
Abstract
Materials displaying metal-insulator transitions (MITs) as a function of external parameters such as temperature, pressure, or composition are most intriguing from the fundamental point of view and also hold high promise for applications. Vanadium dioxide (VO2) is one of the most prominent examples of MIT having prospective applications ranging from intelligent coatings, infrared sensing, or imaging, to Mott memory and neuromorphic devices. The key aspects conditioning possible applications are the controllability and reversibility of the transition. Here we present an intriguing MIT in hydrogenated vanadium dioxide, HxVO2. The transition relies on an increase of the electron occupancy through hydrogenation on the transition metal vanadium, driving the system insulating by a hybrid of two distinct MIT mechanisms. The insulating phase observed in HVO2 with a nominal d2 electronic configuration contrasts with other rutile d2 systems, most of which are metallic. Using spectroscopic tools and state-of-the-art many-body electronic structure calculations, our investigation reveals a correlation-enhanced Peierls and a Mott transition taking place in an orbital-selective manner cooperate to stabilize an insulating phase. The identification of the hybrid mechanism for MIT controlled by hydrogenation opens the way to radically design strategies for future correlated oxide devices by controlling phase reversibly while maintaining high crystallinity.
Similar content being viewed by others

Introduction
Identifying low energy degrees of freedom and their intricate interplay in correlated electron materials is essential to understanding their physical properties. For metal-to-insulator-transitions (MIT) in particular, strong electronic correlations often play a crucial role in the charge localization of systems with non-trivial band fillings. In these systems, the MIT often occurs with either formation of orderings or enhancement of the pre-existing ones, resulting from an intricate interrelationship of lattice, charge, orbital, and spin degrees of freedom. The MITs accompanied by such an emergence or enhancement of orderings have been reported in many transition metal oxides1,2.
Vanadium dioxide (VO2) is one of the most prominent examples of the correlation-driven MIT that has excited the solid-state community for decades3,4,5,6. VO2 undergoes MIT around TMIT ~ 340 K7 with a structural transition from a rutile to a monoclinic phase (M1), involving the formation of V–V dimers. The insulating state can be understood as a combined effect of V–V dimerization and the on-site Coulomb interaction. The former induces a bonding-antibonding splitting within the a1g orbitals with a strong bonding along the dimer direction8. The latter opens a gap by enhancing the bonding-antibonding splitting, resulting in a spin-singlet state in the filled bonding band9,10,11,12.
Recently, the electronic transport and lattice structure of hydrogenated VO2 (HxVO2) have been investigated13,14,15,16,17,18. HxVO2 shows successive phase transitions as a function of hydrogen content x at room temperature: an insulator to metal transition followed by the reentrance of the insulating phase. At low H-content, it shows a bad metallic behavior with a resistivity exceeding the Mott–Ioffe–Regel limit19,20, implying strong electron–electron interactions21. Electronic and optical properties in the metallic phase at low hydrogenation are similar to the oxygen vacant VO2−δ22. As H-doping increases, a structural transition involving dimerization of V-cations occurs18 [Fig. 1a], accompanied by a MIT in the electronic transport. The insulating phase is maintained up to the fully hydrogenated limit (HVO2)23,24. Since the inserted hydrogen is attached to oxygen atoms and hybridizes mainly with O-p states away from the Fermi energy, the primary role of the hydrogenation is changing the V-d occupancy, consistent with density functional theory (DFT) calculations23,24. Thus, the doping-induced change in the valence suggests the presence of a filling-controlled MIT around the V-d2-filling, which is in clear contrast to other d2-filling rutile-based compounds such as CrO2, MoO2, and WO2 showing a low-T metallic phase25,26,27.

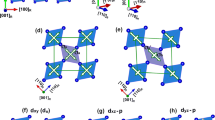
a The primitive unit cell of insulating HVO2 in orthorhombic structure (space group Fdd2). Similar to VO6, the octahedra are edge-sharing in the (001)R direction and corner-sharing in (100)R/(010)R directions, forming chain-like structures with small octahedral rotations. Two inequivalent V sites with four and two hydrogen atoms attached to the VO6 octahedron are defined as V1 and V2, respectively. The V–V bond lengths are 2.98 and 2.78 Å, which are comparable to experimental values of monoclinic VO2 3.10 and 2.77 Å9,42. b Definition of vanadium \({d}_{{x}^{2}-{y}^{2}}\) (a1g-like) and dxz/dyz (\({e}_{g}^{\pi }\)-like) orbitals in this work. The y-axis is set parallel to V-chain direction (001)R.
