
Identification of Cytoprotective Small-Molecule Inducers of Heme-Oxygenase-1
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
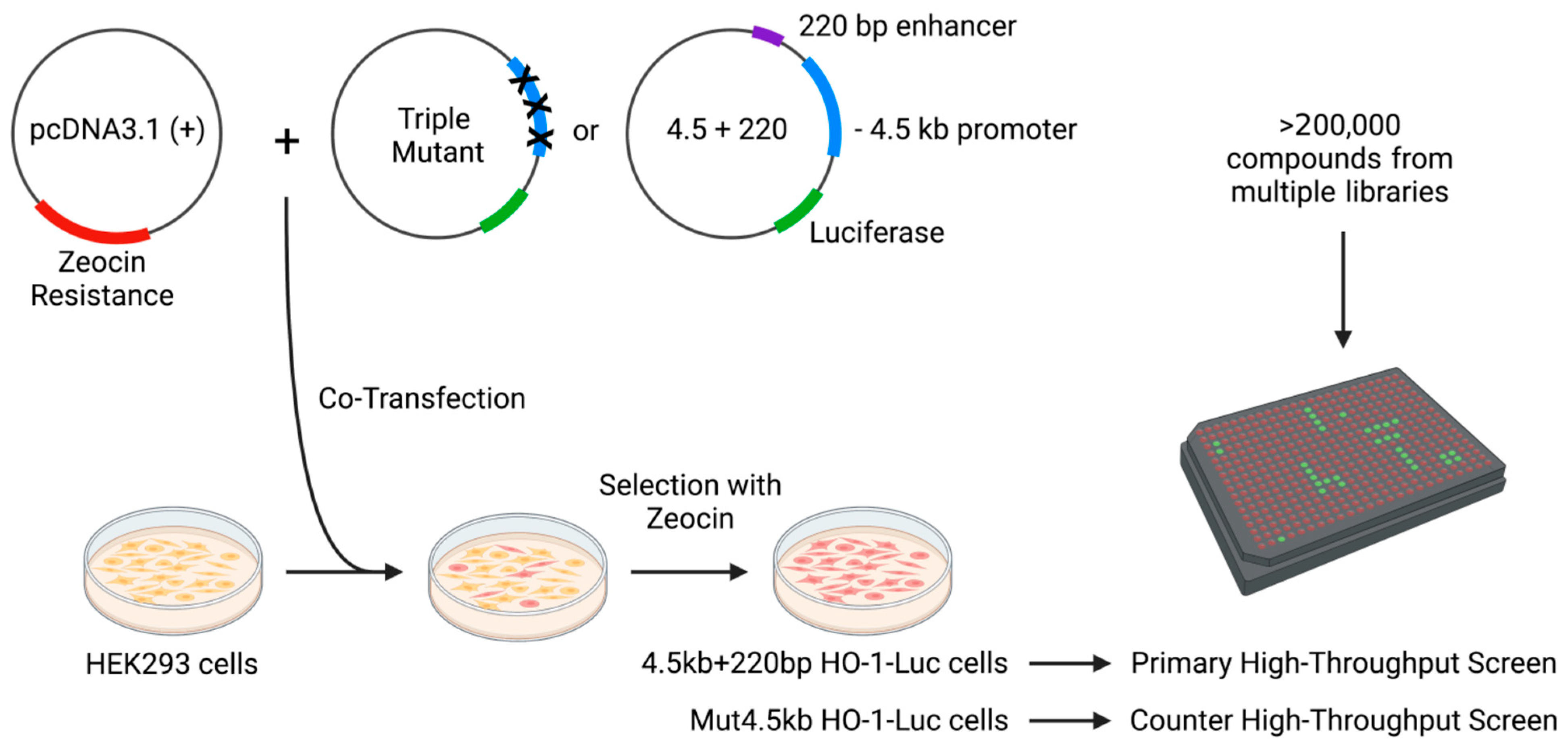
2.1. Stable Cell Lines
2.2. High-Throughput Luciferase Assay
2.3. Western Blotting
2.4. In-Cell Western Assay
2.5. RNA Isolation
2.6. Reverse Transcription and Real-Time PCR
2.7. Bulk RNA Sequencing and Analysis
2.8. siRNA Knockdown
2.9. Cisplatin-Induced Apoptosis Assays
2.10. MTT Assay
2.11. In Vivo Experiments
2.12. Immunohistochemistry
2.13. Statistical Analysis
3. Results
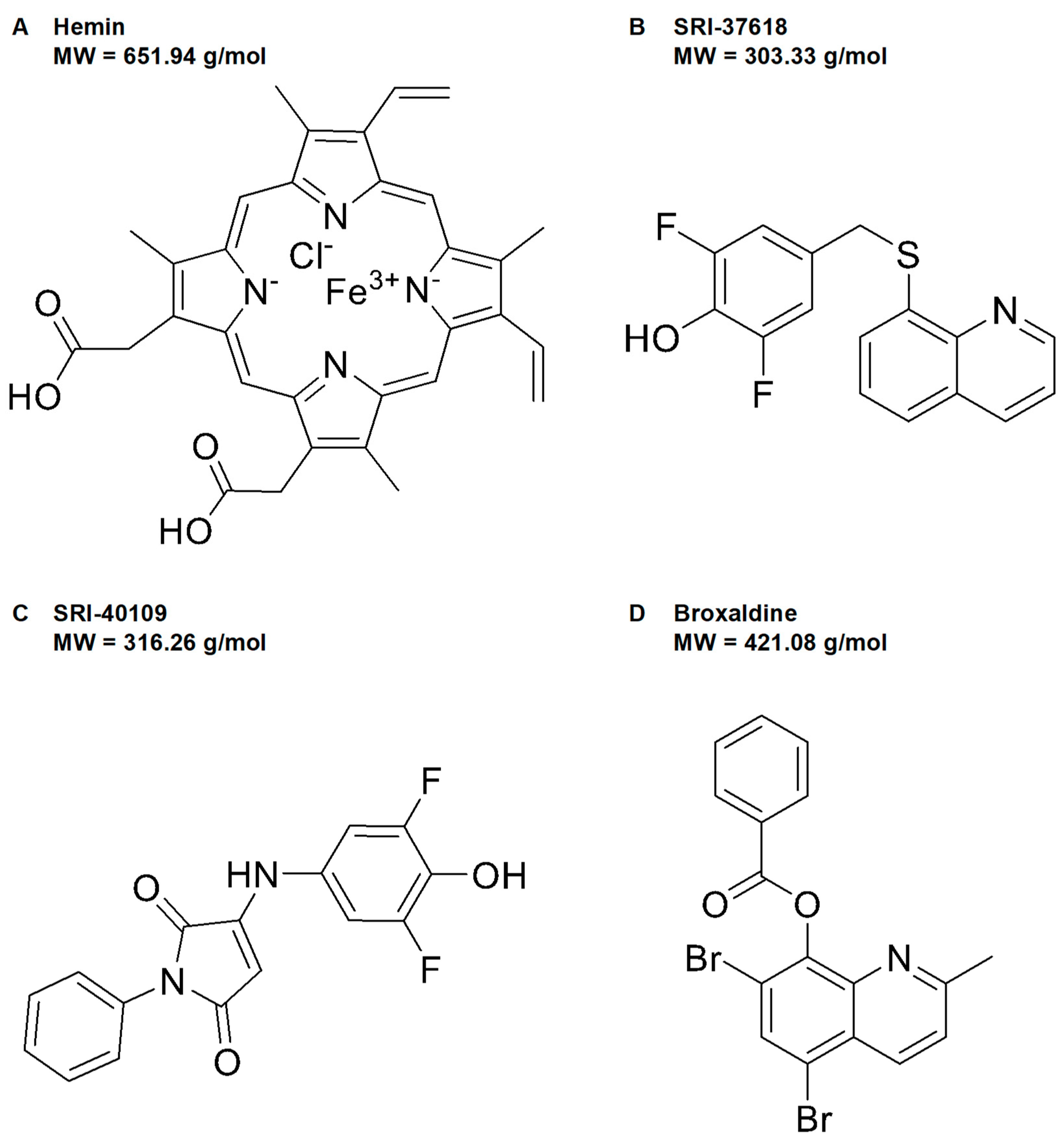
3.1. High-Throughput Screen and Lead Generation
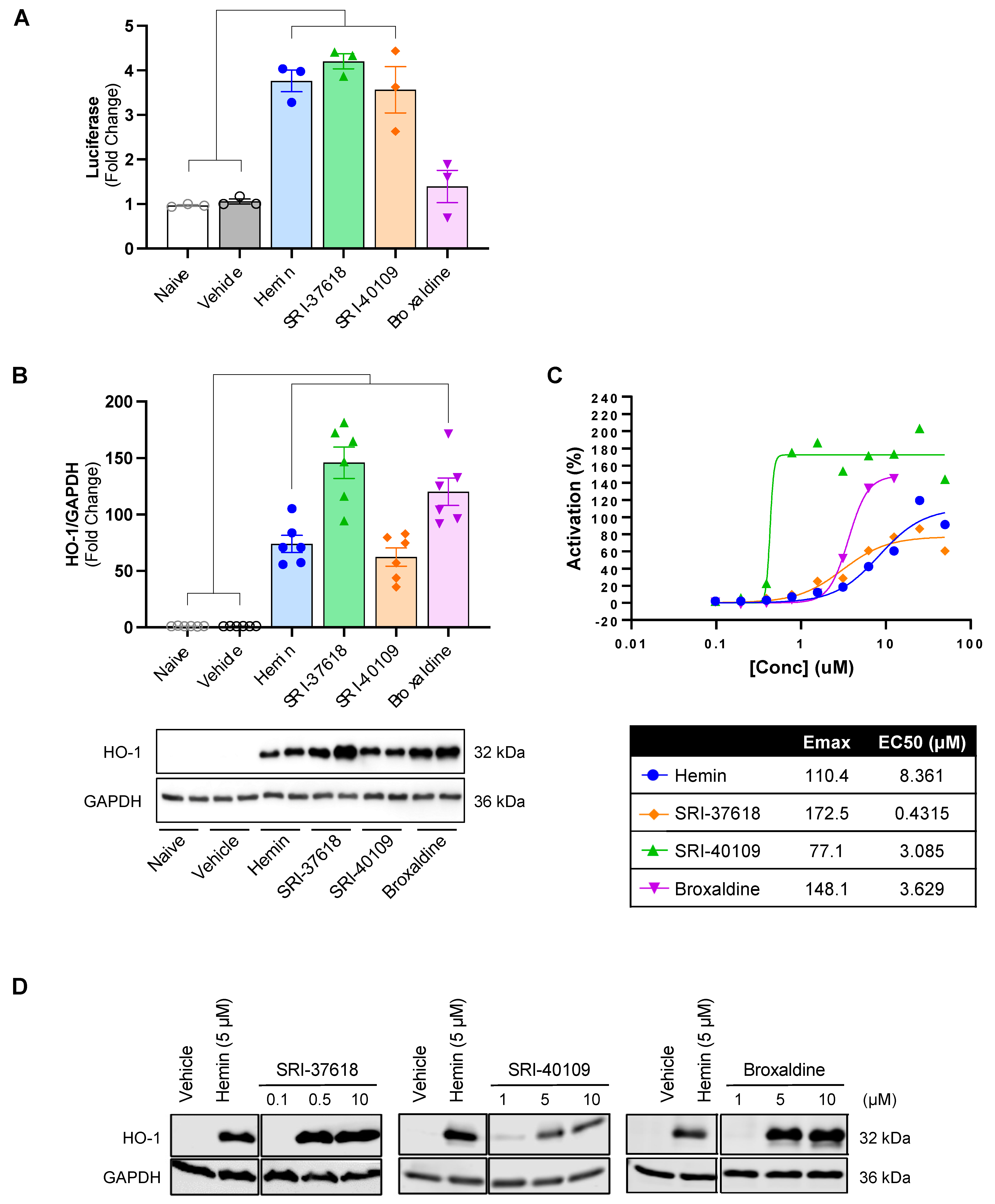
3.2. Induction of Endogenous HO-1 by SRI-37618 and SRI-40109 Does Not Depend on Oxidant Stress
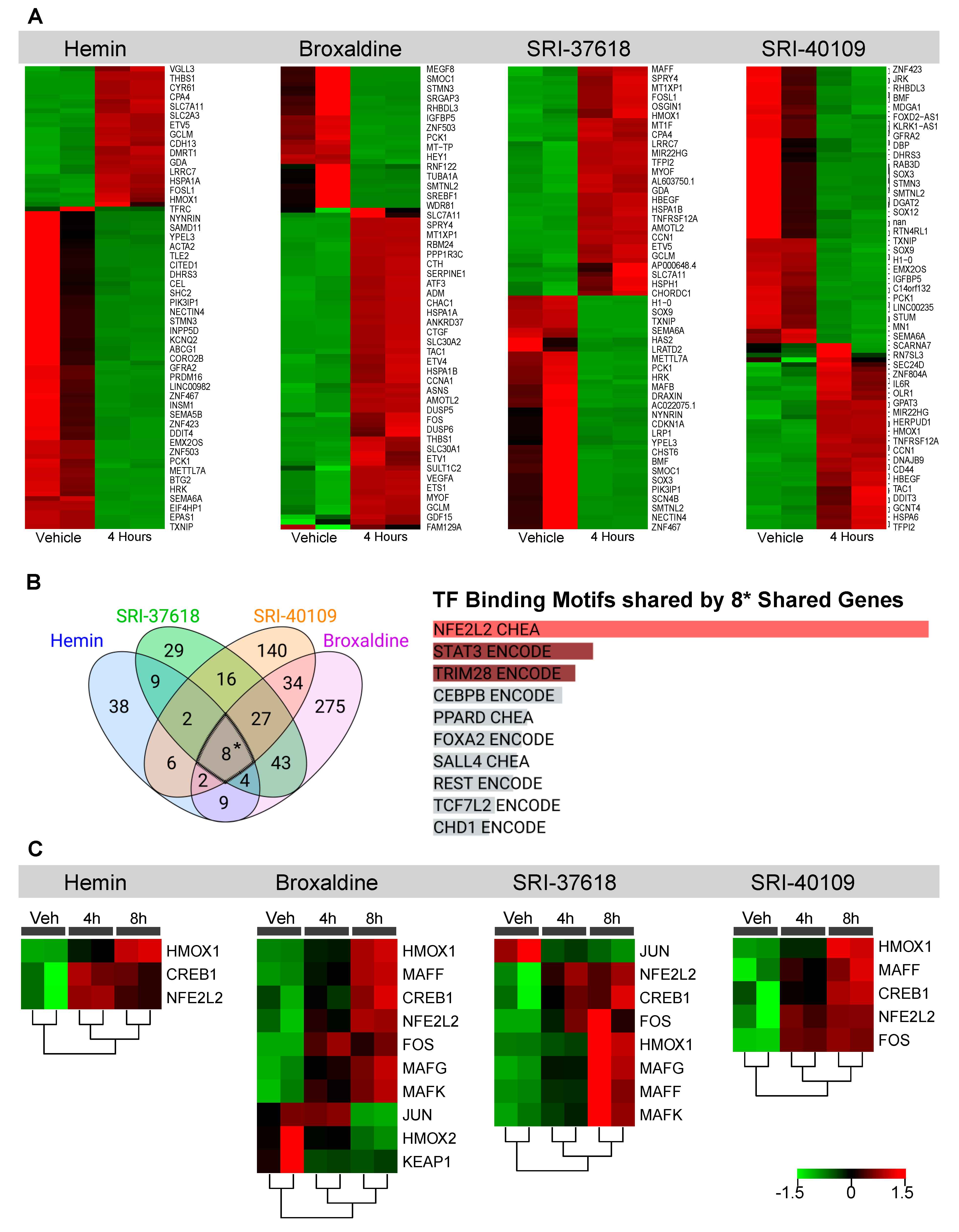
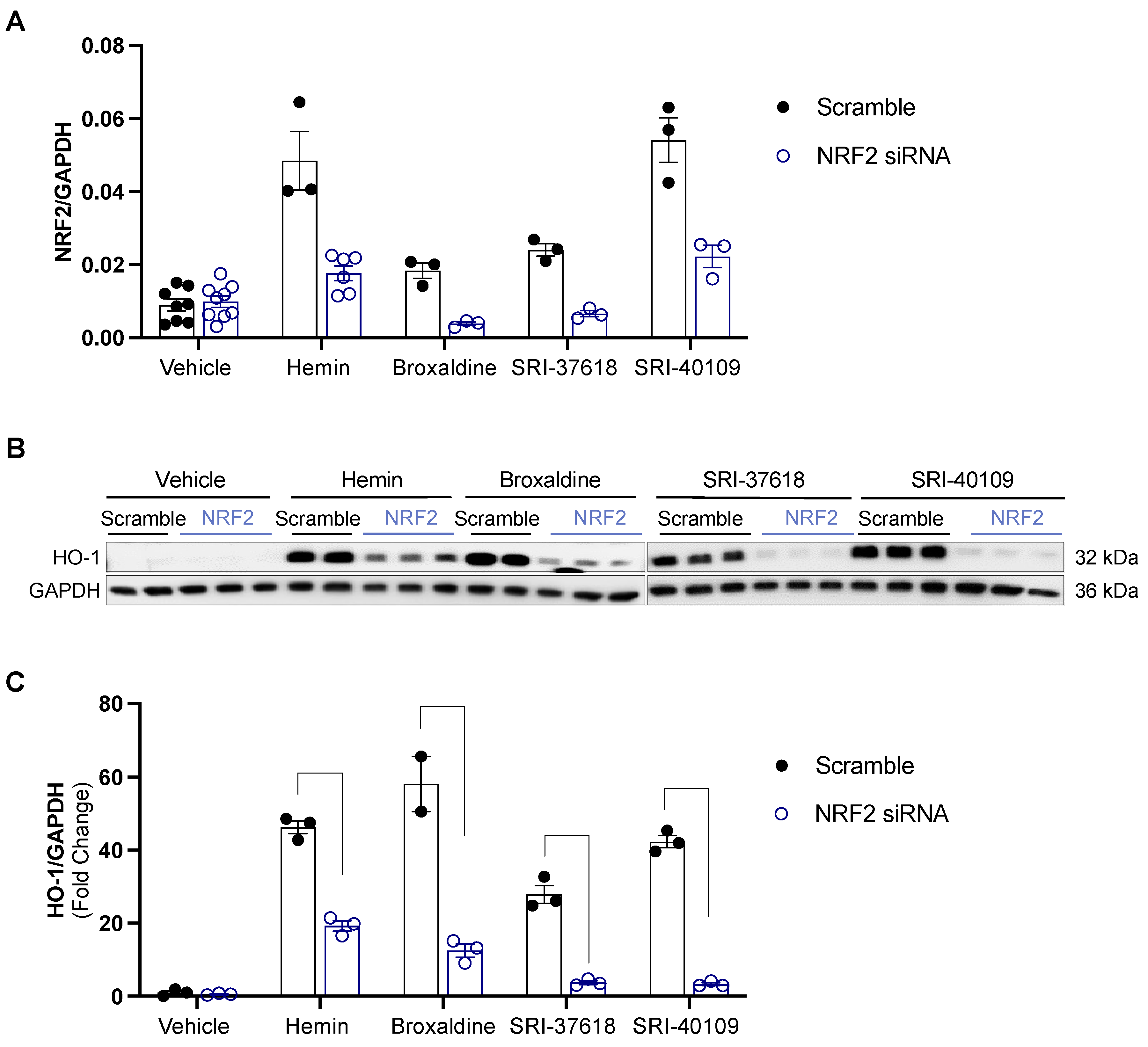
3.3. Genes Upregulated by Hemin, SRI-37618, SRI-40109, and Broxaldine Share Common NRF2-Binding Motifs
3.4. SRI-37618, SRI-40109, and Broxaldine Pretreatment Confer Protection against Cisplatin-Induced Apoptosis In Vitro
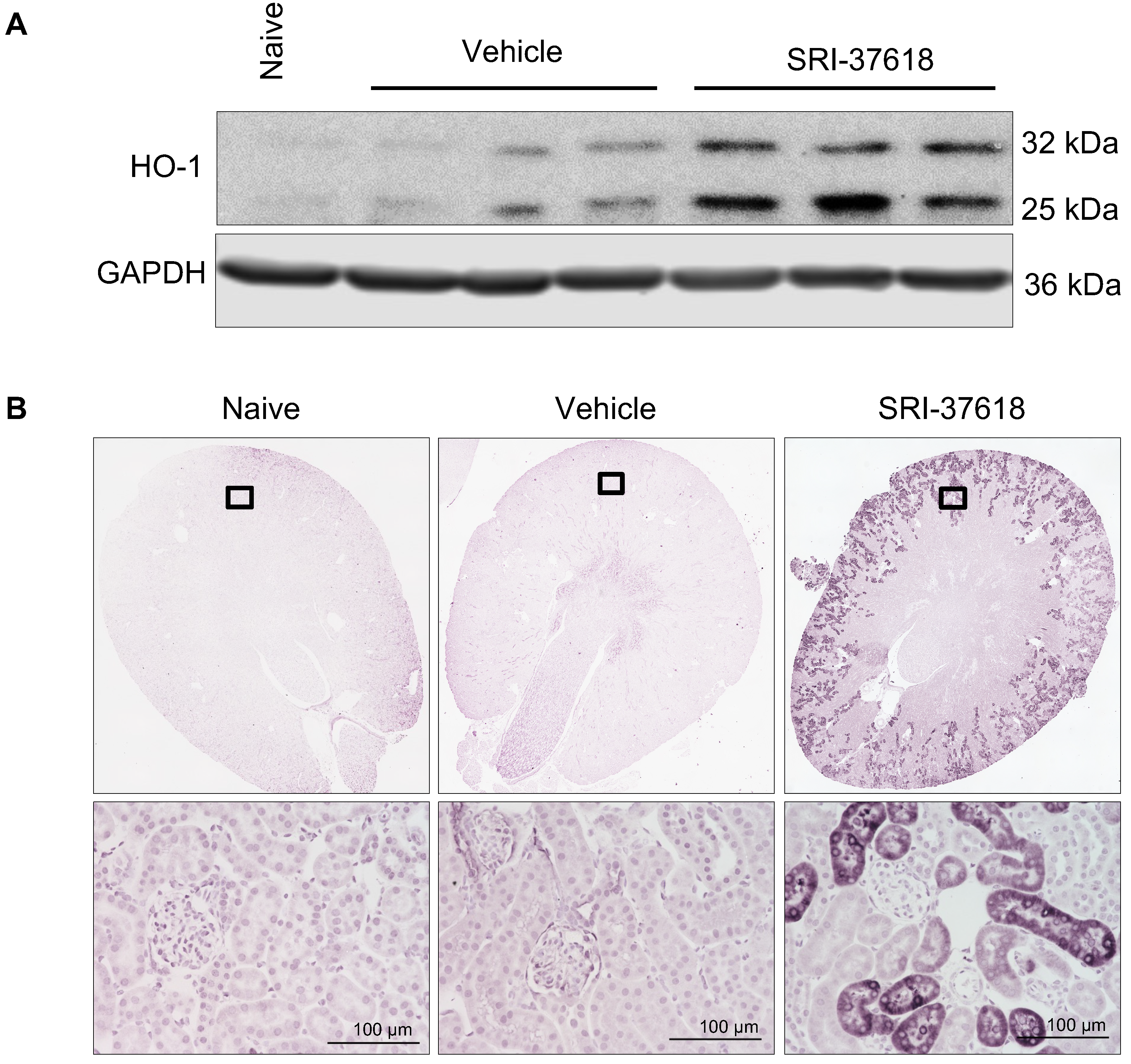
3.5. In Vivo Induction of Endogenous HO-1 in the Kidney
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agarwal, A.; Nick, H.S. Renal response to tissue injury: Lessons from heme oxygenase-1 GeneAblation and expression. J. Am. Soc. Nephrol. 