High detection rate for disease-causing variants in a cohort of 30 Iranian pediatric steroid resistant nephrotic syndrome cases
 Maryam Najafi1,2†
Maryam Najafi1,2†  Korbinian M. Riedhammer3,4†§
Korbinian M. Riedhammer3,4†§  Aboulfazl Rad1,5 Paria Najarzadeh Torbati6 Riccardo Berutti3 Isabel Schüle2 Sophie Schroda2
Aboulfazl Rad1,5 Paria Najarzadeh Torbati6 Riccardo Berutti3 Isabel Schüle2 Sophie Schroda2  Thomas Meitinger3 Jasmina Ćomić3,4
Thomas Meitinger3 Jasmina Ćomić3,4  Simin Sadeghi Bojd7 Tayebeh Baranzehi8 Azadeh Shojaei9 Anoush Azarfar10 Mahmood Reza Khazaei11 Anna Köttgen12,13
Simin Sadeghi Bojd7 Tayebeh Baranzehi8 Azadeh Shojaei9 Anoush Azarfar10 Mahmood Reza Khazaei11 Anna Köttgen12,13  Rolf Backofen13,14 Ehsan Ghayoor Karimiani15,16
Rolf Backofen13,14 Ehsan Ghayoor Karimiani15,16  Julia Hoefele3‡
Julia Hoefele3‡  Miriam Schmidts1,2,13*‡
Miriam Schmidts1,2,13*‡- 1Genome Research Division, Human Genetics Department, Radboud University Medical Center, Nijmegen, Netherlands
- 2Pediatric Genetics Division, Center for Pediatrics and Adolescent Medicine, University Hospital Freiburg, Freiburg University Faculty of Medicine, Freiburg, Germany
- 3Institute of Human Genetics, Klinikum rechts der Isar, Technical University of Munich, School of Medicine, Munich, Germany
- 4Department of Nephrology, Klinikum rechts der Isar, Technical University of Munich, School of Medicine, Munich, Germany
- 5Cellular and Molecular Research Center, Sabzevar University of Medical Sciences, Sabzevar, Iran
- 6Department of Medical Genetics, Next Generation Genetic Polyclinic, Mashhad, Iran
- 7Children and Adolescents Health Research Center, Research Institute of Cellular and Molecular Science in Infectious Diseases, Zahedan University of Medical Sciences, Zahedan, Iran
- 8Department of Biology, University of Sistan and Baluchestan, Zahedan, Iran
- 9Department of Medical Genetics and Molecular Biology, School of Medicine, Iran University of Medical Sciences, Tehran, Iran
- 10Pediatric Nephrology, Kidney Transplantation Complications Research Center, Mashhad University of Medical Sciences, Mashhad, Iran
- 11Department of Pediatrics, Faculty of Medicine, Mashhad Medical Sciences, Islamic Azad University, Mashhad, Iran
- 12Institute of Genetic Epidemiology, Faculty of Medicine and Medical Center - University of Freiburg, Freiburg, Germany
- 13Center for Integrative Biological Signaling Studies, University of Freiburg, Freiburg, Germany
- 14Bioinformatics Group, Department of Computer Science, University of Freiburg, Freiburg, Germany
- 15Next Generation Genetic Polyclinic, Mashhad, Iran
- 16Genetics Research Centre, Molecular and Clinical Sciences Institute, St. George's University, London, United Kingdom
Background: Steroid resistant nephrotic syndrome (SRNS) represents a significant renal disease burden in childhood and adolescence. In contrast to steroid sensitive nephrotic syndrome (SSNS), renal outcomes are significantly poorer in SRNS. Over the past decade, extensive genetic heterogeneity has become evident while disease-causing variants are still only identified in 30% of cases in previously reported studies with proportion and type of variants identified differing depending on the age of onset and ethnical background of probands. A genetic diagnosis however can have implications regarding clinical management, including kidney transplantation, extrarenal disease manifestations, and, in some cases, even causal therapy. Genetic diagnostics therefore play an important role for the clinical care of SRNS affected individuals.