Identification of this newly discovered MIT mechanism requires orbital-sensitive measurements to probe the orbital reconstructions associated with the MIT. Moreover, computational methods incorporating both intra- and inter-site correlations need to be employed to correctly capture the essential many-body effects, such as cluster dynamical mean-field theory (CDMFT), which successfully captures the MIT in VO211. We investigate the mechanism of the MIT by polarization-dependent spectroscopic ellipsometry and X-ray absorption (XAS) measurements, elucidating the orbital-dependent spectral modifications in the insulating phase. The orbital reconstructions accompanied by the MIT are theoretically investigated considering both on-site and inter-site correlation using Δ+DMFT scheme, which successfully capture the orbital structures observed in the experiments. From our analysis, we will show that the MIT is driven by a simultaneous Mott and Peierls transition depending on the orbital character. Our experimental data provides the spectral information of HxVO2 essential for the characterization of electronic structures across the MIT and to the best of our knowledge this is the first proposal of the MIT mechanism involving Mott and Peierls physics with orbital-dependent way, which has not been reported in rutile systems. We expect that the hybrid mechanism along with the continuous control of hydrogenation can provide ways control phase of the matter, which is important to utilize the vanadium-based oxides as various applications27,28,29,30,31 such as hydrogen storage and high-performance smart windows24,32,33,34.
Results and discussion
Effects of the hydrogenation
Figure 2a summarizes our main findings by comparing schematic orbital structures of undoped, partially hydrogenated, and fully hydrogenated HxVO2. Since the average occupancy of V-d orbitals is expected to be in between one and two electrons per V, the low-lying frontier orbitals of HxVO2 are a1g- and \({e}_{g}^{\pi }\)-like orbitals, with lobes extended along and perpendicular to the dimer direction, respectively, as presented in Fig. 1b. Starting from the insulating VO2 system involving the correlation-enhanced bonding-antibonding splitting of \({d}_{{x}^{2}-{y}^{2}}\) (a1g) (left panel), the introduction of a small amount of hydrogen leads to an insulator to metal transition as the added electrons occupy the partially occupied \({e}_{g}^{\pi }\) orbitals (middle panel), consistent with other electron-doped VO2 systems17,27,35.

a Schematic energy band diagrams for the insulating VO2, metallic HxVO2, and insulating HVO2 near Fermi level. b Real part of the optical conductivity of HxVO2 thin film on Al2O3 (0001) substrate measured at room temperature. CT, charge transfer; B, bonding; AB, antibonding; UHB, upper-Hubbard band; LHB, lower-Hubbard band.
As the electron occupancy reaches 2 electrons per vanadium in the fully hydrogenated HVO2, a MIT occurs with a structural transition involving the dimerization of V atoms [Fig. 1a]. Just as the filling the bonding state of \({d}_{{x}^{2}-{y}^{2}}\) opens a gap in dimerized VO2 (left panel), it is strongly suggested that the similar splitting of a1g orbitals is also present in HVO2 as the dimerization occurs simultaneously with the MIT36,37. With a d-occupancy of 2 electrons per V around x = 100%, an additional orbital splitting is expected to induce the band gap. As in Fig. 2, the MIT occurs in which dyz orbitals split to the lower and upper Hubbard bands by a Mott-transition with an orbital ordering between dxz and dyz orbitals, pushing up the energy of dxz orbital relative to dyz. Here, the role of electron correlations is essential, which boosts the orbital ordering and, at the same time, opens a gap by splitting the dyz bands.
To understand the evolution of the electronic structure in HVO2, we examine the structural changes developed by hydrogenation. X-ray and neutron diffraction studies verified the lattice structure of HxVO2 and identified orthorhombic structure (space group Fdd2(43)) for highly hydrogenated and insulating phase18. The structural characterization of our epitaxial films is published elsewhere37. As illustrated in Fig. 1a, it has four V atoms in the primitive unit cell. We note that HVO2 has substantial dimerization along the V-chain with long (short) bonds between vanadium sites with (without) H attached to the bridging oxygens.
Electronic structures and optical conductivity
A pronounced insulator-to-metal transition and reentrance to the insulating phase of HxVO2 at room temperature are well-exhibited in the real part of the optical conductivity σ1(ω) in Fig. 2b. The subsequent transitions observed by increasing x are consistent with transport measurements23,24. At x = 0%, the overall peak structure of our film is in good agreement with previous reports on monoclinic VO238. We can observe an optical gap between filled a1g and \({e}_{g}^{\pi }\) orbitals. The low-intensity peaks near 1.2 and 3 eV are mainly attributed to transitions within V-d orbitals, shown schematically in Fig. 2a. A peak near 3.5 eV marked as CT-1 is the charge transfer peak and corresponds to the excitation from O-p to V-d derived bands39,40.