2000, 11, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A. Heme oxygenase-1: A provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006, 70, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Traylor, A.M.; Kim, J.; Joseph, R.; Ricart, K.; Landar, A.; Agarwal, A. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 1702–1712. [Google Scholar] [CrossRef]
- Hull, T.D.; Kamal, A.I.; Boddu, R.; Bolisetty, S.; Guo, L.; Tisher, C.C.; Rangarajan, S.; Chen, B.; Curtis, L.M.; George, J.F.; et al. Heme Oxygenase-1 Regulates Myeloid Cell Trafficking in AKI. J. Am. Soc. Nephrol. 2015, 26, 2139–2151. [Google Scholar] [CrossRef]
- Stec, D.E.; Hinds, T.D., Jr. Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef]
- Morse, D.; Lin, L.; Choi, A.M.; Ryter, S.W. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic. Biol. Med. 2009, 47, 1–12. [Google Scholar] [CrossRef]
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef]
- Nath, K.A.; Vercellotti, G.M.; Grande, J.P.; Miyoshi, H.; Paya, C.V.; Manivel, J.C.; Haggard, J.J.; Croatt, A.J.; Payne, W.D.; Alam, J. Heme protein-induced chronic renal inflammation: Suppressive effect of induced heme oxygenase-1. Kidney Int. 2001, 59, 106–117. [Google Scholar] [CrossRef]
- Nath, K.A.; Croatt, A.J.; Haggard, J.J.; Grande, J.P. Renal response to repetitive exposure to heme proteins: Chronic injury induced by an acute insult. Kidney Int. 2000, 57, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. Acute Kidney Injury Advisory Group of the American Society of Nephrology: World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Deshane, J.; Kim, J.; Bolisetty, S.; Hock, T.D.; Hill-Kapturczak, N.; Agarwal, A. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J. Biol. Chem. 2010, 285, 16476–16486. [Google Scholar] [CrossRef] [PubMed]
- Hill-Kapturczak, N.; Sikorski, E.; Voakes, C.; Garcia, J.; Nick, H.S.; Agarwal, A. An internal enhancer regulates heme- and cadmium-mediated induction of human heme oxygenase-1. Am. J. Physiol. Ren. Physiol. 2003, 285, F515–F523. [Google Scholar] [CrossRef]
- Hock, T.D.; Liby, K.; Wright, M.M.; McConnell, S.; Schorpp-Kistner, M.; Ryan, T.M.; Agarwal, A. JunB and JunD regulate human heme oxygenase-1 gene expression in renal epithelial cells. J. Biol. Chem. 2007, 282, 6875–6886. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Traylor, A.; Joseph, R.; Zarjou, A.; Agarwal, A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2016, 310, F385–F394. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Lever, J.M.; Hull, T.D.; Boddu, R.; Pepin, M.E.; Black, L.M.; Adedoyin, O.O.; Yang, Z.; Traylor, A.M.; Jiang, Y.; Li, Z.; et al. Resident macrophages reprogram toward a developmental state after acute kidney injury. JCI Insight 2019, 4, e125503. [Google Scholar] [CrossRef]
- Consortium, E.P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef]
- Consortium, E.P. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Vistica, D.T.; Skehan, P.; Scudiero, D.; Monks, A.; Pittman, A.; Boyd, M.R. Tetrazolium-based assays for cellular viability: A critical examination of selected parameters affecting formazan production. Cancer Res. 1991, 51, 2515–2520. [Google Scholar]
