthesis of a control DHFR protein after pre-synthesizing SECΦ27 major capsid protein Gp57 (using a template encoding it), whereas no inhibition was seen for either the L114P or I115F variants of Gp57 (Fig. 3j). We also incubated wild-type or an evolved variant (L114P or I115F) of Gp57 with [γ-32P]ATP and bulk E. coli tRNAs in the presence and absence of purified CapRelSJ46. Wild-type Gp57 strongly stimulated the pyrophosphorylation of tRNAs by CapRelSJ46, similar to the previously characterized toxSAS enzymes FaRel2 and PhRel228 (Fig. 3k). With the L114P or I115F variant of Gp57, tRNA pyrophosphorylation was reduced to the background levels seen with CapRelSJ46 alone. Together, our results demonstrate that Gp57, the major capsid protein of SECΦ27, is both necessary and sufficient to activate CapRelSJ46, enabling it to pyrophosphorylate tRNAs and inhibit translation.
CapRelSJ46 binds SECΦ27 capsid protein
To test whether the SECΦ27 major capsid protein directly binds CapRelSJ46, we first immunoprecipitated CapRelSJ46–Flag from cells infected with wild-type phage or the mutant that produces Gp57(L114P) after verifying the tag does not affect CapRelSJ46 function (Extended Data Fig. 4a). We detected Gp57 that had co-precipitated with CapRelSJ46 by mass spectrometry when cells were infected with wild-type phage, with a significant reduction in the mutant phage (Extended Data Fig. 5a,b). In addition, we co-produced CapRelSJ46–Flag and Gp57–HA and found that wild-type, but not the L114P or I115F variant of the capsid protein, co-precipitated with CapRelSJ46–Flag (Fig. 4a and Extended Data Fig. 5c). Finally, we purified both full-length CapRelSJ46 and Gp57, and used isothermal titration calorimetry (ITC) to show that they interact directly with an affinity of 350 nM in a 1:1 ratio (Fig. 4b). We confirmed that CapRelSJ46 and Gp57 form a complex with 1:1 stoichiometry using size-exclusion chromatography coupled to multi-angle light scattering (SEC–MALS) (Extended Data Fig. 5d). Introducing the escape substitutions L114P and I115F into Gp57 decreased affinity more than 60-fold (Extended Data Fig. 5e).

a, CapRelSJ46–Flag was immunoprecipitated from cells expressing CapRelSJ46–Flag and Gp57–HA (wild-type or indicated mutant) and probed for the presence of the indicated Gp57 variant via the HA tag. Lysates used as input for the immunoprecipitation (IP) were probed as controls for expression levels. Images are representatives of three biological replicates. b, Binding of CapRelSJ46 to Gp57 monitored by ITC. Kd, binding affinity; N, stoichiometry. c, Structural model of the CapRelSJ46–Gp57 complex predicted by AlphaFold. According to the model, the P-domain of Gp57 (pink) recognizes the pseudo-ZFD (orange) and anchor regions (green) of CapRelSJ46. This interaction prevents the recoil of pseudo-ZFD to the active site and activates the enzyme. d, Differential HDX (ΔHDX) between CapRelSJ46 and CapRelSJ46–Gp57 displayed as a difference heat map. Red indicates increased deuteration of CapRelSJ46 in the presence of Gp57; blue indicates lower deuteration. Grey bars indicate peptides identified in mass spectrometry analysis. e, Topological representation of CapRelSJ46, coloured according to the ΔHDX. The active site of the enzyme is marked by a black dashed outline and the catalytic toxSYNTH domain and the phage-recognition pseudo-ZFD are shadowed in light yellow and light orange, respectively. f, Serial dilutions on media containing glucose or arabinose of cells expressing the indicated mutant of CapRelSJ46 from its native promoter and the wild-type Gp57 from an arabinose-inducible promoter. g, Serial dilutions of SECΦ27 phage spotted on cells expressing the indicated mutant of CapRelSJ46 or an empty vector. h, As in a, but with the indicated mutants of CapRelSJ46–Flag. Images shown are representatives of three independent biological replicates.
Consistent with this tight binding interaction, the ab initio AlphaFold prediction of the CapRelSJ46–Gp57 complex has a large contact interface of around 1,800 Å2 (Fig. 4c). Although further structural studies are needed to fully validate this complex structure, the AlphaFold prediction has CapRelSJ46 adopting the same open state seen in our crystal structure (Fig. 2c), with the pseudo-ZFD making extensive contacts with the β-sheet and spine α-helix of the peripheral (P)-domain of Gp57 (Fig. 4c and Extended Data Fig. 5f). Notably, this region of Gp57 contains the residues L114 and I115 identified in our escape mutants. The complex predicted further interactions of pseudo-ZFD β6–β7 loop with the β6–α5 and β8–β9 loops of the axial (A)-domain of Gp57. In this arrangement Gp57 prevents the recoil of pseudo-ZFD to block the active site of the enzyme while stabilizing the YXXY motif in the non-neutralizing hinge conformation.
Results from hydrogen–deuterium exchange (HDX) monitored by mass spectrometry strongly supported the AlphaFold predictions. In the presence of Gp57, the pseudo-ZFD of CapRelSJ46 became more protected with the strongest protection mapping to α10, β8, and the C-terminal α-helical extension (Fig. 4d,e and Extended Data Fig. 5g,h). This overlaps the same region critical for phage specificity (Fig. 2b). The HDX data also confirmed the interface formed between Gp57 P-domain β5 and CapRelSJ46 pseudo-ZFD as well as the Gp57 A-domain β8–β9 loop and CapRelSJ46 β6–β7 loop. Finally, we observed increased deuterium uptake in CapRelSJ46 in residues 110–124 of β2 and 125–130 of α4, which are part of the adenine coordination pocket of toxSYNTH, thus confirming that interaction with Gp57 exposes the active site of the enzyme (Fig. 4d,e and Extended Data Fig. 5g,h).
To further validate the role of the pseudo-ZFD in binding and activating CapRelSJ46, we performed error-prone PCR-based mutagenesis on this domain and screened for mutations that disrupted activation of CapRelSJ46 when it was co-produced with the capsid protein Gp57. The substitutions L280Q and L280P drastically reduced the toxicity of CapRelSJ46 in the presence of wild-type Gp57 (Fig. 4f and Extended Data Fig. 5i), and prevented CapRelSJ46 from protecting against SECΦ27 infection (Fig. 4g and Extended Data Fig. 5j). Importantly, these CapRelSJ46 variants still protected E. coli against phage T2 and T4, indicating that these variants retained structural integrity (Extended Data Fig. 5k). The substitution L307A had similar, but less pronounced, effects on CapRelSJ46 activity (Fig. 4f,g).
The crystal structure of CapRelSJ46 suggested that L280 and L307 in the wild-type protein promote the open, active state, with L280 stabilizing one of the hinge regions involving the pseudo-ZFD and L307 structuring the β6–β7 loop that interacts with Gp57 A-domain. The L280Q and L280P variants of CapRelSJ46 were unable to co-precipitate the major capsid protein of SECΦ27, and the L307A substitution significantly reduced binding in this assay (Fig. 4h). Using ITC, we also showed that the L280P variant of CapRelSJ46 could no longer bind to wild-type Gp57 (Extended Data Fig. 5e). In sum, our findings strongly support a model in which the C-terminal pseudo-ZFD of CapRelSJ46 directly recognizes the major capsid protein of SECΦ27, thereby triggering a relief of autoinhibition of the N-terminal toxSYNTH domain.
Other phage capsids activate CapRelSJ46
The pseudo-ZFD of CapRelSJ46, including residues L280 and L307, is the least well conserved region of the protein (Extended Data Fig. 3a,i). This variability may reflect a ‘Red Queen dynamic’, a hallmark of many host–pathogen interfaces that arises from cycles of selective pressure on pathogens to evade host immunity followed by selection on host immune factors to restore recognition of a pathogen5. As triggers of the CapRel defence system, phage capsid proteins are likely to be under pressure to diversify while retaining the ability to form a capsid, leading to a selective pressure on the pseudo-ZFD of CapRel to diversify and retain its interaction with the capsid proteins. To test this hypothesis, we examined three phages from the BASEL collection37 (Bas4, Bas5 and Bas8) that are closely related to SECΦ27 and contain Gp8, a close homologue of Gp57 (Extended Data Fig. 6a). We first found that co-expressing the major capsid homologues from Bas5 and Bas8—but not that of Bas4—with CapRelSJ46 rendered CapRelSJ46 toxic, as with the SECΦ27 capsid protein (Fig. 5a and Extended Data Fig. 6b). We then tested whether CapRelSJ46 protects against these phages and found that it protected strongly against Bas5 and Bas8, but not Bas4 (Fig. 5b and Extended Data Fig. 6c).

a, Serial dilutions on media containing glucose or arabinose of cells expressing CapRelSJ46 from its native promoter and the major capsid protein homologue from the phage indicated from an arabinose-inducible promoter. b, Serial dilutions of the phages indicated spotted on lawns of cells expressing CapRelSJ46 or an empty vector. c, Serial dilutions of wild-type Bas8 phage or the escape mutants bearing the major capsid mutations indicated spotted on lawns of cells harbouring CapRelSJ46 or an empty vector. d, Alignment of the region of the major capsid protein in SECΦ27, Bas5 and Bas8 that triggers CapRelSJ46, along with Bas4, which has a tyrosine at position 113 instead of phenylalanine. e, Serial dilutions on media containing glucose or arabinose of cells expressing CapRelSJ46 from its native promoter and the Bas4 or SECΦ27 major capsid protein variant indicated from an arabinose-inducible promoter. f, Serial dilutions of wild-type Bas4 or two mutant clones containing Y113F in the major capsid protein Gp8 spotted on lawns of cells expressing CapRelSJ46 or an empty vector. g, Model for the direct activation of CapRelSJ46 by the major capsid protein of SECΦ27 and related phages. After genome injection, the production of the major capsid protein triggers relief of autoinhibition by the C-terminal antitoxin of CapRelSJ46, leading to pyrophosphorylation (PPi) of tRNAs by activated CapRelSJ46, which inhibits translation and restricts viral infection.
To validate that defence against Bas8 requires activation of CapRelSJ46 by the capsid protein homologue of this phage, we isolated spontaneous mutants of Bas8 that escaped defence. Two mutant clones of Bas8 were no longer defended against by CapRelSJ46 and contained either an F120L or I124F substitution in the major capsid homologue Gp8Bas8 (Fig. 5c and Extended Data Fig. 6d). Each substitution significantly reduced the ability of the capsid protein to activate CapRelSJ46 when co-produced (Extended Data Fig. 6e,f). As these Gp8Bas8 variants were not toxic when expressed on their own (Extended Data Fig. 6g), the residual toxicity seen during co-production with CapRelSJ46 probably arises from their accumulation over extended periods of time, despite their reduced binding affinity for CapRelSJ46. By contrast, each escape mutant almost completely evaded CapRelSJ46 defence (Fig. 5c), given the short timescale of phage infection. Notably, the two positions identified in Bas8 escape mutants were close to the positions of the escape mutants identified in SECΦ27 Gp57, further confirming that this region in the major capsid protein is important for activating CapRelSJ46 (Fig. 5d). In addition, we identified another CapRel homologue from E. coli strain HT2012018, called CapRelEcHT, that defends against Bas8 (Extended Data Fig. 6h). Three different substitutions were identified in the same region of the major capsid protein (G111S, L116F and I124N) that enabled Bas8 to escape defence (Extended Data Fig. 6h), suggesting that CapRelEcHT—like CapRelSJ46—is activated by the major capsid protein.