At x = 3%, the optical gap is closed, while the charge transfer peak is almost fully maintained except for a decrease in peak height. We find the gap reopening at x = 100%, reentering the insulating phase. We note here that the composition x is estimated by optical and diffraction characteristics of HxVO2 (see “Methods” and Fig. S2 in the Supplementary Information), which may deviate from exact chemical stoichiometry. The charge transfer peak is shifted higher in energy by 1.3 eV (marked as CT-2) with largely reduced spectral weight over the measured frequency range. At intermediate H-content of x = 46%, σ1(ω) shows two charge transfer peaks. At the same doping, the coexistence of two XRD peaks that correspond to the (100)R lattice of low and high H-compositions (x = 3% and 100%) are observed (see Fig. S2b in the Supplementary Information for details).
We now investigate the electronic structure of the insulating O2 phase in the fully hydrogenated limit (HVO2), including the effect of strong local Coulomb interactions on the V-d orbitals. For this, we employ a Δ+DMFT approach, where we project the DFT Hamiltonian onto localized a1g- and \({e}_{g}^{\pi }\)-like Wannier orbitals for the two inequivalent V atoms in the unit cell as in Fig. 1b. In the reduced model, the on-site correlations of dxz and dyz orbitals are treated by DMFT utilizing a continuous-time Quantum Monte Carlo impurity solver. Our approximation properly respects the fact that the intra- and inter-dimer hopping energies of dxz and dyz orbitals are similar in magnitude and can be considered a local problem, adequately described in a DMFT approach. On the other hand, the inter-site correlations originating from the dimer-forming \({d}_{{x}^{2}-{y}^{2}}\) orbitals are approximated by a static (frequency-independent) dimer-like self-energy of the form of an inter-site orbital-dependent potential Δ as described in ref. 41. This approach effectively captures the correlation-enhanced bonding-antibonding splitting of \({e}_{g}^{\pi }\) orbitals. Such a static treatment of the inter-site correlations has been previously employed in pure VO2 by a LDA+Δ correction41.
Figure 3a presents the orbital-projected spectral function for HVO2 calculated within the Δ+DMFT method. We find a Mott-insulating phase with a gap at the Fermi energy (EF), consistent with our optical spectra [Fig. 2b] and previous transport experiments23,36,42. The O-p derived bands are −4 eV below the EF which is roughly 1.5 eV lower than those of metallic HxVO2 and of pure VO226, which is consistent with the blue shift of the charge transfer peak observed in our optical spectra [CT-2 in Fig. 2b]. The change arises from the formation of O–H bonding during hydrogenation, supposing that the attachment of H+ ion to oxygen lowers the energy of O-p bands (Fig. S4 in the Supplementary Information).

a The spectral function A(ω) of O-p (red), V-da1g/\({e}_{g}^{\pi }\)-like (blue), and V-d \({e}_{g}^{\sigma }\)-like (green) orbital dominated states. b–d Orbital decomposed A(ω) of b \({d}_{{x}^{2}-{y}^{2}}\), c dxz, and d dyz orbitals. The inequivalent V1 and V2 atoms are indicated by solid and dashed lines, respectively. e Experimental optical conductivity σ1(ω) of VO2 measured at 180 K (empty circles) and HVO2 at 300 K (filled squares) grown on Al2O3 (0001) substrate. As the in-plane lattice directions of the sample are not aligned within the beam spot, the optical anisotropy was undetected. f Calculated σ1(ω) of the Δ+DMFT method for insulating VO2 (dotted line) and HVO2 (solid line). The spectra for the two polarizations, E ∥(001)R and E ∥(010)R, is averaged for direct comparison to the experiment.
The orbital-resolved spectral functions clearly demonstrate an orbital-selective mechanism that induces a MIT, as shown in Fig. 3b–d. The spectral function A(ω) of the a1g-like orbital (panel b) exhibits a gap resulting from the enhancement of the bonding-antibonding splitting by the Δ-potential from inter-site correlations. This mechanism corresponds to a correlation-assisted Peierls transition from dimerization, as discussed in VO211,12. The \({e}_{g}^{\pi }\)-like orbitals exhibit an orbital ordering with the dxz orbital shifted above the Fermi energy (panel c) and a Mott gap formed in the dyz orbital (panel d), driven by local correlations. In essence, the pronounced difference in the hopping anisotropy between a1g- and \({e}_{g}^{\pi }\) orbitals leads to two distinct mechanisms: a correlation-assisted Peierls and Mott transition for a1g and \({e}_{g}^{\pi }\) orbitals, respectively, which we define as an orbital-selective Mott and Peierls transition.