- Zarjou, A.; Bolisetty, S.; Joseph, R.; Traylor, A.; Apostolov, E.O.; Arosio, P.; Balla, J.; Verlander, J.; Darshan, D.; Kuhn, L.C.; et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J. Clin. Investig. 2013, 123, 4423–4434. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Balla, G.; Vercellotti, G.M.; Balla, J.; Jacob, H.S.; Levitt, M.D.; Rosenberg, M.E. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Investig. 1992, 90, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Shiraishi, F.; Curtis, L.M.; Truong, L.; Poss, K.; Visner, G.A.; Madsen, K.; Nick, H.S.; Agarwal, A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am. J. Physiol. Ren. Physiol. 2000, 278, F726–F736. [Google Scholar] [CrossRef]
- Agarwal, A.; Balla, J.; Alam, J.; Croatt, A.J.; Nath, K.A. Induction of heme oxygenase in toxic renal injury: A protective role in cisplatin nephrotoxicity in the rat. Kidney Int. 1995, 48, 1298–1307. [Google Scholar] [CrossRef]
- Nath, K.A.; Grande, J.P.; Croatt, A.J.; Likely, S.; Hebbel, R.P.; Enright, H. Intracellular targets in heme protein-induced renal injury. Kidney Int. 1998, 53, 100–111. [Google Scholar] [CrossRef]