Unlike Bas8, Bas4 was not defended against by CapRelSJ46, and its capsid homologue did not activate CapRelSJ46 despite being 98% identical to SECΦ27 Gp57, with just 5 amino acid differences between the two. However, one difference is at position 113, near the region that is likely to bind to CapRelSJ46. This residue is a phenylalanine in the capsid proteins of SECΦ27, Bas5 and Bas8, but a tyrosine in the Bas4 capsid homologue (Fig. 5d). We tested whether this residue is critical for activation by making a Y113F substitution in the Bas4 capsid homologue and found that it gained the ability to activate CapRelSJ46 when co-produced (Fig. 5e and Extended Data Fig. 6i). By contrast, a F113Y substitution in the SECΦ27 capsid protein abolished its ability to activate CapRelSJ46. Additionally, we mutated Bas4 phage such that it produces major capsid protein with the Y113F substitution. This mutant phage could still produce mature virions, but was defended against by CapRelSJ46 (Fig. 5f and Extended Data Fig. 6j). These results support the notion of a Red Queen dynamic between the pseudo-ZFD of CapRel and the phage capsid proteins that directly bind and activate CapRel.
Conclusions
We propose the following model for CapRelSJ46 activation by SECΦ27 (Fig. 5g). Without phage infection, CapRelSJ46 adopts an inactive, closed conformation in cells with its C-terminal antitoxin domain autoinhibiting the N-terminal toxin domain. Upon infection, the phage’s major capsid protein is produced and directly binds to CapRelSJ46 to stabilize the active, open state. This open state enables CapRelSJ46 to pyrophosphorylate tRNAs and inhibit translation, which is likely to prevent mature virion production, leading to an abortive infection that prevents propagation of phage through a population of cells. Notably, our results imply that type II toxin–antitoxin systems, which feature protein antitoxins, can be activated without proteolysis of the antitoxin, which is often asserted as their primary means of activation. Although our work focused on a fused type II toxin–antitoxin system, activation by the direct binding of phage proteins could also be a common mechanism for canonical, non-fused type II toxin–antitoxin systems. As noted above, although antitoxins are frequently less stable than their cognate toxins, their turnover may not occur quickly enough to respond to phage infection22, whereas direct binding to phage-encoded triggers could allow rapid activation.
Major capsid proteins, such as Gp57 from SECΦ27, may be a common trigger for toxin–antitoxin systems and other anti-phage defence systems. Prior studies found that a short peptide called Gol within the major capsid protein Gp23 of T4 can activate the Lit protease in E. coli, which cleaves EF-Tu, if both Gol and Lit protease are overproduced38,39. For PifA, which allows the F plasmid to exclude T7, escape mutants mapped to the major capsid protein, but this interaction has not been studied biochemically40. Recent work reported that mutations in the major capsid protein of T5 allow it to overcome Pycsar-mediated defence, but the capsid protein alone is insufficient to activate Pycsar41. Finally, mutations in the gene encoding a major capsid protein of a Pseudomonas aeruginosa phage enables escape from CBASS-mediated defence, but whether the capsid protein is an activator of CBASS is not yet known42. Nevertheless, we anticipate that major capsid proteins may be common direct triggers for a diverse range of anti-phage defence systems. Other structural components of phages may also serve as triggers; for example, antiviral STAND defence systems were recently found to be directly triggered by terminase or portal proteins43. Although structural proteins may be effective triggers, the intense coevolution of phages and bacteria may also drive defence systems to rely on yet other triggers44.
As with PAMPs in eukaryotes, relying on essential, abundant components of phages for activation may help ensure that an immune response is mounted only following an infection. Notably, the capsid proteins of some eukaryotic viruses stimulate mammalian innate immune pathways. For instance, HIV capsid protein is directly detected in the host cell cytoplasm and nucleus by TRIM5 and NONO, respectively, to trigger innate immune activation7,9. Thus, our results suggest that similar principles of pathogen detection underlie the function and molecular basis of innate immunity in both bacteria and eukaryotes.
Methods
Strains and growth conditions
All bacterial and phage strains used in this study are listed in Supplementary Table 2. Escherichia coli strains were routinely grown at 37 °C in Luria broth (LB) medium for cloning and maintenance. Phages were propagated by infecting a culture of E. coli MG1655 at an A600 of 0.1–0.2 with a MOI of 0.1. Cleared cultures were pelleted by centrifugation to remove residual bacteria and filtered through a 0.2-μm filter. Chloroform was then added to phage lysates to prevent bacterial growth. All phage infection experiments in liquid media and phage spotting experiments were performed in LB medium at 25 °C, except for spotting of T2 and T4 on strains producing CapRelSJ46 open reading frames were (6.4 g l−1 Na2HPO4-7H2O, 1.5 g l−1 KH2PO4, 0.25 g l−1 NaCl, 0.5 g l−1 NH4Cl medium supplemented with 0.1% casamino acids, 0.4% glycerol, 0.4% glucose, 2 mM MgSO4, and 0.1 mM CaCl2) at 30 °C. For liquid induction experiments from pBAD33 vectors, bacterial cells were grown in M9 medium. Antibiotics were used at the following concentrations (liquid; plates): carbenicillin (50 μg ml−1; 100 μg ml−1) and chloramphenicol (20 μg ml−1; 30 μg ml−1).
Plasmid construction
All plasmids are listed in Supplementary Table 3. All primers and synthesized gene sequences are listed in Supplementary Table 4.
pBR322-capRel constructs: DNA encoding CapRelSJ46, CapRelEbc, CapRelKp or CapRelEcHT open reading frames were expressed in E. coli and 100–200 bp of the upstream region from the source organism was added in each case for native expression (TZ-1 to TZ-5, TZ-92 and TZ-93). DNA was commercially synthesized by Integrated DNA Technology as gBlocks and assembled into a promoter-less backbone of pBR322 amplified with TZ-6 and TZ-7 by Gibson assembly. Mutations that produce the single amino acid substitutions CapRelSJ46(Y155A), CapRelEbc(Y153A), CapRelSJ46(L280Q), CapRelSJ46(L280P) and CapRelSJ46(L307A) were generated by site-directed mutagenesis using primers TZ-8 to TZ-11 and TZ-49 to TZ-54. To add an N-terminal His6-tag or a C-terminal Flag tag to CapRelSJ46, primers TZ-41 and TZ-42 or TZ-45 and TZ-46 were used to PCR-amplify pBR322-capRelSJ46 followed by Gibson assembly. pBR322-capRel-chimera was constructed by inserting capRelEbc(270–339) that had been PCR-amplified with TZ-22 and TZ-23 into pBR322-capRelSJ46 linearized with TZ-20 and TZ-21 using Gibson assembly. To add a C-terminal Flag tag to CapRelEbc or the CapRel chimera, primers TZ-90 and TZ-91, or TZ-45 and TZ-46 were used to PCR-amplify corresponding pBR322 vectors followed by Gibson assembly.
pBAD33-capRelSJ46 constructs: capRelSJ46(1–272) or full-length capRelSJ46 was PCR-amplified with TZ-14 and TZ-15, or TZ-14 and TZ-24, respectively, and inserted into pBAD33 linearized with TZ-12 and TZ-13 using Gibson assembly. pBAD33-capRelSJ46 variants (A77K, R116A, V338A, L339A, A341K, A351K, Y352A or Y355A) were constructed by site-directed mutagenesis using primers TZ-25 to TZ-40. pBAD33-capRelSJ46 variants (R78A, K311A, R314A, E319A and K346A) were constructed by site-directed mutagenesis using primers TZ-80 to TZ-89.
pEXT20-capRelSJ46 construct: capRelSJ46(273–373) was PCR-amplified with primers TZ-18 and TZ-19, and then inserted into linearized pEXT20 with TZ-16 and TZ-17 using Gibson assembly.
pBAD33-gp57 constructs: wild-type or mutant variant (L114P or I115F) gp57 was PCR-amplified from the corresponding wild-type or escape-mutant SECΦ27 phage using primers TZ-43 and TZ-44, and inserted into linearized pBAD33 using Gibson assembly. A C-terminal HA tag was added to wild-type or mutant gp57 using primers TZ-47 and TZ-48 to PCR-amplify the corresponding construct followed by Gibson assembly. The F113Y variant of gp57 was generated by site-directed mutagenesis using primers TZ-63 and TZ-64.
pBAD33-gp8: the genes encoding the major capsid protein homologues Gp8Bas4, Gp8Bas5 and Gp8Bas8 were PCR-amplified from the corresponding phage using primers TZ-55 to TZ-60 and inserted into linearized pBAD33 by Gibson assembly. The Y113F variant of gp8Bas4 was generated by site-directed mutagenesis using primers TZ-61 and TZ-62. The F120L and I124F variants of gp8Bas8 were cloned from the corresponding phage escape mutants using primers TZ-59 and TZ-60.
pET-gp57 constructs: a gp57 fragment was PCR-amplified with primers TZ-65 and TZ-66 and the TZ-67 template using Gibson assembly, the resulting linear DNA fragment was inserted into linearized pET24d (without tag) using TZ-68 and TZ-69. Template TZ-67 was synthesized as a gBlock by Integrated DNA Technology. To construct L114P and I115F-substituted gp57 variants, DNA fragments were PCR-amplified with primer pairs TZ-70 + TZ-71 and TZ-72 + TZ-73 (for L114P) or with pairs TZ-74 + TZ-71 and TZ75 + TZ-73 (for I115F) and pET-gp57 template. The resulting linear DNA fragments were assembled using Gibson assembly.
pET24d-His10-SUMO-capRelSJ46 constructs: the capRelSJ46 open reading frame was PCR-amplified using primers TZ-76 and TZ-77 as well as pBAD-capRelSJ46 as template, and, using Gibson assembly, inserted into a linearized pET24d-His10-SUMO plasmid using primers TZ-78 and TZ-79.
Strain construction
Plasmids described above were introduced into E. coli MG1655 or BW27783 by TSS transformation or electroporation.
A single copy of capRelSJ46, capRelSJ46(Y155A), His6-capRelSJ46, gp57 or gp57-HA with its native promoter was inserted onto the MG1655 chromosome at the HK022 attachment site using the CRIM system45, using the pAH69 helper plasmid with the pAH144 vector containing the desired insert.
Bas4 mutant phage were generated using a CRISPR–Cas system for targeted mutagenesis as described46. In brief, sequences for RNA guides to target Cas9-mediated cleavage were designed using the toolbox in Geneious Prime 2021.2.2 and selected for targeting of gp8Bas4 but nowhere else in the Bas4 genome. The guides were inserted into the pCas9 plasmid and tested for their ability to restrict Bas4. An efficient guide was selected and the pCas9 guide plasmid was co-transformed into E. coli MG1655 with a high copy number repair plasmid containing gp8Bas4(Y113F) with the guide mutated to prevent self-cutting. The wild-type Bas4 phage was plated onto a strain containing both the pCas9 guide and the repair plasmid, and single plaques were screened by Sanger Sequencing. Two clones that produce the Y113F substituted Gp8 were propagated twice on strains containing only pCas9 guide for further selection and genomes were sequence verified by Illumina sequencing as described below.