We note that there is no qualitative difference between the inequivalent V1 and V2 sites defined in Fig. 1a. The major features of the orbital orderings without correlations (defined with non-magnetic Wannier Hamiltonian), such as bonding-antibonding splitting of \({d}_{{x}^{2}-{y}^{2}}\) orbitals and energy of dxz orbital higher than dyz orbitals are the same for both sites with the octahedral distortions and attached hydrogen ion. Given the non-interacting orbital structure with the bonding-antibonding splitting of \({d}_{{x}^{2}-{y}^{2}}\) orbitals and the energy difference between dxz/dyz orbitals, the Mott and Peierls and Mott transition occurring by the inclusion of the inter-site and on-site correlations enhances the orbital splitting. The resulting orbital splitting is much larger (in the order of U) than that from structural effects (<0.4 eV), and the enhanced splittings dominate the orbital structures. The orbital energies from the non-magnetic Wannier tight-binding Hamiltonian are discussed in Sec. VII in the Supplementary Information.
The calculated optical conductivity matches with the important characteristics of the spectral transfer observed in the experiment, which strongly supports the orbital selective phase transition in HVO2. Figure 3e shows the experimental σ1(ω) of the VO2 extracted from thin film grown on an Al2O3 substrate before and after hydrogenation. The σ1(ω) of VO2 calculated by using Δ-potential to include inter-site correlations [Fig. 3f] is consistent with that calculated by CDMFT43 and also with the experimental data; this supports the validity of employing a static intra-dimer Δ-potential in our approach. Moreover, we find that the calculated optical conductivity of HVO2 from Δ+DMFT successfully reproduces the essential features such as the optical gap, suppression of the spectral weight, and the blue shift of the charge transfer peak about 1.5 eV coming from the down-shift of O-p states (for details see Fig. S4 in the Supplementary Information). These results provide a strong support to the Mott and Peierls insulating phase as the ground state of HVO2.
Polarization-dependent optical responses
An interesting feature resulting from the orbital-selective Mott and Peierls transition is orbital ordering, by which polarization-dependent responses are expected. We employ two different measurements, polarization-dependent X-ray absorption and ellipsometry, to further confirm the proposed Mott-Peierls insulating phase. We prepare an additional set of VO2 films of 10-nm thickness grown epitaxially on TiO2 (100) substrate, which allows measuring the polarization-dependent responses36.
To obtain the orbital-dependent responses using polarized light, alignment of the VO2 on the substrate is crucial37. The VO2 films epitaxially grown on Al2O3 have a significant advantage due to the large band gap (~7 eV), allowing for the optical measurements up to the visible/UV ranges, but the VO2 films have domains oriented in different directions inhibiting the polarization-dependent experiments. In contrast, the VO2 films grown in TiO2 are oriented in either (100)R or (001)R36,37, making it possible to perform polarization-dependent measurements up to the visible ranges.
Figure 4a presents the measurement geometry where the s- and p-polarized light is mainly parallel and perpendicular to (001)R (or y-axis) in Fig. 1, respectively. We find that the linear dichroism of O K-edge (O 1s → 2p transition) of VO2 (Fig. S3) before hydrogenation is in good agreement with that of single crystal VO244,45,46. This shows that our film on TiO2 is oriented well-enough to detect the dichroism signal.

a Hydrogenated 10-nm-thickness VO2/TiO2 (100)R thin film and measurement geometry in XAS and ellipsometry. The (001)R direction is parallel to the y-axis defined for the calculations in Fig. 1b. b O K-edge XAS spectra with E ∥(001)R (filled circles) and E ⊥(001)R (empty circles) for HVO2 taken at 250 K. c–e Decomposed A(ω) calculated for c dxz and dyz, d \({d}_{{x}^{2}-{y}^{2}}\), and e dxz and \({d}_{{z}^{2}}\) orbitals. The intensity for inequivalent V1 and V2 atoms, total 4 atoms in unit cell, are summed up. f, g Optical conductivity σ1(ω) spectra from (f) experiment and (g) Δ+DMFT. h The deviation of σ1(ω) in E ∥(001)R, σ∥(ω), and E ⊥(001)R, σ⊥(ω). The experimental spectrum in symbols is multiplied by 6. Pol., polarization.
The X-ray absorption of fully hydrogenated HVO2 in Fig. 4b confirms the calculated orbital polarization of an orbital selective Mott and Peierls insulator. We define the case of an intensity of E ∥(001)R greater (less) than E ⊥(001)R as positive (negative) dichroism. There are three characteristic features in the XAS: (1) a peak at 528.5 eV with negative dichroism, (2) a small peak at 529.5 eV with positive dichroism (marked with a star), and (3) peak around 532 eV with positive dichroism. The dichroism can be understood from the orbital-projected spectral function of each d-orbitals summed over all V-atoms in the unit cell presented in Fig. 4c–e. The negative dichroism of (1) can be understood from the dxz-derived states located right above the Fermi energy [Fig. 4c], which hybridize mainly with O-px/pz orbitals and thus have large absorption for the polarization in the xz [E ⊥(001)R] plane. The feature of (2) is associated with the transition to the \({d}_{{x}^{2}-{y}^{2}}\)-derived antibonding states [Fig. 4d] in which the \({d}_{{x}^{2}-{y}^{2}}\)-py hybridization prefers the absorption of E ∥(001)R (parallel to y-axis in Fig. 1b), resulting in overall positive dichroism and the peak position well matches with the transition energy to the antibonding \({d}_{{x}^{2}-{y}^{2}}\) state. We find the positive dichroism of (3) is common for both VO2 and HVO2, due to the similar \({e}_{g}^{\sigma }\) orbital structures between them.