- Morimoto, K.; Ohta, K.; Yachie, A.; Yang, Y.; Shimizu, M.; Goto, C.; Toma, T.; Kasahara, Y.; Yokoyama, H.; Miyata, T.; et al. Cytoprotective role of heme oxygenase (HO)-1 in human kidney with various renal diseases. Kidney Int. 2001, 60, 1858–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Johnson, A.C.; Becker, K. Plasma and urinary heme oxygenase-1 in AKI. J. Am. Soc. Nephrol. 2012, 23, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Askenazi, D.J.; Halloran, B.; Patil, N.; Keeling, S.; Saeidi, B.; Koralkar, R.; Ambalavanan, N. Genetic Polymorphisms of Heme-oxygenase 1 (HO-1) may Impact on Acute Kidney Injury, Bronchopulmonary Dysplasia and Mortality in Premature Infants. Pediatr. Res. 2015, 77, 793–798. [Google Scholar] [CrossRef]
- Baan, C.; Peeters, A.; Lemos, F.; Uitterlinden, A.; Doxiadis, I.; Claas, F.; Ijzermans, J.; Roodnat, J.; Weimar, W. Fundamental role for HO-1 in the self-protection of renal allografts. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2004, 4, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Leaf, D.E.; Body, S.C.; Muehlschlegel, J.D.; McMahon, G.M.; Lichtner, P.; Collard, C.D.; Shernan, S.K.; Fox, A.A.; Waikar, S.S. Length Polymorphisms in Heme Oxygenase-1 and AKI after Cardiac Surgery. J. Am. Soc. Nephrol. 2016, 27, 3291–3297. [Google Scholar] [CrossRef]
- Katavetin, P.; Inagi, R.; Miyata, T.; Shao, J.; Sassa, R.; Adler, S.; Eto, N.; Kato, H.; Fujita, T.; Nangaku, M. Erythropoietin induces heme oxygenase-1 expression and attenuates oxidative stress. Biochem. Biophys. Res. Commun. 2007, 359, 928–934. [Google Scholar] [CrossRef]