External data
Previously published structures are available from the Protein Data Bank (PDB) with IDs 5DEC, 6S2T and 2LVH. The UniRef90 database is publicly available. Reference phage genomes are publicly available: SECΦ27 (NC_047938.1), Bas04 (MZ501069.1), Bas08 (MZ501059.1).
Toxicity assays on solid media
For producing the CapRelSJ46 N- and C-terminal domains, single colonies of E. coli MG1655 containing pBAD33-capRelSJ46(1–272) and pEXT20-capRelSJ46(273–373) or the corresponding empty vectors were grown for 6 h at 37 °C in LB-glucose to saturation. 200 μl of each saturated culture was then pelleted by centrifugation at 4,000g for 10 min, washed once in 1× phosphate-buffered saline (PBS), and resuspended in 400 μl 1× PBS. Cultures were then serially diluted 10-fold in 1× PBS and spotted on M9L plates (M9 medium supplemented with 5% LB (v/v)) further supplemented with 0.4% glucose, 0.2% arabinose or 0.2% arabinose and 100 μM IPTG. Plates were then incubated at 37 °C overnight before imaging.
For producing full-length CapRelSJ46, E. coli MG1655 containing pBAD33-capRelSJ46 or a mutant form of capRelSJ46 were grown to saturation and processed as above. Cultures were plated onto 0.4% glucose and 0.2% arabinose and incubated at 37 °C overnight.
For co-producing CapRelSJ46 and the major capsid proteins from SECΦ27, Bas4, Bas5, or Bas8, E. coli MG1655 harbouring pBR322-capRelSJ46 or MG1655 containing genomic capRelSJ46 and pBAD33-capsid protein were grown to saturation and processed as above. Cultures were plated onto 0.4% glucose and 0.2% arabinose and incubated at 37 °C overnight.
For co-producing CapRelSJ46 and variants of the major capsid protein from Bas8, E. coli BW27783 harbouring pBR322-capRelSJ46 and pBAD33-gp8Bas8 (wild-type or a mutant variant) were grown to saturation and processed as above. Cultures were plated onto 0.4% glucose and 0.0002% arabinose and incubated at 37 °C overnight.
Phage spotting assays and EOP measurements
Phage stocks isolated from single plaques were propagated in E. coli MG1655 at 37 °C in LB. To titre phage, dilutions of stocks were mixed with E. coli MG1655 and melted LB + 0.5% agar and spread on LB + 1.2% agar plates and incubated at 37 °C overnight. For phage spotting assays, 40 μl of a bacterial strain of interest was mixed with 4 ml LB + 0.5% agar and spread on an LB + 1.2% agar + antibiotic plate. Phage stocks were then serially diluted in 1× FM buffer (20 mM Tris-HCl pH 7.4, 100 mM NaCl, 10 mM MgSO4), and 2 μl of each dilution was spotted on the bacterial lawn. Plates were then incubated at 25 °C overnight before imaging. EOP was calculated by comparing the ability of the phage to form plaques on an experimental strain relative to the control strain. Experiments were replicated 3 times independently and representative images are shown.
For spotting phage T2 and T4 on strains producing CapRelSJ46 variants, 40 μl of a bacterial strain of interest was mixed with 4 ml M9 + 0.5% agar and spread on an M9 + 1.2% agar + antibiotic plate. Phage were serially diluted and spotted as described above. Plates were then incubated at 30 °C overnight before imaging.
Growth curves following phage infection in liquid culture
Single colonies of E. coli MG1655 pBR322 empty vector or pBR322-capRelSJ46 or pBR322-capRelSJ46(Y155A) were grown in LB overnight. Cultures were then back-diluted to A600 = 0.1 in fresh LB and 100 μl of cells were added into each well of a 96-well plate. Ten microlitres of serial diluted T4 phage were added to each well at the indicated MOI and growth following phage infection was measured at 15 min intervals with orbital shaking at 25 °C on a plate reader (Biotek). Data reported are the mean and standard deviation of eight plate replicates and the growth curve experiment was replicated three times independently.
One-step growth curves
Single colonies of E. coli MG1655 pBR322 empty vector or pBR322-capRelSJ46 were grown overnight in LB. Overnight cultures were back-diluted to A600 = 0.05 in 25 ml fresh LB and grown to A600 ~0.3 at 25 °C. Ten millilitres of each culture was infected with T4 phage at an MOI of 0.05 or SECΦ27 phage at an MOI of 0.01 in LB at 25 °C and phages were allowed to adsorb for 10 min before serial dilution in LB three times (1:100, 1:10, 1:10 serial dilution) to three flasks. Then, at indicated time points, 100 μl of infected cells from the corresponding dilution flask were mixed with 100 μl of indicator cells MG1655 pBR322 empty vector (A600 ~0.3), and the mixtures were mixed with 4 ml of LB + 0.5% agar and spread on LB + 1.2% agar plates. Plates were incubated overnight at 25 °C and plaques were enumerated the following day. PFU was calculated based on the dilution flask samples were taken from. Data reported are the mean and individual data points from 3 biological replicates.
Adsorption assays
Single colonies of E. coli MG1655 pBR322-capRelSJ46 or pBR322 empty vector were grown overnight in LB. Overnight cultures were back-diluted to A600 = 0.05 in 25 ml of fresh LB and grown to A600 = 0.2 at 25 °C. Cells or a control containing only media were infected with phage T4 or SECΦ27 at MOI = 0.1 and incubated at 25 °C for 10 min to allow for adsorption. Cells were pelleted at 9,000g for 2 min and unadsorbed phages from the supernatant were serial diluted, mixed with indicator cells MG1655 pBR322 empty vector + top agar and plated. Plates were incubated overnight at 25 °C and plaques were enumerated the following day. Percent adsorption was calculated by normalizing unadsorbed phages from each sample to media control. Data reported are the mean and individual data points from 3 biological replicates.
Western blot of CapRelSJ46 and Gp57 after phage infection
For immunoblotting of CapRelSJ46, single colonies of E. coli MG1655 pBR322-His6-capRelSJ46 or E. coli MG1655 genomic His6-capRelSJ46 under its native promoter were grown overnight in LB. Overnight cultures were back-diluted to A600 = 0.05 in 25 ml fresh LB and grown to A600 = 0.2 at 25 °C. Cells were infected with phage SECΦ27 at MOI = 100, and incubated at 25 °C during the experiment. At each indicated time point (0, 10, 20, 40, 60 min), A600 was measured and 1 ml of cells was pelleted at 21,000g for 2 min at 4 °C. Supernatant was removed and pellets were flash-frozen in liquid nitrogen. Pellets were thawed and resuspended in 1× Laemmli sample buffer (Bio-Rad) supplemented with 2-mercaptoethanol with A600 normalized. Samples were then boiled at 95 °C and analysed by 12% SDS–PAGE and transferred to a 0.45 μm PVDF membrane. Anti-His6 antibody (Invitrogen) was used at a final concentration of 1:1000, and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher) was used to develop the blots. Blots were imaged by a ChemiDoc Imaging system (Bio-Rad). Blots were stained with Coomassie stain and imaged as loading control. Image shown is a representative of two independent biological replicates.
For immunoblotting of Gp57, single colonies of E. coli MG1655 containing genomic gp57-HA under its native promoter were grown overnight in LB. Overnight cultures were back-diluted to A600 = 0.05 in 25 ml of fresh LB and grown to A600 = 0.2 at 25 °C. Cells were infected with phage SECΦ27 at MOI = 100, and incubated at 25 °C during the experiment. At each indicated time point (0, 10, 20, 30, 40, 50 min), A600 was measured and 1 ml of cells was pelleted at 21,000g for 2 min at 4 °C. Samples were processed as described above with A600 normalized. Samples were then boiled at 95 °C and analysed by 12% SDS–PAGE and transferred to a 0.45 μm PVDF membrane. Anti-HA antibody (Cell Signaling Technology) was used at a final concentration of 1:1000, and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher) was used to develop the blots. Blots were imaged by a ChemiDoc Imaging system (Bio-Rad), then stained with Coomassie stain and imaged as loading control. The experiment was performed three times independently and band intensities were quantified using Fiji. Relative band intensities were calculated by normalizing the summed intensity of both Gp57 bands to the intensity of total proteins by Coomassie stain.
Western blot of CapRelSJ46, CapRelEB or chimera expression levels
Single colonies of E. coli MG1655 pBR322-capRelSJ46, pBR322-capRelSJ46-Flag, pBR322-capRelEbc-Flag or pBR322-chimera-Flag were grown overnight in LB. Overnight cultures were back-diluted to A600 = 0.05 in 5 ml fresh LB and grown to A600 = 0.2 at 37 °C. A600 was measured and 5 ml of cells was pelleted at 4,000g for 5 min. Supernatant was removed and pellets were resuspended in 1× Laemmli sample buffer (Bio-Rad) supplemented with 2-mercaptoethanol with A600 normalized. Samples were then boiled at 95 °C and analysed by 12% SDS–PAGE and transferred to a 0.45 μm PVDF membrane. Anti-Flag antibody (Cell Signaling Technology) and anti-GyrA antibody (Inspiralis) were used at a final concentration of 1:1,000, and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher) was used to develop the blots. Blots were imaged by a ChemiDoc Imaging system (Bio-Rad). Image shown is a representative of 2 independent biological replicates.
Error-prone PCR mutagenesis of CapRelSJ46
The C terminus of CapRelSJ46 was mutagenized using error-prone PCR-based mutagenesis as described previously47. In brief, primers TZ-94 and TZ-95 were used to amplify the C terminus of CapRelSJ46 using Taq polymerase (NEB) and 0.5 mM MnCl2 was added to the reaction as the mutagenic agent. PCR products were treated with Dpn I, column purified, and inserted into pBR322-CapRelSJ46 backbone amplified with primer TZ-96 and TZ-97 using Gibson assembly. Gibson products were transformed into DH5α and grown overnight in LB at 37 °C. Overnight cultures were miniprepped to obtain the mutagenized library. Individual colonies were Sanger sequenced to assess the number of mutations. To perform the selection, mutagenized library was electroporated into E. coli MG1655 pBAD33-gp57, and plated onto LB plates containing 0.2% arabinose to select for survivors. Colonies were picked and sequenced to identify mutations in CapRelSJ46, and further validated by constructing plasmid with only single mutants.
Isolation of phage escape mutants to infect CapRelSJ46
The phage evolution experiment was conducted as described previously48. In brief, 5 independent populations were evolved in a 96-well plate containing a sensitive host E. coli MG1655 pBR322 empty vector and a resistant host E. coli MG1655 pBR322-capRelSJ46. One control population was evolved with only the sensitive host. Overnight bacterial cultures were back-diluted to A600 = 0.1 in LB and 100 μl were seeded into each well. Cells were infected with tenfold serial dilutions of SECΦ27 phage with MOI from 102 to 10−4, with one well uninfected to monitor for contamination. Plates were sealed with breathable plate seals and incubated at 25 °C for 6 h in a plate shaker at 1,000 rpm. Cleared wells from each population were pooled, pelleted at 4,000g for 20 min to remove bacteria, and the supernatant lysates were transferred to a 96 deep-well block with 40 µl chloroform added to prevent bacterial growth. Lysates were spotted onto both sensitive and resistant hosts to check the defence phenotype. Thirteen rounds of evolution were performed to allow all five populations to overcome CapRelSJ46 defence. Evolved clones from each evolved population were isolated by plating to single plaques on lawns of resistant host, and control clones from the control population were isolated on a lawn of the sensitive host. Two clones from each population were propagated using the corresponding host and sequenced as described below.