The polarization-dependent optical conductivity σ1(ω) provides further evidence of the proposed orbital structures of HVO2. In both experiment and calculation, we find that the difference in σ1(ω) between E ∥(001)R and E ⊥(001)R gradually increases up to 3 eV [Fig. 4f, g). We note that σ1(ω) above 3 eV becomes unreliable due to a strong TiO2 signal and thus not shown. Below 1 eV, the optical response is contributed mainly from the transitions from occupied dyz and \({d}_{{x}^{2}-{y}^{2}}\) states to unoccupied dxz states, having small matrix elements and dichroism. Above 1 eV, the transitions between bonding and antibonding \({d}_{{x}^{2}-{y}^{2}}\) start to be significant, having large matrix elements for E ∥(001)R compared with E ⊥(001)R. This, in turn, results in a significant increase of the dichroism [Fig. 4h] at around 2.5 eV corresponding to the bonding-antibonding splitting of \({d}_{{x}^{2}-{y}^{2}}\) bands. We find that the measured dichorism in the experiment is roughly 6 times smaller than in the calculation. This is partially from the magnitude of the experimental optical conductivity being about half of the calculated one, resulting in about 50% reduction of the dichroism signal. The non-ideal sample may be another reason, such as oxygen vacancies formation on the surface during annealing or the coexistence of other remnant phases (e.g., mixed domains). Overall, the consistent trend of the experimental and theoretical dichroism in the optical conductivity further supports the orbital structures resulting from a Mott and Peierls transition.
We note that the inter-site correlations included with the Δ+DMFT approach are crucial for predicting the correct orbital reconstructions across the MIT. To confirm the role of the inter-site correlations, we have additionally performed single-site DMFT calculations that neglect these nonlocal effects (see Fig. S6 in the Supplementary Information). In this case we find that the orbital ordering pattern is not consistent with the polarization-dependent XAS. Moreover, the optical conductivity and its polarization show marked differences around the low-frequency region (see Supplementary Information for further discussion).
Outlook and conclusion
Several properties of the MIT in HxVO2 need further investigation. For example, since two different mechanisms are involved in the MIT, it is an essential question whether the two transitions occur synchronously or asynchronously, and how the hydrogens percolate into the films. Unveiling the dynamics of the intermediate state can answer such questions. Theoretically, a temperature-dependent self-energy calculated for the O1 and O2 structures by DFT plus cellular DMFT methods including both a1g and \({e}_{g}^{\pi }\) orbitals could predict whether two transitions occur simultaneously. This would be an important future direction to pursue in the future. Experimentally, systematic temperature-dependent measurements of the orbital polarization by polarized X-ray absorption, dimerization by X-ray diffraction, and conductivity by transport or optical absorption measurements can be used. These experiments would allow investigating the orbital occupation, structural parameters, and band gap as the function of temperature. A key to achieving systematic results here is to maintain the hydrogen content at different temperatures, which is challenging because the x of the HxVO2 films changes at a different rate at different temperatures and chamber pressure. Therefore, systematic studies and surrounding hydrogen gas pressure conditions need to be preceded in HxVO2.
To conclude, we have investigated the electronic and optical properties of HxVO2 across the MIT occurring in the highly hydrogenated limit. Our main finding is that the insulating phase is induced in an orbital selective way, in which the a1g-like orbital undergoes a correlation-assisted Peierls transition due to the relatively large dimer hopping, in a similar fashion as in pure VO2. On the other hand, the \({e}_{g}^{\pi }\)-like orbitals undergo a Mott-insulator transition after orbital ordering. We identify this scenario as an orbital-selective hybrid Mott and Peierls transition. The resulting orbital structure from the MIT is supported by polarization-dependent XAS and ellipsometry, showing energy-dependent dichroism, as well as Δ+DMFT calculations which successfully reproduce the experimental data. To the best of our knowledge, this is the first study uncovering the nature of the insulating phase under hydrogenation by providing solid evidence of orbital reconstructions across the MIT.