- Zou, L.; Sato, N.; Attuwaybi, B.O.; Kone, B.C. Delayed administration of alpha-melanocyte-stimulating hormone or combined therapy with BAY 11-7085 protects against gut ischemia-reperfusion injury. Shock 2003, 20, 469–475. [Google Scholar] [CrossRef]
- Lam, C.W.; Getting, S.J.; Perretti, M. In vitro and in vivo induction of heme oxygenase 1 in mouse macrophages following melanocortin receptor activation. J. Immunol. 2005, 174, 2297–2304. [Google Scholar] [CrossRef]
- Lee, T.S.; Chau, L.Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 2002, 8, 240–246. [Google Scholar] [CrossRef]
- Mori, K.; Lee, H.T.; Rapoport, D.; Drexler, I.R.; Foster, K.; Yang, J.; Schmidt-Ott, K.M.; Chen, X.; Li, J.Y.; Weiss, S.; et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J. Clin. Investig. 2005, 115, 610–621. [Google Scholar] [CrossRef]
- Uddin, M.J.; Li, C.S.; Joe, Y.; Chen, Y.; Zhang, Q.; Ryter, S.W.; Chung, H.T. Carbon Monoxide Inhibits Tenascin-C Mediated Inflammation via IL-10 Expression in a Septic Mouse Model. Mediat. Inflamm. 2015, 2015, 613249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Dhar, G.J.; Bossenmaier, I.; Cardinal, R.; Petryka, Z.J.; Watson, C.J. Transitory renal failure following rapid administration of a relatively large amount of hematin in a patient with acute intermittent porphyria in clinical remission. Acta Med. Scand. 1978, 203, 437–443. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Carraway, M.S.; Suliman, H.B. Carbon monoxide, oxidative stress, and mitochondrial permeability pore transition. Free Radic. Biol. Med. 2006, 40, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, E.M.; Hock, T.; Hill-Kapturczak, N.; Agarwal, A. The story so far: Molecular regulation of the heme oxygenase-1 gene in renal injury. Am. J. Physiol. Ren. Physiol. 2004, 286, F425–F441. [Google Scholar] [CrossRef]
- Yang, H.; Magilnick, N.; Lee, C.; Kalmaz, D.; Ou, X.; Chan, J.Y.; Lu, S.C. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol. Cell. Biol. 2005, 25, 5933–5946. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hsiao, L.D.; Wang, C.Y.; Lin, W.N.; Shih, Y.F.; Chen, Y.W.; Cho, R.L.; Tseng, H.C.; Yang, C.M. HO-1 Upregulation by Kaempferol via ROS-Dependent Nrf2-ARE Cascade Attenuates Lipopolysaccharide-Mediated Intercellular Cell Adhesion Molecule-1 Expression in Human Pulmonary Alveolar Epithelial Cells. Antioxidants 2022, 11, 782. [Google Scholar] [CrossRef]
- Wright, M.M.; Kim, J.; Hock, T.D.; Leitinger, N.; Freeman, B.A.; Agarwal, A. Human haem oxygenase-1 induction by nitro-linoleic acid is mediated by cAMP, AP-1 and E-box response element interactions. Biochem. J. 2009, 422, 353–361. [Google Scholar] [CrossRef]
- Jagadeesh, A.S.V.; Fang, X.; Kim, S.H.; Guillen-Quispe, Y.N.; Zheng, J.; Surh, Y.J.; Kim, S.J. Non-canonical vs. Canonical Functions of Heme Oxygenase-1 in Cancer. J. Cancer Prev. 2022, 27, 7–15. [Google Scholar] [CrossRef]