Bas8 escape mutants were isolated by plating a population of phage onto CapRelSJ46- or CapRelEcHT-containing cells. Twenty microlitres of 1011 PFU ml−1 Bas8 phage mixed with 40 µl overnight culture of E. coli MG1655 pBR322-capRelSJ46 or pBR322-capRelEcHT were added to 4 ml LB + 0.5% agar and spread onto LB + 1.2% agar. Plates were incubated at 25 °C overnight. Single plaques were isolated and propagated using the same strain in LB at 25 °C. Amplified phage lysates were pelleted to remove bacteria, and then plated to single plaques and propagated similarly for a second round of isolation to improve purity and sequenced.
Phage DNA extraction and Illumina sequencing
To extract phage DNA, high-titre phage lysates (>106 PFU μl−1) were treated with DNase I (0.001 U µl−1) and RNase A (0.05 mg ml−1) at 37 °C for 30 min. 10 mM EDTA was used to inactivate the nucleases. Lysates were then incubated with proteinase K at 50 °C for 30 min to disrupt capsids and release phage DNA. Phage DNA was isolated by ethanol precipitation. In brief, sodium acetate pH 5.2 was added to 300 mM followed by 100% ethanol to a final volume fraction of 70%. Samples were incubated at −80 °C overnight, pelleted at 21,000g for 20 min and supernatant removed. Pellets were washed with 100 µl isopropanol and 200 µl 70% (v/v) ethanol, and then aired dried at room temperature and resuspended in 25 µl 1× TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8). Concentrations of extracted DNA were measured by NanoDrop (Thermo Fisher Scientific).
To prepare Illumina sequencing libraries, 100–200 ng of genomic DNA was sheared in a Diagenode Bioruptor 300 sonicator water bath for twenty 30 s cycles at maximum intensity. Sheared genomic DNA was purified using Ampure XP beads, followed by end repair, 3′ adenylation, and adapter ligation. Barcodes were added to both 5′ and 3′ ends by PCR with primers that anneal to the Illumina adapters. The libraries were cleaned by Ampure XP beads using a double cut to elute fragment sizes matching the read lengths of the sequencing run. Libraries were sequenced on an Illumina MiSeq at the MIT BioMicro Center. Illumina reads were assembled to the reference genomes using Geneious Prime 2021.2.2.
Mass spectrometry of phages
Wild-type or mutant (L114P in Gp57, evolved clone 1 from population 3) SECΦ27 phage were propagated in E. coli MG1655 for high-titre stocks. In brief, E. coli MG1655 (A600 = 0.2) in LB were infected with phages at MOI = 0.1 and incubated at 37 °C for 4 h. Cells were pelleted at 4,000g for 10 min and supernatant lysates were filtered through 0.2-μm filters. Five hundred microlitres of phage stocks (1010 PFU μl−1) were further concentrated with Amicon Ultra filter (MW 100 kDa) and washed twice with 1× FM buffer (20 mM Tris-HCl pH 7.4, 100 mM NaCl, 10 mM MgSO4). Concentrated phage lysates were boiled to denature virions and run on 4–20% SDS–PAGE. Each lane from the gel was excised. Proteins were reduced with 10 mM dithiothreitol (Sigma) for 1 h at 56 °C and then alkylated with 20 mM iodoacetamide (Sigma) for 1 h at 25 °C in the dark. Proteins were then digested with 12.5 ng µl−1 modified trypsin (Promega) in 50 μl 100 mM ammonium bicarbonate, pH 8.9 at 25 °C overnight. Peptides were extracted by incubating the gel pieces with 50% acetonitrile/5% formic acid then 100 mM ammonium bicarbonate, repeated twice followed by incubating the gel pieces with 100% acetonitrile then 100 mM ammonium bicarbonate, repeated twice. Each fraction was collected, combined, and reduced to near dryness in a vacuum centrifuge. Peptides were desalted using Pierce Peptide Desalting Spin Columns (Thermo) and then lyophilized. The tryptic peptides were separated by reverse phase HPLC (Thermo Ultimate 3000) using a Thermo PepMap RSLC C18 column over a 90 min gradient before nano-electrospray using an Exploris mass spectrometer (Thermo). Solvent A was 0.1% formic acid in water and solvent B was 0.1% formic acid in acetonitrile. Detected peptides were mapped to SECΦ27 protein sequences and the abundance of proteins were estimated by number of spectrum counts/molecular mass to normalize for protein sizes.
Co-immunoprecipitation analysis
For immunoprecipitation of CapRelSJ46 after phage infection, E. coli MG1655 containing pBR322-capRelSJ46-Flag were grown overnight in LB. Overnight cultures were back-diluted to A600 = 0.05 in 175 ml of LB and grown to A600 ~0.3 at 25 °C. Cells were infected with wild-type or mutant (L114P in Gp57, evolved clone 1 from population 3) SECΦ27 at MOI = 100 and incubated at 25 °C. At the indicated time points (15 min or 40 min), A600 was measured and 50 ml of cells were pelleted at 6,000g for 5 min at 4 °C. Uninfected cells were collected at 0 min before phage infection. Supernatant was removed and cells were resuspended in 900 μl lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 and 5% glycerol) supplemented with protease inhibitor (Roche), 1 μl per ml Ready-Lyse Lysozyme Solution (Lucigen) and 1 μl per ml benzonase nuclease (Sigma). Samples were lysed by two freeze-thaw cycles, and lysates were normalized by A600. Lysates were pelleted at 21,000g for 10 min at 4 °C, and 850 μl of supernatant were incubated with pre-washed anti-Flag M2 magnetic beads (Sigma) for 1 h at 4 °C with end-over-end rotation. Beads were then washed 3 times with lysis buffer containing 350 mM NaCl but free of detergent. On-bead reduction, alkylation and digestion were performed. Proteins were reduced with 10 mM dithiothreitol (Sigma) for 1 h at 56 °C and then alkylated with 20 mM iodoacetamide (Sigma) for 1 h at 25 °C in the dark. Proteins were then digested with modified trypsin (Promega) at an enzyme/substrate ratio of 1:50 in 100 mM ammonium bicarbonate, pH 8 at 25 °C overnight. Trypsin activity was halted by addition of formic acid (99.9 %, Sigma) to a final concentration of 5 %. Peptides were desalted using Pierce Peptide Desalting Spin Columns (Thermo) then lyophilized. The tryptic peptides were subjected to LC–MS/MS as described above. Experiments were performed two times independently and spectral counts are reported. Ratio of spectral counts between Gp57 and CapRelSJ46 were calculated and plotted for normalization.
For co-producing CapRelSJ46 and Gp57, E. coli MG1655 containing pBR322-capRelSJ46 or pBR322-capRelSJ46-Flag (wild type or mutants) and pBAD33-gp57-HA (wild type or mutants) were grown overnight in M9-glucose. Overnight cultures were back-diluted to A600 = 0.05 in 50 ml of M9 (no glucose) and grown to A600 ~0.3 at 37 °C. Cells were induced with 0.2% arabinose for 30 min at 37 °C, then A600 was measured and cells were pelleted at 4,000 g for 10 min at 4 °C. Supernatant was removed and cells were resuspended in 900 μl lysis buffer as described above. Samples were lysed by two freeze-thaw cycles, and lysates were normalized by A600. Lysates were pelleted at 21,000g for 10 min at 4 °C, and 850 μl of supernatant were incubated with pre-washed anti-Flag M2 magnetic beads (Sigma) for 1 h at 4 °C with end-over-end rotation. Beads were then washed 3 times with lysis buffer containing 350 mM NaCl. Laemmli sample buffer (Bio-Rad) supplemented with 2-mercaptoethanol was added to beads directly to elute proteins. Samples were boiled at 95 °C and analysed by 12% SDS–PAGE and transferred to a 0.45 μm PVDF membrane. Anti-Flag and anti-HA antibodies (Cell Signaling Technology) were used at a final concentration of 1:1,000, and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher) was used to develop the blots. Blots were imaged by a ChemiDoc Imaging system (Bio-Rad). Images shown are representatives of 3 independent biological replicates.
Incorporation assays
For co-producing CapRelSJ46 and Gp57, the SECΦ27 major capsid protein, single colonies of E. coli MG1655 containing pBR322-capRelSJ46 and pBAD33-gp57 (wild-type or L114P variant) or corresponding empty vectors were grown overnight in M9-glucose. Overnight cultures were back-diluted to A600 = 0.05 in 25 ml M9-glucose and grown to A600 ~0.3 at 37 °C. Cells were pelleted at 4,000g for 5 min at 4 °C and washed once with M9 (no glucose), and then back-diluted to A600 = 0.1 in 15 ml M9 (no glucose) and recovered for 45 min at 37 °C. At the beginning of the experiment, cells were induced with 0.2% arabinose. At the indicated time points (0, 10, 20, 30, 40 min), A600 was measured and an aliquot of 250 μl of cells was transferred to microcentrifuge tube containing [5,6-3H]uridine (PerkinElmer) (4 μCi ml−1) for transcription measurements or EasyTag EXPRESS-35S Protein Labeling Mix, [35S] (PerkinElmer) at 44 μCi ml−1 for translation measurements. Tubes were incubated at 37 °C for 2 min, then quenched by addition of nonradioactive uridine (1.5 mM) or cysteine and methionine (15 mM each) and incubated for an additional 2 min. Samples were then added to ice cold trichloroacetic acid (TCA) (10% w/v) and incubated at least 30 min on ice to allow for precipitation. Resulting samples were vacuum filtered onto a glass microfibre filter (Whatman, 1820-024) that had been pre-wetted with 5% w/v TCA. Filters were washed with 35× volume of 5% w/v TCA, then with 5× volume of 100% ethanol. Air dried filters were placed in tubes with scintillation fluid and measured in a scintillation counter (PerkinElmer). CPM (Counts Per Million) was normalized to A600 and percent incorporation at each time point was calculated by normalizing to t = 0. Data reported are the mean and individual data points from three independent biological replicates.
For producing the CapRelSJ46 N-terminal toxin domain, single colonies of E. coli MG1655 containing pBAD33-capRelSJ46(1–272) or an empty vector were grown overnight in M9-glucose. Transcription and translation experiments were done as described above. Data reported are the mean and individual data points from three independent biological replicates.
For phage infection experiments, single colonies of E. coli MG1655 harbouring pBR322 empty vector or pBR322-capRelSJ46 were grown overnight in LB. Overnight cultures were back-diluted to A600 = 0.05 in 25 ml fresh LB and grown to A600 ~0.3 at 25 °C. Cells were then diluted to A600 = 0.1 in 10 ml LB and infected with wild-type or mutant (L114P in Gp57, evolved clone 1 from population 3) SECΦ27 at MOI = 100 and incubated at 25 °C. At the indicated time points (0, 15, 30, 45 and 60 min), A600 was measured and an aliquot of 250 μl of cells was transferred to a microcentrifuge tube containing [5,6-3H]uridine (PerkinElmer) (32 μCi ml−1) for transcription measurements or EasyTag EXPRESS-35S Protein Labeling Mix, [35S] (PerkinElmer) at 88 μCi ml−1 for translation measurements. Tubes were incubated at 25 °C for 4 min, then quenched by addition of nonradioactive uridine (1.5 mM) or cysteine and methionine (15 mM) and incubated for an additional 2 min. Samples were then processed same as above. Data reported are the mean and individual data points from three independent biological replicates. Statistical significance was determined by unpaired, two-tailed Student’s t-test (P < 0.05).