Methodology and results: Here, we performed NPHS2 Sanger sequencing and subsequent exome sequencing in 30 consanguineous Iranian families with a child affected by SRNS with a mean age of onset of 16 months. We identified disease-causing variants and one variant of uncertain significance in 22 families (73%), including variants in NPHS1 (30%), followed by NPHS2 (20%), WT1 (7%) as well as in NUP205, COQ6, ARHGDIA, SGPL1, and NPHP1 in single cases. Eight of these variants have not previously been reported as disease-causing, including four NPHS1 variants and one variant in NPHS2, ARHGDIA, SGPL1, and NPHP1 each.
Conclusion: In line with previous studies in non-Iranian subjects, we most frequently identified disease-causing variants in NPHS1 and NPHS2. While Sanger sequencing of NPHS2 can be considered as first diagnostic step in non-congenital cases, the genetic heterogeneity underlying SRNS renders next-generation sequencing based diagnostics as the most efficient genetic screening method. In accordance with the mainly autosomal recessive inheritance pattern, diagnostic yield can be significantly higher in consanguineous than in outbred populations.
Introduction
Mammalian kidneys fulfill a tremendous job by filtrating enormous amounts of blood without loss of essential components such as proteins in the urine. The glomerular filtration barrier consisting of three major layers [podocyte foot processes and the slit diaphragm, the glomerular basement membrane (GBM), and glomerular endothelial cells (GEC)] hereby ensures that proteins larger than 60 kD remain within the blood vessels. Smaller filtrated proteins are subsequently largely re-absorbed within the tubular system. Failure of this fine-tuned system results in proteinuria. Loss of large amounts of protein in the urine can result in nephrotic syndrome, characterized by proteinuria of more than >3.5 g per 1.73 m2 body surface area per day, hypoproteinemia (serum albumin is reduced to <2.5 g/dl), hyperlipoproteinemia, and edema. Acute complications include thrombosis, pleural effusions and ascites. Over time, growth restriction in children as well as progressive loss of podocytes with subsequent decline in renal function become evident (1). Two main clinical entities of nephrotic syndrome are distinguished: steroid sensitive nephrotic syndrome (SSNS) and steroid resistant nephrotic syndrome (SRNS). Steroid resistant nephrotic syndrome is defined as persistent nephrotic-range proteinuria after 4 weeks of oral prednisone at 60 mg/m2 per day (2). Steroid resistant nephrotic syndrome is a rare condition, however represents one of the most common causes of childhood-onset kidney failure, occurring as frequently as 1:10,000 in some populations (3). Histopathological findings are most often focal segmental glomerulosclerosis (FSGS) (4). Disease-causing variants in over 60 genes have been described to date to cause monogenic forms of SRNS, explaining approximately one-third of all SRNS cases with an age of onset <25 years (5). Many disease-associated genes encode for proteins essential for proper podocyte function. Most commonly, disease-causing variants affecting the slit diaphragm proteins Nephrin (NPHS1) and Podocin (NPHS2) or the transcription factor WT1 or LAMB2 are identified (6). Hereditary SRNS usually does not respond to immunosuppressive treatment and, in contrast to SSNS, has a poor prognosis with regards to renal survival: 10 years after onset of proteinuria, SSNS patients experience end-stage kidney failure (ESKF) in <5% of cases while ESKF occurs in 50% of individuals with sporadic SRNS and 80% of genetically proven SRNS cases (4, 7). However, disease re-occurrence after kidney transplantation is less likely in most hereditary SRNS cases compared to sporadic SRNS cases (8). Identification of an underlying genetic cause in SRNS individuals therefore has direct therapeutic consequences. Childhood-onset SRNS occurs more frequently in consanguineous populations as it is often autosomal recessively inherited (9, 10).
In this study, we describe the genetic findings using exome sequencing (ES) in 29 Iranian children with SRNS from consanguineous parents with as well as one Iranian parent pair where only insufficient DNA of the affected child was available.