Our results provide a potential pathway to stabilize an insulating phase by combining Peierls and Mott physics coexisting in orbital space. This is in close analogy to the M2 phase in VO2 having alternating antiferromagnetic and dimerized edge-sharing chains47. The present compound, HVO2, is the first example of a compound displaying coexistence of Mott and Peierls behavior without spatial separation, differentiated only in orbital space. The proposed hybrid mechanism for a MIT provides a route for devising functionalities of correlated oxides from the two main aspects. The broken orbital symmetry with large energy separation in the presence of the two distinct ordering gives polarization-dependent responses varying with light frequencies and the lowered crystalline symmetry can be utilized for the optical control of the MIT by exciting the dimer oscillation allowed by the distinct V1 and V2 sites. Moreover, we expect that the transition temperature of the MIT could be controlled by the uniaxial strain along the chain direction. Utilizing hydrogenation, we expect to control the MIT with a small electric field, which could be achieved by tuning the hydrogen doping to the MIT boundary such that modulating the orbital occupation by small applied electric fields could induce the MIT.
Methods
Sample preparation
We prepared 30-nm-thick VO2 films epitaxially grown on the Al2O3 (0001) substrate, and 10-nm-thick VO2 on the TiO2 substrate (100)R and (001)R using pulsed laser deposition. The top surface of the films was then deposited with nano-sized Pt-islands to implement hydrogen spillover method as described in refs. 23,36,37.
Spectroscopic ellipsometry
For the optical conductivity measurements, we used spectroscopic ellipsometry (M-2000) and implemented fitting program (V-VASE) of J. A. Woollam Co. The hydrogenation was controlled on a hot plate (annealing T ~ 400 K) under the flow of H2 (5%)/Ar (95%) in air. Each measurement was immediately followed by the XRD to characterize the lattice constant, from which we applied the linear relation between hydrogenation and lattice constants18,23 to obtain doping content x.
To obtain the anisotropic optical conductivity, we scanned the VO2/TiO2 (100)R and the TiO2 (100)R substrate twice for each sample, with s-polarized light parallel to (010)R and (001)R by rotating the film in 90∘ in-plane. After fitting the substrate with experimental data, we modeled the anisotropic film on the anisotropic substrate and fitted the optical conductivity with experimental film data. The photon energy ranges of the thin film spectra are limited by the band gap of the substrates, which is around 3 eV for TiO2 and 7 eV for Al2O3 (see Fig. S4).
NEXAFS
NEXAFS data were obtained at the beamline 2A of Pohang Accelerator Laboratory (PAL). The incident angle [θi in Fig. 4a] was about 67.5∘. To compare the XAS data with different light polarization, a constant offset below 510 eV has been subtracted. The resulting dichroism signal of VO2 at the V L3-edge are consistent with previous reports46,48.
DFT+DMFT calculations
We performed first-principles DFT calculations within the generalized gradient approximation (GGA) using the Vienna ab-initio simulation package49,50. We used the projector augmented wave method51 and Perdew–Becke–Erzenhof parametrization52 for the exchange-correlation functional. The experimental lattice parameters18 have been used for the O1 and O2 structures in which the 8 × 8 × 16 and 8 × 8 × 8k-point grids are used, respectively. The energy cut-off of 500 eV is used. For the insulating O2 phase, the effective tight-binding Hamiltonian consisting of the V-d-derived frontier orbitals (\({d}_{{x}^{2}-{y}^{2}},{d}_{xz},{d}_{yz}\)) is obtained for four V-sites of the primitive unit cell of HVO2 using the Wannier90 package53, based on the non-magnetic DFT-GGA calculation. To this Hamiltonian a local Coulomb interaction term is added with interaction parameters similar to those of the undoped parent compound with U = 3.5 eV and J = 0.7 eV and solved by Δ+DMFT method in which \({d}_{{x}^{2}-{y}^{2}}\) orbitals have been treated with static inter-site correlations Δ of −1.0 eV within the Hubbard dimer approximation (for detail see the Supplementary Information). For dxz and dyz orbitals, we have used a continuous-time quantum Monte-Carlo impurity solver in the hybridization expansion with a density–density form of the interaction as implemented in the ALPS package54. The optical conductivity is calculated by upfolding the band structures43. The spectral functions from DMFT are upfolded to the p-d energy window. The optical matrix elements are calculated in the energy range containing O-p and V-d bands by upfolding the band structure43. The matrix elements were evaluated using the real space position operators in the Wannier basis.
Data availability
All relevant data that support the findings of this study are available from the corresponding author on request.
Code availability
The codes used are VASP, ALPS, and Wannier90 (http://www.wannier.org). Additional scripts and modifications of the last are available from the corresponding author upon reasonable request.
References
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Khomskii, D. I. Transition Metal Compounds (Cambridge University Press, 2009).
Shi, R. et al. Phase management in single-crystalline vanadium dioxide beams. Nat. Commun. 12, 4214 (2021).
Budai, J. D. et al. Metallization of vanadium dioxide driven by large phonon entropy. Nature 515, 535–539 (2014).
Zhou, Y. & Ramanathan, S. Mott memory and neuromorphic devices. Proc. IEEE 103, 1289–1310 (2015).