Homology search, alignment, and conservation analysis
CapRelSJ46 was identified in the sequence database from our previous bioinformatic survey of RSH proteins24 that included gene neighbourhood analysis to identify toxin–antitoxin systems49. Bacterial strains containing CapRelSJ46, CapRelEbc or CapRelKp with 100% amino acid identity were found on NCBI database. Local genomic regions (±10 kb of CapRel) were extracted and annotated for all coding sequences. Prophage genes and intact prophage regions were identified by PHASTER50. Additional homologues of CapRelSJ46 were identified by ConSurf51 using PSI-BLAST (default settings) to search UniRef90 database, yielding 44 homologues. For Extended Data Fig. 1, sequences were aligned with MAFFT L-INS-i v7.453 (ref. 52) with manual curation of the C-terminal region guided by homology modelling of the stand-alone Phrann Gp30 antitoxin using Swiss-Model53, and with our CapRelSJ46 predicted structure as a template. For ConSurf analysis, 52 homologues were used to generate the multiple sequence alignment by MAFFT and used as input. Conservation scores were calculated using the Bayesian method and default settings. For gene neighbourhood analysis, previously identified CapRel and PhRel sequences24 were reduced to representatives with 65% amino acid identity using MMSeqs v13.45111 easy-cluster (default settings54), while retaining proteins of interest. The associated accession numbers were then used as input to FlaGs v1.2.6 (one flanking gene either side of the query and otherwise default settings)49.
Homologues of the major capsid proteins in BASEL phages were identified by BLASTp55 searches against each phage genome. Homologues of Gp57 (Gp8Bas4, Gp8Bas5 and Gp8Bas8) were aligned by MUSCLE56.
CapRelSJ46 preparation for crystallization and HDX-MS
For the production of His10–SUMO-tagged CapRelSJ46 and CapRelSJ46 variants, E. coli BL21 (DE3) cells were transformed with pET24d plasmids containing the gene of interest and grown in LB medium to A600 of 0.6. Expression of the protein of interest was induced by addition of 0.5 mM IPTG, and cells were grown for 3 h at 30 °C. The culture was then centrifuged, and pellet was resuspended in resuspension buffer (50 mM Tris-HCl pH 8.0, 1.5 M KCl, 2 mM MgCl2, 1 mM TCEP, 0,002% mellitic acid and 1 pastil of protease inhibitors cocktail (Roche)).
Cells were disrupted using a high-pressure homogenizer (Emulsiflex) and the supernatant was separated from the pellet by centrifugation and filtered through 0.45 μm filters. Protein extracts were loaded onto a gravity-flow column (Cytiva) packed with HisPur Nickel resin (Thermo Fisher Scientific), washed with buffer A (50 mM Tris-HCl pH 8, 500 mM NaCl, 500 mM KCl, 1 mM TCEP, 0.002% Melitic acid) and stepwise eluted in buffer A supplemented with 500 mM imidazole. To remove remaining contaminants and imidazole, the elution fraction was immediately transferred to a size-exclusion chromatography (SEC) column Superdex 200 pgcolumn (Cytiva), previously equilibrated in the SEC buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 500 mM KCl, 2 mM MgCl2, 1 mM TCEP, 0.002% mellitic acid). The fractions containing the protein were concentrated to around 1 mg ml−1 and the His tag was removed by incubating with UlpI protease (1:50 molar ratio) at 4 °C for 30 min. The His10–SUMO tag and the protease were then removed by passing the samples over a gravity-flow column (Cytiva) packed with HisPur Nickel resin (Thermo Fisher Scientific). Purity of the sample preparation was assessed spectrophotometrically and by SDS–PAGE. For all the purified protein samples, A260/A280 ratio was below 0.6. Samples were stored at −20 °C or concentrated to 7 mg ml−1 and used directly in crystallization experiments.
For the purification of the complex containing His10–SUMO–CapRelSJ46 and His10–SUMO–Gp57, E. coli BL21 (DE3) strain containing freshly transformed pET24d-His10-SUMO-capRelSJ46(Y155A) and pET21a-His10-SUMO-gp57 were grown in LB medium to A600 of 0.2. This culture was then diluted in fresh LB media and grown until A600 of 0.6. Expression of the protein of interest was induced by addition of 0.5 mM IPTG, and cells were grown for overnight at 16 °C. The subsequent purification, Sumo tag cleavage and purity assessment steps were identical to the workflow described above for the all the CapRelSJ46 protein variants.
Crystallization of CapRelSJ46
The screening of crystallization conditions of CapRelSJ46 was carried out using the sitting-drop vapour-diffusion method. The drops were set up in Swiss (MRC) 96-well two-drop UVP sitting-drop plates using the Mosquito HTS system (TTP Labtech). Drops of 0.1 μl protein and 0.1 μl precipitant solution were equilibrated to 80 μl precipitant solution in the reservoir. Commercially available screens LMB and SG1 (Molecular Dimensions) were used to test crystallization conditions. The condition resulting in protein crystals (LMB screen position C9 for CapRelSJ46: 26% w/v PEG 2000 MME 0.1 M Bis-Tris 5.8) were repeated as 2 µl drops. The final crystallization drop was made by mixing in a 1:1 ratio the crystallization mother liquor at pH 5.8 and CapRelSJ46 at pH 8.0. Crystals were collected using suitable cryo-protecting solutions (mother liquor supplemented with 20% glycerol) and vitrified in liquid N2 for transport and storage before X-ray exposure. X-ray diffraction data was collected at the SOLEIL synchrotron (Gif-sur-Yvette, Paris, France) on the Proxima 1 (PX1) and Proxima 2A (PX2A) beamlines using an Eiger-X 16M detector. Because of the high anisotropic nature of the data from all the crystals we performed anisotropic cut-off and correction of the merged intensity data as implemented on the STARANISO server (http://staraniso.globalphasing.org/) using the DEBYE and STARANISO programs. The analysis of the data suggested a resolution of 2.31 Å (with 2.31 Å in a*, 2.85 Å in b* and 2.72 Å in c*).
Structure determination
The data were processed with the XDS suite57 and scaled with Aimless. In all cases, the unit cell content was estimated with the program MATTHEW COEF from the CCP4 program suite58. Molecular replacement was performed with Phaser59. The crystals of CapRelSJ46 diffracted on average to ~2.3 Å. We used the coordinates of RelTtNTD (PDB ID 6S2T) as a search model for the toxSYNTH domain60. The MR solution from Phaser was used in combination with Rosetta as implemented in the MR-Rosetta61 suit from the Phenix package62. After several iterations of manual building with Coot63 and maximum likelihood refinement as implemented in Buster/TNT64, the model was extended to cover all the residues (R/Rfree of 21.5/26.0). Extended Data Table 1 details all the X-ray data collection and refinement statistics.
Isothermal titration calorimetry
For all ITC measurements CapRelSJ46 samples were prepared from the pET24d-His10-SUMO-capRelSJ46 as detailed above. In the case of Gp57, E. coli BL21 (DE3) cells were transformed with pET21a-His10-SUMO-gp57 and grown in LB medium to A600 of 0.2. This culture was then diluted in fresh LB media and grown until A600 of 0.6. Expression of His10–SUMO–Gp57 was induced by addition of 0.1 mM IPTG, and cells were grown for overnight at 16 °C. The subsequent purification, SUMO-tag cleavage and purity assessment steps were identical to the workflow described above for the all the CapRelSJ46 protein variants. After removing the SUMO tag, samples were concentrated to 10 μM and used directly for ITC immediately after purification.
All titrations were performed with an Affinity ITC (TA instruments) at 25 °C. For the titration, CapRelSJ46 was loaded in the instrument syringe at 150 μM and Gp57 was used in the cell at 10 μM. The titration was performed in 50 mM HEPES pH 7.5; 500 mM KCl; 500 mM NaCl; 150 mM imidazole; 10 mM MgCl2; 1 mM TCEP; 0.002% mellitic acid. Final concentrations were verified by the absorption using a Nanodrop One (ThermoScientific). All ITC measurements were performed by titrating 2 µl of CapRelSJ46 (or a CapRelSJ46 variant) into Gp57 (or a Gp57 variant) using a constant stirring rate of 75 rpm. All data were processed, buffer-corrected and analysed using the NanoAnalyse and Origin software packages.
Multiwavelength light scattering
The sample of the CapRelSJ46-Gp57 complex used for the MALS measurements was prepared in the same way as the samples used for hydrogen–deuterium exchange mass spectrometry (HDX-MS): 50 mM HEPES pH 7.5; 500 mM KCl; 500 mM; NaCl; 10 mM MgCl2; 1 mM TCEP; 0.002% mellitic acid at a concentration of 5 mg ml−1. The measurement was performed in an HPLC Alliance system (Waters) connected to a 2998 PDA detector (Waters), a TREOS II MALS detector (Wyatt Technology) and a RI-501 refractive index detector (Shodex). The data were analysed with Astra 7 suite (Wyatt technology).
HDX-MS
HDX-MS experiments were performed on an HDX platform composed of a Synapt G2-Si mass spectrometer (Waters Corporation) connected to a nanoAcquity UPLC system. Samples of CapRelSJ46 and CapRelSJ46 complexed with Gp57 were prepared at a concentration of 20 to 50 µM. For each experiment 5 µl of sample (CapRelSJ46 or CapRelSJ46-Gp57) were incubated for 1 min, 5 min, 15 min or 60 min in 95 µl of labeling buffer L (50 mM HEPES, 500 mM KCl, 500 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 0.002% mellitic acid, pH 7.5) at 20 °C. The non-deuterated reference points were prepared by replacing buffer L by equilibration buffer E (50 mM HEPES, 500 mM KCl, 500 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 0.002% mellitic acid, pH 7.5). After labelling, the samples are quenched by mixing with 100 µl of pre-chilled quench buffer Q (1.2 % formic acid, pH 2.4). Seventy microlitres of the quenched samples are directly transferred to the Enzymate BEH Pepsin Column (Waters Corporation) at 200 µl min−1 and at 20 °C with a pressure 8.5 kPSI. Peptic peptides were trapped for 3 min on an Acquity UPLC BEH C18 VanGuard Pre-column (Waters Corporation) at a 200 µl min−1 flow rate in water (0.1% formic acid in HPLC water pH 2.5) before eluted to an Acquity UPLC BEH C18 Column for chromatographic separation. Separation was done with a linear gradient buffer (7–40% gradient of 0.1% formic acid in acetonitrile) at a flow rate of 40 µl min−1. Peptides identification and deuteration uptake analysis was performed on the Synapt G2-Si in ESI ± HDMSE mode (Waters Corporation). Leucine enkephalin was applied for mass accuracy correction and sodium iodide was used as calibration for the mass spectrometer. HDMSE data were collected by a 20–30 V transfer collision energy ramp. The pepsin column was washed between injections using pepsin wash buffer (1.5 M guanidinium HCl, 4% (v/v) methanol, 0.8% (v/v) formic acid). A blank run was performed between each sample to prevent significant peptide carry-over. Optimized peptide identification and peptide coverage for all samples was performed from undeuterated controls (five replicates). All deuterium time points were performed in triplicate.