Methods
Study population
The clinical diagnosis of SRNS was defined as absence of complete remission after 4 weeks of daily prednisone therapy at a dose of 60 mg/m2 per day for all affected individuals. Informed consent for genetic diagnostics was obtained from all participants of this study or their legal guardians and genetic testing was conducted in accordance with the standards of the 2013 Helsinki declaration. This study was approved by the local ethics committees of Nijmegen, Netherlands, Freiburg und Munich, Germany and samples processed under the Radboudumc Diagnostics Innovation programme (CMO2006-048) to establish a genetic diagnosis underlying the patient's health disturbances.
DNA extraction
Genomic DNA was extracted from whole blood, using standard salting out method. The concentration of DNA was measured by Qubit 2.0 (Life Technologies, Carlsbad, CA, USA).
PCR and Sanger sequencing
Samples were first tested for the presence of previously reported recurrent (likely) disease-causing variants in NPHS2 exons 5 and 7 (11) using PCR followed by Sanger sequencing. Likewise, segregation of variants identified was verified by PCR and Sanger sequencing in parents unless trio (i.e., case-parent) exome analysis had been performed. Conventional PCR was performed by Taq polymerase (Roche, Mannheim, Germany) based on manufacturer's instruction. Primer sequences are available upon request.
Exome sequencing
Two to five micrograms of DNA were subjected to ES and exome capture was performed using Agilent SureSelect Human All Exon V6 Kit. Paired-end sequencing on a HiSeq 2500 Genome Analyzer (Illumina, San Diego, CA, USA) was performed and build hg19 of the UCSC Genome Browser was used as a reference genome. VarScan version 2.2.5, Mutect, and GATK Somatic Indel Detector were used to detect SNVs and indels, respectively. Filtering was performed as previously described (12). In brief, a minor allele frequency cutoff of 1% in gnomAD (gnomAD, http://gnomad.broadinstitute.org) was applied and the remaining variants were filtered first for genes in which SRNS disease-causing variants had been previously published (13). If no disease-causing variants could be identified using this strategy, BAM files were manually inspected for homozygous CNVs in these genes and exome data further analyzed for disease-causing variants in other genes not previously associated with SRNS. Variants rated as “likely pathogenic” or “pathogenic” as per ACMG criteria and current amendments (14) are summarized as “disease-causing” throughout the text.
Results
Molecular genetic diagnostics were undertaken for 30 Iranian unrelated consanguineous families with at least one child clinically diagnosed with SRNS. In one family (sample 3), insufficient DNA of the affected child was available for genetic testing by ES, therefore both parents were tested by ES instead, followed by Sanger sequencing of the child.
In total, we identified an underlying genetic cause in 21/30 families and a variant of uncertain significance (VUS) in one family (diagnostic yield: 73%) (Table 1). All identified disease-causing variants locate to genes known to cause monogenic SRNS when dysfunctional with the exception of one case carrying a variant in a nephronophthisis gene (NPHP1) instead. The gene most frequently affected was NPHS1 (MIM# 602716; nine families: 38% of solved families and 30% of all families), followed by NPHS2 (MIM# 604766; six families: 29% of solved families, 20% of all families). Two cases were found to be caused by WT1 variants (MIM# 607102; 10% of solved families, 7% of all families), while variants in NUP205 (MIM# 614352), COQ6 (MIM# 614647), ARHGDIA (MIM# 601925), and SGPL1 (MIM# 603729) were identified in single families (5% of solved families, 3% of all families). In addition, we identified a disease-causing deletion in NPHP1 [MIM# 607100; gene previously associated with nephronophthisis (NPHP)] in one family (5% of solved families, 3% of all families; Figure 1).