Lee, S. et al. Anomalously low electronic thermal conductivity in metallic vanadium dioxide. Science 355, 371–374 (2017).
Morin, F. J. Oxides which show a metal-to-insulator transition at the Neel temperature. Phys. Rev. Lett. 3, 34–36 (1959).
Goodenough, J. B. The two components of the crystallographic transition in VO2. J. Solid State Chem. 3, 490–500 (1971).
Zylbersztejn, A. & Mott, N. F. Metal-insulator transition in vanadium dioxide. Phys. Rev. B 11, 4383–4395 (1975).
Sommers, C. & Doniach, S. First principles calculation of the intra-atomic correlation energy in VO2. Solid State Commun. 28, 133–135 (1978).
Biermann, S., Poteryaev, A., Lichtenstein, A. I. & Georges, A. Dynamical singlets and correlation-assisted peierls transition in VO2. Phys. Rev. Lett. 94, 026404 (2005).
Tomczak, J. M., Aryasetiawan, F. & Biermann, S. Effective bandstructure in the insulating phase versus strong dynamical correlations in metallic VO2. Phys. Rev. B 78, 115103 (2008).
Li, Z. et al. Hydrogen treatment for superparamagnetic VO2 nanowires with large room-temperature magnetoresistance. Angew. Chem. Int. Ed. 55, 8018–8022 (2016).
Shi, R. et al. Axial modulation of metal-insulator phase transition of VO2 nanowires by graded doping engineering for optically readable thermometers. J. Phys. Chem. C 121, 24877–24885 (2017).
Andreev, V. N., Kapralova, V. M. & Klimov, V. A. Effect of hydrogenation on the metal-semiconductor phase transition in vanadium dioxide thin films. Phys. Solid State 49, 2318–2322 (2007).
Wu, C. et al. Hydrogen-incorporation stabilization of metallic VO2(R) phase to room temperature, displaying promising low-temperature thermoelectric effect. J. Am. Chem. Soc. 133, 13798–13801 (2011).
Wei, J., Ji, H., Guo, W., Nevidomskyy, A. H. & Natelson, D. Hydrogen stabilization of metallic vanadium dioxide in single-crystal nanobeams. Nat. Nanotechnol. 7, 357–362 (2012).
Filinchuk, Y. et al. In situ diffraction study of catalytic hydrogenation of VO2: stable phases and origins of metallicity. J. Am. Chem. Soc. 136, 8100–8109 (2014).
Ioffe, A. & Regel, A. Non-crystalline, amorphous and liquid electronic semiconductors. Prog. Semiconductors 4, 237–291 (1960).
Mott, N. F. Conduction in non-crystalline systems IX. the minimum metallic conductivity. Philos. Mag. 26, 1015–1026 (1972).
Hardy, W. J., Ji, H., Paik, H., Schlom, D. G. & Natelson, D. Mesoscopic quantum effects in a bad metal, hydrogen-doped vanadium dioxide. J. Phys. Condens. Matter. 29, 185601 (2017).
Zhang, Z. et al. Evolution of metallicity in vanadium dioxide by creation of oxygen vacancies. Phys. Rev. Appl. 7, 034008 (2017).
Yoon, H. et al. Reversible phase modulation and hydrogen storage in multivalent VO2 epitaxial thin films. Nat. Mat. 15, 1113–1119 (2016).
Chen, S. et al. Sequential insulator-metal-insulator phase transitions of VO2 triggered by hydrogen doping. Phys. Rev. B 96, 125130 (2017).
Dissanayake, M. & Chase, L. L. Optical properties of CrO2 and MoO2 from 0.2–6 eV. Phys. Rev. B 10, 6872–6879 (1978).
Eyert, V. The metal-insulator transitions of VO2: a band theoretical approach. Ann. Phys. 11, 650–704 (2002).
Hiroi, Z. Structural instability of the rutile compounds and its relevance to the metal–insulator transition of VO2. Prog. Solid State Chem. 43, 47–69 (2015).
Oh, S. et al. Energy-efficient Mott activation neuron for full-hardware implementation of neural networks. Nat. Nanotechnol. 16, 680–687 (2021).
Tang, K. et al. Millikelvin-resolved ambient thermography. Sci. Adv. 6, abd8688 (2020).
Brahlek, M. et al. Opportunities in vanadium-based strongly correlated electron systems. MRS Commun. 7, 27–52 (2017).
Kim, S. Y. et al. Spectroscopic studies on the metal-insulator transition mechanism in correlated materials. Adv. Mater. 30, 1704777 (2018).
Wang, S. et al. Scalable thermochromic smart windows with passive radiative cooling regulation. Science 374, 1501–1504 (2021).
Jo, M. et al. Gate-induced massive and reversible phase transition of VO2 channels using solid-state proton electrolytes. Adv. Funct. Mater. 28, 1802003 (2018).