Data treatment and statistical analysis of HDX-MS
The non-deuterated references points were analysed by PLGS (ProteinLynx Global Server 2.5.1, Waters) to identify the peptic peptides belonging to CapRelSJ46 or Gp57. Then, all the HDMSE data including reference and deuterated samples were processed by DynamX 3.0 (Waters) for deuterium uptake determination. We chose the following filtering parameters: minimum intensity of 1,000, minimum and maximum peptide sequence length of 5 and 20, respectively, minimum MS/MS products of 3, minimum products per amino acid of 0.27, minimum score of 5, and a maximum MH+ error threshold of 15 p.p.m. Data were analysed at peptidic and overall level and manually curated by visual inspection of individual spectra. The overall level is based on the relative fractional uptake (RFUa,t), which can be calculated by the following formula:
where \({\gamma }_{a,t}\) is the deuterium uptake for peptide a at incubation time t, and \({{\rm{M}}{\rm{a}}{\rm{x}}{\rm{U}}{\rm{p}}{\rm{t}}{\rm{a}}{\rm{k}}{\rm{e}}}_{a}\times D\) is the theorical maximum uptake in deuterium value that peptide a can take. The ΔRFU compared RFU value between two different experimental conditions, in this case, this is the comparison between CapRelSJ46 and CapRelSJ46 + Gp57. Heat maps have been generated in DynamX. All the raw data can be accessed at: https://doi.org/10.6084/m9.figshare.19745089.
CapRelSJ46 expression and purification for biochemical assays
Full-length capRelSJ46 was overexpressed in freshly transformed E. coli BL21(DE3) pET24d-N-His10-SUMO-capRelSJ46 pMG25-paSpo co-transformed with the plasmid encoding PaSpo small alarmone hydrolase (SAH) from Salmonella phage SSU5, which has been shown to neutralize the toxicity of other toxSAS toxins24. Fresh transformants were used to inoculate 800 ml of LB medium (final A600 of 0.03) supplemented with 50 μg ml−1 kanamycin, 20 μg ml−1 chloramphenicol and 0.2% arabinose. Bacterial cultures were grown at 37 °C until an A600 of 0.4–0.5 and protein expression was induced with 0.1 mM IPTG (final concentration). Cells were grown for additional 1 h at 30 °C and the biomass was collected by centrifugation (10,000 rpm, for 5 min, JLA-10.500 rotor (Beckman Coulter)).
Cell mass was resuspended in buffer A (750 mM KCl, 500 mM NaCl, 5 mM MgCl2, 40 μM MnCl2, 40 μM zinc acetate, 1 mM mellitic acid, 20 mM imidazole, 10% glycerol, 4 mM β-mercaptoethanol and 25 mM HEPES:KOH pH 8) supplemented with 0.1 mM PMSF and 1 U ml−1 of DNase I, and lysed by one passage through a high-pressure cell disrupter (Stansted Fluid Power, 150 MPa). Mellitic acid was added to buffers as it was earlier shown to stabilize Thermus thermophilus Rel stringent factor65. Cell debris was removed by centrifugation (25,000 rpm for 1 h at 4 ºC, JA-25.50 rotor (Beckman Coulter)), the clarified lysate was filtered through a 0.22-μm syringe filter and loaded onto a HisTrap 5 ml HP column (Cytiva) pre-equilibrated in buffer A. The column was washed with 5 column volumes of buffer A, and the protein was eluted using a combination of stepwise and linear gradient (5 column volumes with 0–100% buffer B) of buffer B (750 mM KCl, 500 mM NaCl, 5 mM MgCl2, 40 μM MnCl2, 40 μM Zn(OAc)2, 1 mM mellitic acid, 1 M imidazole, 10% glycerol, 4 mM β-mercaptoethanol, 25 mM HEPES:KOH pH 8). Fractions enriched in CapRelSJ46 (approximately 40% buffer B) were pooled, totalling approximately 5 ml. The sample was loaded on a HiLoad 16/600 Superdex 200 pg column pre-equilibrated with a high-salt buffer (buffer C; 2 M NaCl, 5 mM MgCl2, 10% glycerol, 4 mM β-mercaptoethanol, 25 mM HEPES:KOH pH 8). The fractions containing CapRelSJ46 were pooled and applied on a HiPrep 10/26 desalting column (Cytiva) pre-equilibrated with storage buffer (buffer D; 720 mM KCl, 5 mM MgCl2, 40 mM arginine, 40 mM glutamic acid, 10% glycerol, 4 mM β-mercaptoethanol, 25 mM HEPES:KOH pH 8). Fractions containing CapRelSJ46 were collected (about 14 ml in total) and the His10–SUMO tag was cleaved off by addition of 10 µg of His6–Ulp1 per 1 mg CapRelSJ46 followed by a 30-min incubation on ice. After the His10–SUMO tag was cleaved off, the protein was passed through a 5 ml HisTrap HP pre-equilibrated with buffer D supplemented with 20 mM imidazole. Fractions containing CapRelSJ46 in the flow-through were collected and concentrated on an Amicon Ultra (Millipore) centrifugal filter device with a 10 kDa cut-off. The purity of protein preparations was assessed by SDS–PAGE. Protein preparations were aliquoted, frozen in liquid nitrogen and stored at –80 °C. Individual single-use aliquots were discarded after the experiment.
Cell-free translation
Experiments with PURExpress in vitro protein synthesis kit (NEB, E6800) were performed as per the manufacturer’s instructions. All reactions were supplemented with 0.8 U µl−1 RNase Inhibitor Murine (NEB, M0314S). Purified CapRelSJ46 protein was used at a final concentration of 250 nM, with gp57, gp57(L114P) or gp57(I115F) as template plasmid at 10 ng µl−1. As a mock control CapRelSJ46 was substituted for equal volume of HEPES:Polymix buffer66, pH 7.5. After a 10-min incubation at 37 °C, a 1.34 µl aliquot of the reaction mixture was taken and quenched by addition of 13.66 µl of 2× sample buffer (100 mM Tris:HCl pH 6.8, 4% SDS, 0.02% bromophenol blue, 20% glycerol, 20 mM DTT and 4% β-mercaptoethanol), and DHFR template plasmid was added to the remaining reaction mixture at a final concentration of 20 ng µl−1. After further incubation at 37 °C for 1 h, the reaction mixture was mixed with ninefold volume of 2× sample buffer, boiled at 98 ºC for 5 min, and 5 µl of the mixture was resolved by 18% SDS–PAGE. The SDS–PAGE gel was fixed by incubating for 5 min at room temperature in 50% ethanol solution supplemented with 2% phosphoric acid, washed three times with water for 20 min at room temperature, and stained with ‘blue silver’ solution (0.12% Brilliant Blue G250 (Sigma-Aldrich, 27815), 10% ammonium sulfate, 10% phosphoric acid, and 20% methanol) overnight at room temperature. After washing with water for 3 h at room temperature, the gel was imaged on an Amersham ImageQuant 800 (Cytiva) imaging system. For tRNA pyrophosphorylation experiments, Gp57, Gp57(L114P), or Gp57(I115F) was produced in a similar reaction mixture without CapRelSJ46 and DHFR template at 37 °C for 2 h.
tRNA pyrophosphorylation by CapRelSJ46
The reaction mixture containing 5 µM tRNA from E. coli MRE600 (Sigma-Aldrich, 10109541001), 500 µM [γ32P]ATP, 250 nM CapRelSJ46 and 1/10 volume of either wild-type Gp57, Gp57(L114P), or Gp57(I115F) product from the PUREsystem in HEPES:Polymix buffer, pH 7.5 (5 mM Mg2+ final concentration) supplemented with 1 mM DTT was incubated at 37 °C for 10 min. To visualize phosphorylated tRNA, the reaction sample was mixed in 2 volumes of RNA dye (98% formamide, 10 mM EDTA, 0.3% bromophenol blue and 0.3% xylene cyanol), tRNA was denatured at 37 °C for 10 min and resolved on urea-PAGE in 1× TBE (8 M urea, 8% PAGE). The gel was stained with SYBR Gold (Life technologies, S11494) and exposed to an imaging plate overnight. The imaging plate was imaged by a FLA-3000 (Fujifilm).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Structural data from this study are available from the Protein Data Bank (PDB) under accession 7ZTB. HDX raw data can be accessed at https://doi.org/10.6084/m9.figshare.19745089. Sequencing data are available in the Sequence Read Archive (SRA) under BioProject PRJNA837951. Materials including strains and plasmids are available upon reasonable request. Source data are provided with this paper.
References
Bernheim, A. & Sorek, R. The pan-immune system of bacteria: antiviral defence as a community resource. Nat. Rev. Microbiol. 18, 113–119 (2020).
Hampton, H. G., Watson, B. N. J. & Fineran, P. C. The arms race between bacteria and their phage foes. Nature 577, 327–336 (2020).
Rostøl, J. T. & Marraffini, L. (Ph)ighting phages: how bacteria resist their parasites. Cell Host Microbe 25, 184–194 (2019).
Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of immunity. Cell 180, 1044–1066 (2020).
Daugherty, M. D. & Malik, H. S. Rules of engagement: molecular insights from host–virus arms races. Annu. Rev. Genet. 46, 677–700 (2012).
Fletcher, A. J. et al. Trivalent RING assembly on retroviral capsids activates TRIM5 ubiquitination and innate immune signaling. Cell Host Microbe 24, 761–775.e6 (2018).
Lahaye, X. et al. NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation. Cell 175, 488–501.e22 (2018).
Lin, Y.-T., Chen, Y.-P., Fang, C.-H., Huang, P.-Y. & Liang, S.-M. Capsid proteins of foot-and-mouth disease virus interact with TLR2 and CD14 to induce cytokine production. Immunol. Lett. 223, 10–16 (2020).
Pertel, T. et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365 (2011).
Shepardson, K. M. et al. Induction of antiviral immune response through recognition of the repeating subunit pattern of viral capsids is toll-like receptor 2 Dependent. mBio 8, e01356–17 (2017).
Rousset, F. et al. Phages and their satellites encode hotspots of antiviral systems. Cell Host Microbe 30, 740–753.e5 (2022).
Doron, S. et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120 (2018).
Millman, A. et al. An expanding arsenal of immune systems that protect bacteria from phages. Preprint at bioRxiv https://doi.org/10.1101/2022.05.11.491447 (2022).
Vassallo, C. N., Doering, C. R., Littlehale, M. L., Teodoro, G. I. C. & Laub, M. T. A functional selection reveals previously undetected anti-phage defence systems in the E. coli pangenome. Nat. Microbiol. 7, 1568–1579 (2022).
Gao, L. et al. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077–1084 (2020).
Lopatina, A., Tal, N. & Sorek, R. Abortive infection: bacterial suicide as an antiviral immune strategy. Annu. Rev. Virol. 7, 371–384 (2020).
LeRoux, M. & Laub, M. T. Toxin–antitoxin systems as phage defense elements. Annu. Rev. Microbiol. 76, 21–43 (2022).
Song, S. & Wood, T. K. A primary physiological role of toxin/antitoxin systems is phage inhibition. Front. Microbiol. 11, 1895 (2020).
Guegler, C. K. & Laub, M. T. Shutoff of host transcription triggers a toxin–antitoxin system to cleave phage RNA and abort infection. Mol. Cell 81, 2361–2373.e9 (2021).