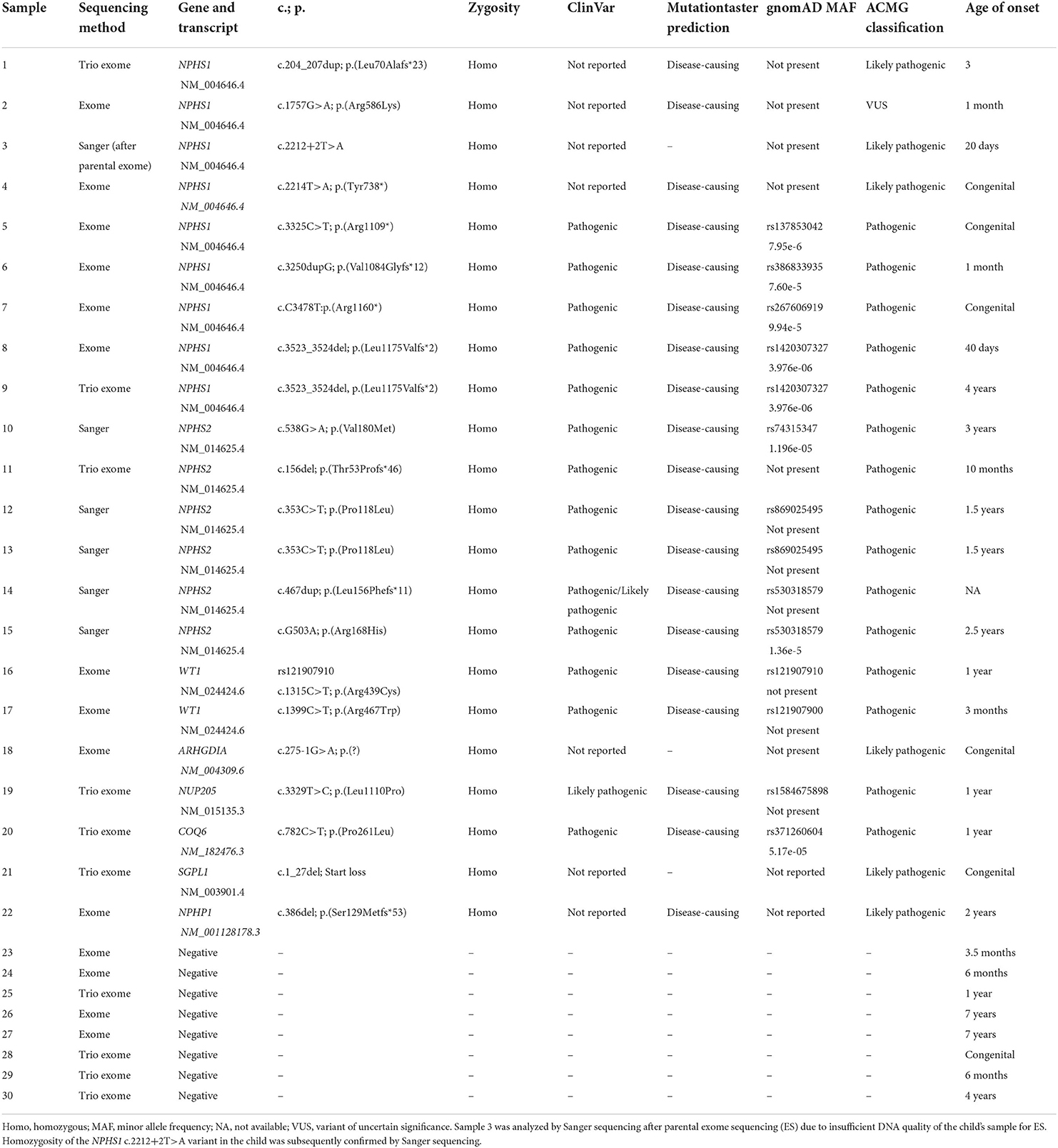
Table 1. Summary of genetic findings.