Chen, S. et al. Gate-controlled VO2 phase transition for high-performance smart windows. Sci. Adv. 5, eaav6815 (2019).
Cui, Y., Shi, S., Chen, L., Luo, H. & Gao, Y. Hydrogen-doping induced reduction in the phase transition temperature of VO2: a first-principles study. Phys. Chem. Chem. Phys. 17, 20998–21004 (2015).
Yoon, H., Park, J., Choi, S.-Y., Lee, D. & Son, J. Facet-dependent phase control by band filling and anisotropic electron-lattice coupling in HVO2 epitaxial films. Adv. Electron. Mater. 4, 1800128 (2018).
Park, J., Yoon, H., Sim, H., Choi, S.-Y. & Son, J. Accelerated hydrogen diffusion and surface exchange by domain boundaries in epitaxial VO2 thin films. ACS Nano 14, 2533–2541 (2020).
Qazilbash, M. M. et al. Electrodynamics of the vanadium oxides VO2 and V2O3. Phys. Rev. B 77, 115121 (2008).
Tomczak, J. M. & Biermann, S. Optical properties of correlated materials—or why intelligent windows may look dirty. Phys. Status Solidi B 246, 1996–2005 (2009).
Tomczak, J. M. & Biermann, S. Materials design using correlated oxides: optical properties of vanadium dioxide. EPL 86, 37004 (2009).
Tomczak, J. M. & Biermann, S. Effective band structure of correlated materials: the case of VO2. J. Condens. Matter Phys. 19, 365206 (2007).
Chen, L. et al. Tuning the phase transition temperature, electrical and optical properties of VO2 by oxygen nonstoichiometry: insights from first-principles calculations. RSC Adv. 6, 73070–73082 (2016).
Tomczak, J. M. & Biermann, S. Optical properties of correlated materials: generalized Peierls approach and its application to VO2. Phys. Rev. B 80, 085117 (2009).
Abbate, M. et al. Soft-x-ray-absorption studies of the electronic-structure changes through the VO2 phase transition. Phys. Rev. B 43, 7263–7266 (1991).
Koethe, T. C. et al. Transfer of spectral weight and symmetry across the metal-insulator transition in VO2. Phys. Rev. Lett. 97, 116402 (2006).
Haverkort, M. W. et al. Orbital-assisted metal-insulator transition in VO2. Phys. Rev. Lett. 95, 196404 (2005).
Pouget, J. P. et al. Dimerization of a linear Heisenberg chain in the insulating phases of V1−xCrxO2. Phys. Rev. B 10, 1801–1815 (1974).
Aetukuri, N. B. et al. Control of the metal insulator transition in vanadium dioxide by modifying orbital occupancy. Nat. Phys. 9, 661–666 (2013).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Mostofi, A. A. et al. An updated version of wannier90: a tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 185, 2309–2310 (2014).
Bauer, B. et al. The ALPS project release 2.0: open source software for strongly correlated systems. J. Stat. Mech. Theory Exp. 2011, P05001 (2011).
Acknowledgements
We acknowledge insightful discussions with D. Khomskii, A. J. Millis, B. C. Park, and J. Tomczak. This work was supported by the Institute for Basic Science (IBS) in Korea (Grant No. IBS-R009-D1), a Consolidator Grant of the European Research Council (Project CorrelMat-617196), and IDRSI-GENCI (Orsay) under project number t2020091393. S.Y.P. was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2021R1C1C1009494) and by Basic Science Research Program through the NRF funded by the Ministry of Education (No. 2021R1A6A1A03043957). H.Y. and J.S. were supported by the Basic Science Program through the NRF grant funded by the Ministry of Science and ICT (No. 2020R1A4A1018935). C.S. was supported by the National Research Foundation (NRF) funded by the Ministry of Science and ICT(2020R1C1C1008734). S.K. was supported by the Global Ph.D. Fellowship Program through the NRF funded by the Ministry of Education (No. 2015H1A2A1034943).
Author information
Authors and Affiliations
Contributions
S.K., S.B., S.B., T.W.N., and S.Y.P. designed research. S.K., S.B., H.Y., W.K., C.S., J.S., and S.Y.P. performed research. S.K., S.B., S.B., T.W.N., and S.Y.P. wrote the paper with contributions from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, S., Backes, S., Yoon, H. et al. Orbital-selective Mott and Peierls transition in HxVO2. npj Quantum Mater. 7, 95 (2022). https://doi.org/10.1038/s41535-022-00505-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41535-022-00505-y
This article is cited by
-
Thermally Induced Switching of Electrical and Optical Characteristics of VO2 Thin Nanostructured Films Fabricated by Radio Frequency Magnetron Sputtering
Arabian Journal for Science and Engineering (2024)
-
Orbital-selective charge-density wave in TaTe4
npj Quantum Materials (2023)