Fineran, P. C. et al. The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc. Natl Acad. Sci. USA 106, 894–899 (2009).
Short, F. L., Akusobi, C., Broadhurst, W. R. & Salmond, G. P. C. The bacterial type III toxin–antitoxin system, ToxIN, is a dynamic protein–RNA complex with stability-dependent antiviral abortive infection activity. Sci. Rep. 8, 1013 (2018).
LeRoux, M., Culviner, P. H., Liu, Y. J., Littlehale, M. L. & Laub, M. T. Stress can induce transcription of toxin–antitoxin systems without activating toxin. Mol. Cell 79, 280–292.e8 (2020).
Bobonis, J. et al. Bacterial retrons encode phage-defending tripartite toxin–antitoxin systems. Nature 609, 144–150 (2022).
Jimmy, S. et al. A widespread toxin–antitoxin system exploiting growth control via alarmone signaling. Proc. Natl Acad. Sci. USA 117, 10500–10510 (2020).
Anderson, B. W., Fung, D. K. & Wang, J. D. Regulatory themes and variations by the stress-signaling nucleotide alarmones (p)ppGpp in bacteria. Annu. Rev. Genet. 55, 115–133 (2021).
Bange, G., Brodersen, D. E., Liuzzi, A. & Steinchen, W. Two P or not two P: understanding regulation by the bacterial second messengers (p)ppGpp. Annu. Rev. Microbiol. 75, 383–406 (2021).
Ahmad, S. et al. An interbacterial toxin inhibits target cell growth by synthesizing (p)ppApp. Nature 575, 674–678 (2019).
Kurata, T. et al. RelA–SpoT homolog toxins pyrophosphorylate the CCA end of tRNA to inhibit protein synthesis. Mol. Cell 81, 3160–3170 (2021).
Kurata, T. et al. A hyperpromiscuous antitoxin protein domain for the neutralization of diverse toxin domains. Proc. Natl Acad. Sci. USA 119, e2102212119 (2022).
Dedrick, R. M. et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2, 16251 (2017).
Owen, S. V. et al. Prophages encode phage-defense systems with cognate self-immunity. Cell Host Microbe 29, 1620–1633.e8 (2021).
Steinchen, W. et al. Structural and mechanistic divergence of the small (p)ppGpp synthetases RelP and RelQ. Sci. Rep. 8, 2195 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Van Melderen, L., Bernard, P. & Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 11, 1151–1157 (1994).
Koga, M., Otsuka, Y., Lemire, S. & Yonesaki, T. Escherichia coli rnlA and rnlB compose a novel toxin–antitoxin system. Genetics 187, 123–130 (2011).
Duda, R. L. & Teschke, C. M. The amazing HK97 fold: versatile results of modest differences. Curr. Opin. Virol. 36, 9–16 (2019).
Maffei, E. et al. Systematic exploration of Escherichia coli phage–host interactions with the BASEL phage collection. PLoS Biol. 19, e3001424 (2021).
Kao, C., Gumbs, E. & Snyder, L. Cloning and characterization of the Escherichia coli lit gene, which blocks bacteriophage T4 late gene expression. J. Bacteriol. 169, 1232–1238 (1987).
Bergsland, K. J., Kao, C., Yu, Y. T., Gulati, R. & Snyder, L. A site in the T4 bacteriophage major head protein gene that can promote the inhibition of all translation in Escherichia coli. J. Mol. Biol. 213, 477–494 (1990).
Molineux, I. J., Schmitt, C. K. & Condreay, J. P. Mutants of bacteriophage T7 that escape F restriction. J. Mol. Biol. 207, 563–574 (1989).
Tal, N. et al. Cyclic CMP and cyclic UMP mediate bacterial immunity against phages. Cell 184, 5728–5739.e16 (2021).
Huiting, E. et al. Bacteriophages antagonize cGAS-like immunity in bacteria. Preprint at bioRxiv https://doi.org/10.1101/2022.03.30.486325 (2022).
Gao, L. A. et al. Prokaryotic innate immunity through pattern recognition of conserved viral proteins. Science 377, eabm4096 (2022).
Stokar-Avihail, A. et al. Discovery of phage determinants that confer sensitivity to bacterial immune systems. Preprint at bioRxiv https://doi.org/10.1101/2022.08.27.505566 (2022).
Haldimann, A. & Wanner, B. L. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J. Bacteriol. 183, 6384–6393 (2001).
Duong, M. M., Carmody, C. M., Ma, Q., Peters, J. E. & Nugen, S. R. Optimization of T4 phage engineering via CRISPR/Cas9. Sci. Rep. 10, 18229 (2020).
Cadwell, R. C. & Joyce, G. F. Randomization of genes by PCR mutagenesis. Genome Res. 2, 28–33 (1992).
Srikant, S., Guegler, C. K. & Laub, M. T. The evolution of a counter-defense mechanism in a virus constrains its host range. eLife 11, e79549 (2022).
Saha, C. K., Sanches Pires, R., Brolin, H., Delannoy, M. & Atkinson, G. C. FlaGs and webFlaGs: discovering novel biology through the analysis of gene neighbourhood conservation. Bioinformatics 37, 1312–1314 (2021).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21 (2016).
Ashkenazy, H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Hauser, M., Steinegger, M. & Söding, J. MMseqs software suite for fast and deep clustering and searching of large protein sequence sets. Bioinformatics 32, 1323–1330 (2016).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410 (1990).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–W279 (2022).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 (1994).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Tamman, H. et al. A nucleotide-switch mechanism mediates opposing catalytic activities of Rel enzymes. Nat. Chem. Biol. 16, 834–840 (2020).
Terwilliger, T. C. et al. phenix.mr_rosetta: molecular replacement and model rebuilding with Phenix and Rosetta. J. Struct. Funct. Genomics 13, 81–90 (2012).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68, 352–367 (2012).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Smart, O. S. et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D 68, 368–380 (2012).
Van Nerom, K., Tamman, H., Takada, H., Hauryliuk, V. & Garcia-Pino, A. The Rel stringent factor from Thermus thermophilus: crystallization and X-ray analysis. Acta Crystallogr. F 75, 561–569 (2019).
Takada, H. et al. The C-terminal RRM/ACT domain is crucial for fine-tuning the activation of ‘long’ RelA-SpoT homolog enzymes by ribosomal complexes. Front. Microbiol. 11, 277 (2020).
Acknowledgements
We thank A. Harms for sharing the BASEL phage collection; the MIT BioMicro Center and its staff for their support in sequencing; the MIT Biopolymers and Proteomics Core and its staff for their help in mass spectrometry experiments; W. Verseés for allowing the use of the biophysics facilities at the VUB; the Mass Spectrometry Laboratory at ULiège and Thomas Tilmant for support and assistance regarding MS data acquisition; K. Gozzi and B. Wang for comments on the manuscript; and all members of the Laub laboratory for helpful discussions. G.C.A. and V.H. were supported by the Swedish Research council (grant 2018-00956 within the RIBOTARGET consortium under the framework of JPIAMR, project grants 2017-03783 and 2021-01146 to V.H., project grant 2019-01085 to G.C.A.), the Knut and Alice Wallenberg Foundation (2020.0037 to G.C.A.), the Ragnar Söderberg Foundation (M23/14 to V.H.), the European Regional Development Fund through the Centre of Excellence for Molecular Cell Technology (V.H.), and the Estonian Science Foundation (project grant PRG335 to V.H.). A.G.-P. was supported by Fonds National de Recherche Scientifique (FRFS-WELBIO CR-2017S-03, FNRS CDR J.0068.19, FNRS-EQP UN.025.19 and FNRS-PDR T.0090.22), the European Research Council (CoG DiStRes, no. 864311), the Joint Programming Initiative on Antimicrobial Resistance (JPI-EC-AMR-R.8004.18), the Programme Actions de Recherche Concerté 2016-2021, Fonds Jean Brachet and the Fondation Van Buuren, Chargé de Recherches fellowship from the FNRS no. CR/DM-392 (H.T.). A.C. and K.C.W. are fellows of the FRIA, C.M. is supported as a Research Associate of the FRS–FNRS. C.M. was supported by grant F.4532.22 from the FRS–FNRS. The authors acknowledge the use of the PROXIMA 1 and 2A beamlines at the Soleil synchrotron (Gif-sur-Yvette, France). M.T.L. is an Investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
Experiments were conceived and designed by T.Z., T.K., G.C.A., V.H., A.G.-P. and M.T.L. Phage and bacterial experiments, as well as incorporation and co-immunoprecipitation assays, were done by T.Z. with assistance from M.L. and S.S. Metabolic labelling experiments were done by T.Z. and T.B. Cell-free translation and tRNA pyrophosphorylation assays were done by T.Z. and T.K. CapRel and Gp57 purification was done by T.Z., T.K., H.T., A.C. and A.T. ITC was performed by H.T. and A.C. MALS was done by A.C. and A.T. HDX-MS data acquisition and analysis was performed by C.M. and K.C.W. X-ray data collection and analyses was performed by H.T., A.T., A.C. and A.G.-P. Bioinformatic analyses were performed by T.Z. and G.C.A. Structural modelling was done by T.Z., A.T. and A.G.-P. Figure design, manuscript writing, and editing was done by T.Z., T.K., G.C.A., V.H., A.G.-P. and M.T.L. Project supervision and funding was provided by G.C.A., V.H., A.G.-P. and M.T.L.
Corresponding authors
Ethics declarations
Competing interests
A.G.-P. is co-founder and stockholder of Santero Therapeutics. the other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Philip Kranzusch and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 CapRel is broadly distributed in different bacteria, and is usually a fused TA system.
FlaGs output for CapRel and PhRel representatives (black arrows). Those proteins studied in this paper are in bold and underlined. Most CapRel systems are fused, with the exception of Actinobacteria where the TA pair is usually unfused. The unfused state is the most common form of the system in the closely related PhRel subfamily of toxSASs, and fusion and/or fission events appear to have occurred independently multiple times. Conserved flanking open reading frames are colored and numbered by homologous clusters: 1 (red): antitoxin homologous to the CapRel C-terminus, and 2 (purple): Panacea/SocA (DUF4065) domain-containing proteins. For other coloured open reading frames see the legend on the lower left. Arrows with no fill colour and with blue and green outlines are, respectively, pseudogenes and non-coding RNA genes. Red numbers on branches show Maximum Likelihood bootstrap support on a scale of 0-1, where 1 is 100% support.
Extended Data Fig. 2 Analysis of CapRel homologs.
(a) Sequence alignment comparing fused CapRel systems with related, unfused systems. Alignment of toxSAS PhRel and ATphRel from the Mycobacterium phage Phrann, non-fused CapRel and ATcapRel from Mycobacterium terramassilience, and the three fused systems CapRelSJ46, CapRelEbc and CapRelKp. The N-terminal region of fused CapRel systems is a toxSAS toxin domain, while the C-terminal region is homologous to the antitoxins of the PhRel and unfused CapRel TA systems. Substituted sites that rendered CapRelSJ46 toxic (see Extended Data Fig. 3j) are indicated with black arrowheads. The inset diagram summarises the homologous regions of the bicistronic toxin-antitoxin and fused toxin-antitoxin systems considered here. (b) Genome maps of native locations of CapRelSJ46, CapRelEbc and CapRelKp (+/− 10 kb) with predicted flanking prophage and phage genes. (c) Summary of 3 independent replicates of cell viability assay in Fig. 1b. Asterisks indicate p = 0.007 (unpaired two-tailed t-test). (d) Serial dilutions of the phages indicated spotted on lawns of cells producing CapRelSJ46, CapRelEbc, or CapRelKp or harboring an empty vector (EV). (e) Summary of 3 independent replicates of phage spotting assay in Fig. 1d. Asterisks indicate p = 10−10 (T2, SECΦ27), 10−6 (T4) (unpaired two-tailed t-test). (f) Serial dilutions of the phages indicated spotted on lawns of cells containing genomic CapRelSJ46, CapRelSJ46 (Y155A) or His6-CapRelSJ46. (g) Summary of 3 independent replicates of phage spotting assay in Fig. 1g. Asterisk indicates p = 10−22 (unpaired two-tailed t-test).