- Lerolle, N.; Nochy, D.; Guerot, E.; Bruneval, P.; Fagon, J.Y.; Diehl, J.L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensiv. Care Med. 2010, 36, 471–478. [Google Scholar] [CrossRef]
- Aslan, A.; van den Heuvel, M.C.; Stegeman, C.A.; Popa, E.R.; Leliveld, A.M.; Molema, G.; Zijlstra, J.G.; Moser, J.; van Meurs, M. Kidney histopathology in lethal human sepsis. Crit. Care 2018, 22, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarjou, A.; Agarwal, A. Sepsis and acute kidney injury. J. Am. Soc. Nephrol. 2011, 22, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghajar-Rahimi, G.; Traylor, A.M.; Mathew, B.; Bostwick, J.R.; Nebane, N.M.; Zmijewska, A.A.; Esman, S.K.; Thukral, S.; Zhai, L.; Sambandam, V.; et al. Identification of Cytoprotective Small-Molecule Inducers of Heme-Oxygenase-1. Antioxidants 2022, 11, 1888. https://doi.org/10.3390/antiox11101888
Ghajar-Rahimi G, Traylor AM, Mathew B, Bostwick JR, Nebane NM, Zmijewska AA, Esman SK, Thukral S, Zhai L, Sambandam V, et al. Identification of Cytoprotective Small-Molecule Inducers of Heme-Oxygenase-1. Antioxidants. 2022; 11(10):1888. https://doi.org/10.3390/antiox11101888
Chicago/Turabian StyleGhajar-Rahimi, Gelare, Amie M. Traylor, Bini Mathew, James R. Bostwick, N Miranda Nebane, Anna A. Zmijewska, Stephanie K. Esman, Saakshi Thukral, Ling Zhai, Vijaya Sambandam, and et al. 2022. "Identification of Cytoprotective Small-Molecule Inducers of Heme-Oxygenase-1" Antioxidants 11, no. 10: 1888. https://doi.org/10.3390/antiox11101888