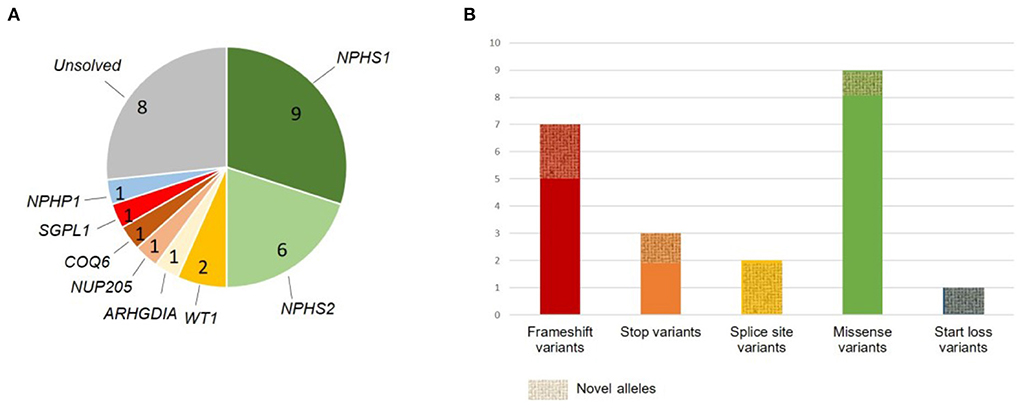
Figure 1. Summary of genetic findings. (A) Gene-specific distribution of identified disease-causing variants and one variant of uncertain significance; unsolved cases are shown in gray. (B) Variant type distribution. Alleles not previously reported are marked as novel alleles.
In total, we detected seven variants not previously reported in ClinVar or HGMD, including four different NPHS1 variants (three variants classified as disease-causing and one VUS) and one novel variant in ARHGDIA, SGPL1, and NPHP1 each (all classified as disease-causing). Amongst the known SRNS disease alleles, we identified NPHS2 c.353C>T (p.Pro118Leu) as well as NPHS1 c.3523_3524del (p.Leu1175Valfs*2) in two unrelated families each. Three of the four novel NPHS1 variants as well as variants in ARHGDIA, SGPL1, and NPHP1 all represent novel disease-causing variants as defined by ACMG criteria.
All identified variants except the two variants identified in WT1 were present in a homozygous state in affected children. Parents were heterozygous carriers of the respective variant. Nine variants represent stop or frameshift variants in NPHS1, NPHS2, and NPHP1 (45%), one allele was a −1 canonical splice site variant in ARHGDIA, one a +2 splice site variant in NPHS1 classified as likely pathogenic according to ACMG criteria, one small deletion causing a start loss in SGPL1, and the remaining variants were missense variants. All but 1 variant (NPHS1 c.1757G>A, p.Arg586Lys, rs730880174; classified as VUS) were classified as disease-causing according to the recommendations of ACMG. Interestingly, rs730880174 in NPHS1 affecting likewise amino acid 586 but changing it into a glycine instead of a lysine has been reported as pathogenic in ClinVar. Likewise, NPHP1 c.386del (p.Ser129Metfs*53) affects the same amino acid position as rs757139057 (c.385_386del), reported pathogenic in ClinVar.
Both heterozygous missense variants we identified in WT1 have been reported as pathogenic before with WT1 c.1315C>T, p.Arg439Cys identified in Denys-Drash syndrome and Meacham syndrome as well as in a Japanese patient with SRNS (15). Variant information for all identified variants and localization of variants in COQ6, NUP205, ARHGDIA, and SGPL1 on protein level are shown in Table 1 and Figures 1, 2. A graphical summary of subcellular localisations and functions of proteins encoded for by genes in which we detected disease-causing variants in this study is shown in Figure 3.

Figure 2. Location of disease-causing variants in COQ6, NUP205, ARHGDIA, and SGPL1 on protein level. (A) COQ6, (B) NUP205, (C) ARHDIA, and (D) SGPL1 variants. Previously reported variants are shown in blue, variants first reported in this publication marked in red.