Extended Data Fig. 3 Structural analysis of CapRelSJ46.
(a) Alignment of CapRelSJ46 and diverse fused CapRel homologs, with labels indicating the pseudo-ZFD and location of substitutions that render CapRelSJ46 constitutively active or unable to be activated by Gp57, the SECΦ27 major capsid protein. (b) Summary of 3 independent replicates of phage spotting assay in Fig. 2b. (c) Immunoblot of untagged CapRel chimera or FLAG-tagged CapRelSJ46, CapRelEbc or CapRel chimera. GyrA is included as a loading control. Image shown is a representative of 2 biological replicates. (d) Topology of CapRelSJ46. The toxSYNTH domain is colored in light yellow, the pseudo-ZFD in dark gold and the regions that anchor pseudo-ZFD to toxSYNTH are in green. The adenine coordinating R79 and R116 are shown as red dots and the G-loop is colored in red. (e) Superposition of the toxSYNTH domain of CapRelSJ46 (colored in light yellow) onto RelQ (PDB ID: 5DEC, colored in light orange) from Bacillus subtilis. (f) Superposition of the pseudo-ZFD of CapRelSJ46 (colored in dark gold) onto the ZFD transcription factor of Acidianus hospitalis (2LVH, colored in purple). (g) Analysis of asymmetric unit and symmetry-related partners of CapRelSJ46 crystal packing. Black arrow indicates steric clash that would arise if CapRelSJ46 were in a closed conformation. (h) Superposition of the crystal structure of CapRelSJ46 (colored in light yellow) onto the structure of the open state predicted by AlphaFold (colored in green). (i) Structures of the open (left; from crystal structure) or closed (right; AlphaFold prediction) conformations of CapRelSJ46 color coded by the conservation score of each amino acid calculated by ConSurf. Substitutions that render CapRelSJ46 constitutively active or unable to be activated by Gp57 are labeled as spheres. (j) Serial dilutions of cells expressing the indicated variant of CapRelSJ46 from an arabinose-inducible promoter on media containing glucose (left) or arabinose (right). (k) Summary of 3 independent replicates of cell viability assay in Extended Data Fig. 3j under arabinose induction. Asterisks indicate p = 0.007 (unpaired two-tailed t-test).
Extended Data Fig. 4 Gp57 from SECΦ27 triggers CapRelSJ46 to inhibit translation, not transcription.
(a) Serial dilutions of phage SECΦ27 spotted on lawns of cells producing CapRelSJ46, His6-CapRelSJ46, or CapRelSJ46-FLAG, or harboring an empty vector (EV). (b) Loading control for Fig. 3a shown as total protein levels stained by Coomassie stain. Image shown is a representative of 2 biological replicates. (c) Immunoblot of His6-CapRelSJ46 expressed from the bacterial genome or a low-copy number plasmid following infection with SECΦ27 (MOI = 100) compared to an uninfected control. Total protein levels stained by Coomassie stain is included as a loading control. Images shown are representatives of 2 biological replicates. (d) Summary of 3 independent replicates of phage spotting assay in Fig. 3c. Asterisk indicates p = 10−10 (unpaired two-tailed t-test). (e) Serial dilutions on media containing glucose (left) or arabinose (right) of cells expressing CapRelSJ46 from its native promoter in the bacterial genome and expressing Gp57 from an arabinose-inducible promoter or an empty vector. (f) Summary of 3 independent replicates of cell viability assay in Fig. 3g under arabinose induction. Asterisk indicates p = 10−30 (unpaired two-tailed t-test). (g) Serial dilutions on media containing glucose (left) or arabinose (right) of cells expressing CapRelSJ46(Y155A) from its native promoter or an empty vector and expressing the indicated variant of Gp57 from an arabinose-inducible promoter. (h) Cells harboring CapRelSJ46 and producing the wild-type or L114P variant of Gp57 (expressed from an arabinose-inducible promoter) or harboring an empty vector were pulse-labeled with 3H-uridine at the times indicated post-addition of arabinose. (i) Cells producing the CapRelSJ46 N-terminal toxin domain (expressed from an arabinose-inducible promoter) or harboring an empty vector were pulse-labeled with 35S-Cys/Met (left) or 3H-uridine (right) at the times indicated post-addition of arabinose. (j) Same as (h) but for cells carrying CapRelSJ46 or an empty vector and at times post-infection with SECΦ27 at MOI = 100. (k) Adsorption of T4 and SECΦ27 with cells containing CapRelSJ46 or an empty vector. Data reported are the mean +/− SD from 3 biological replicates. (l) Immunoblot of Gp57-HA expressed from its native promoter in the bacterial genome following infection with SECΦ27. Total protein level stained by Coomassie stain is included as a loading control (left). Quantification of the relative intensities of both Gp57-HA bands normalized to the loading control from 3 independent replicates (right). Asterisks indicate p = 0.004 (30 min), 0.0003 (40 min), 0.0002 (50 min) (unpaired two-tailed t-test).
Extended Data Fig. 5 Characterization of the CapRelSJ46-Gp57 interaction.
(a) Immunoprecipitation of CapRelSJ46-FLAG from cells infected with wild-type SECΦ27 or mutant phage that produces Gp57(L114P), followed by mass spectrometry. Spectrum counts (SC) of Gp57 that had co-precipitated with CapRelSJ46 were normalized to the spectrum counts of CapRelSJ46. Data reported are 2 biological replicates. (b) Same as in (a) but showing spectrum counts of CapRelSJ46 and Gp57 in two independent replicates. (c) Serial dilutions on media containing glucose (left) or arabinose (right) of cells producing CapRelSJ46 or CapRelSJ46-FLAG, each expressed from its native promoter, and the indicated variant of untagged or HA-tagged version of Gp57, expressed from an arabinose-inducible promoter. (d) SEC-MALS analysis of CapRelSJ46-Gp57 complex, revealing a molecular weight of 74 kDa. The monomers of CapRelSJ46 and Gp57 are predicted to be 42 and 36 kDa, respectively. (e) Binding of CapRelSJ46 to Gp57 (L114P I115F) (left) or CapRelSJ46(L280P) to Gp57 (right) monitored by isothermal titration calorimetry (ITC). (f) Topology and cartoon representation of SECΦ27 Gp57. The P-domain is colored in pink and the A-domain in violet. (g) Heat maps representing the HDX of CapRelSJ46 (top) and CapRelSJ46-Gp57 complex (center) and the ΔHDX (bottom). Regions involved in strong uptake such as residues 115-145 and 225-235 (which includes the active site β-strand β2 and the G-loop) are shaded in red and regions involved in strong protection 240-268 and 288-366 (which include both anchors and the pseudo-ZFD) are shaded in blue. (h) Heat map representing the HDX of Gp57 in the complex with CapRelSJ46. Shaded regions highlight areas of variable HDX signal that indicate these regions are involved in the CapRelSJ46-Gp57 interface. (i) Summary of 3 independent replicates of cell viability assay in Fig. 4f under arabinose induction. (j) Summary of 3 independent replicates of phage spotting assay in Fig. 4g. (k) Serial dilutions of T2 and T4 phage spotted on cells producing the indicated mutant of CapRelSJ46 or harboring an empty vector.
Extended Data Fig. 6 The major capsid proteins from multiple, related phages activate CapRelSJ46.
(a) Multiple sequence alignment of the major capsid proteins from phages SECΦ27, Bas4, Bas5 and Bas8. (b) Summary of 3 independent replicates of cell viability assay in Fig. 5a under arabinose induction. Asterisks indicate p = 10−34 (unpaired two-tailed t-test). (c) Summary of 3 independent replicates of phage spotting assay in Fig. 5b. (d) Summary of 3 independent replicates of phage spotting assay in Fig. 5c. (e) Serial dilutions on media containing glucose (left) or arabinose (right) of cells expressing CapRelSJ46 from its native promoter or harboring an empty vector and producing the indicated variant of the Bas8 major capsid protein (Gp8Bas8) from an arabinose-inducible promoter. (f) Summary of 3 independent replicates of cell viability assay in Extended Data Fig. 6e under arabinose induction. (g) Serial dilutions on media containing glucose (left) or arabinose (right) of cells containing an empty vector and producing the indicated variant of the Bas8 major capsid protein from an arabinose-inducible promoter. (h) Serial dilutions of the phages indicated spotted on lawns of cells harboring CapRelEcHT or an empty vector. (i) Summary of 3 independent replicates of cell viability assay in Fig. 5e under arabinose induction. (j) Summary of 3 independent replicates of phage spotting assay in Fig. 5f. Asterisks indicate p = 10−6 (unpaired two-tailed t-test).
Supplementary information
Supplementary Information
This file contains Supplementary Fig. 1 and Tables 1–4. Supplementary Fig. 1: uncropped blots from main figures and extended data figures. a, Immunoblot of His6–CapRelSJ46 for Fig. 3a. b, Coomassie stain shown in Extended Data Fig. 4b as a loading control for Fig. 3a. c, Blue silver stain of in vitro transcription–translation assays for Fig. 3j. d, Autoradiography and SYBR Gold stain for Fig. 3k. e, Immunoblots of lysates and IP samples for Fig. 4a. f, Immunoblots of lysates and IP samples for Fig. 4h. g, Immunoblot for Extended Data Fig. 3c. h, Immunoblot of His6–CapRelSJ46 for Extended Data Fig. 4c and the corresponding loading control. i, Immunoblots of Gp57–HA for Extended Data Fig. 4l and the corresponding loading controls. 3 independent replicates are shown. Supplementary Table 1: list of all mutations present in the phage genomes of 10 evolved clones from 5 independent populations or 2 clones from the control population. Supplementary Table 2: list of bacterial and phage strains used in this study. Supplementary Table 3: list of plasmids used in this study. Supplementary Table 4: list of primers and synthesized gene sequences used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, T., Tamman, H., Coppieters ’t Wallant, K. et al. Direct activation of a bacterial innate immune system by a viral capsid protein. Nature 612, 132–140 (2022). https://doi.org/10.1038/s41586-022-05444-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05444-z
This article is cited by
-
Bacteriophages avoid autoimmunity from cognate immune systems as an intrinsic part of their life cycles
Nature Microbiology (2024)
-
Conservation and similarity of bacterial and eukaryotic innate immunity
Nature Reviews Microbiology (2024)
-
Anti-phage defence through inhibition of virion assembly
Nature Communications (2024)
-
The highly diverse antiphage defence systems of bacteria
Nature Reviews Microbiology (2023)
-
Comparative analysis of effectiveness for phage cocktail development against multiple Salmonella serovars and its biofilm control activity
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