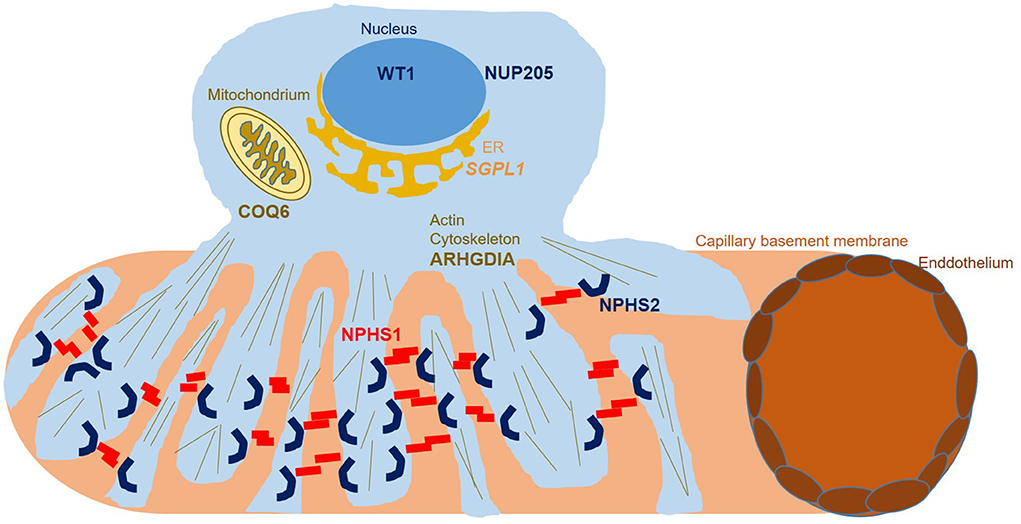
Figure 3. Podocyte subcellular localization of proteins encoded for by genes found to carry disease alleles in this study. While WT1 functions as a transcription factor in the nucleus, NUP205 functions within the nuclear pore complex, enabling transport between the nucleus and the cytoplasm. SGPL1 represents an endoplasmic reticulum based enzyme catalyzing sphingolipid breakdown resulting in cleavage of the lipid-signaling molecule sphingosine-1-phosphate. COQ6 represents a flavin-dependent monooxygenase essential for biosynthesis of coenzyme Q10 which serves as a redox carrier in the mitochondrial respiratory chain as well as an antioxidant protecting cells from reactive oxygen damage. ARHGDIA sequesters Rho-GTPases in an inactive state in the cytosol, controlling RhoA, Rac1, and Cdc42 levels and influencing actin dynamics. NPHS1 and NPHS2 localize to the podocyte foot process membrane, enabling proper function of the glomerular slit diaphragm.
Discussion
We identified disease-causing variants according to ACMG criteria in genes previously reported to cause childhood SRNS in 21 out of 30 families (70%) and a VUS in one out of 30 families (3%), exceeding the diagnostic yield reported in other larger NGS studies by far (4, 16–18) (Table 2). However, in contrast to outbred study population described in those studies, all of the families referred to us for genetic diagnostics were consanguineous. In addition, it has been previously shown that the diagnostic yield in SRNS is highest in children with congenital nephrotic syndrome and significantly lower with increasing age at first manifestation during the first 6 years of life (18). In line with this, average age at first manifestation in our cohort was 17.5 months and all of our genetically solved cases were below 5 years of age when showing first SRNS symptoms while two of the genetically unsolved cases were 7 years old (Tables 1, 2).

Table 2. Comparison of genetic findings in different SRNS cohorts.
Congenital cases are often caused by disease-causing variants in NPHS1 and we identified such variants in two cases as well as disease-causing variants in SGPL1 or ARHGDIA in one case, respectively. Age of onset was overall lower in NPHS1 cases compared to NPHS2 cases, as expected. In total, we most frequently detected biallelic variants in NPHS1 (38% of solved families and 30% of all families) followed by NPHS2 (29% of solved families and 20% of all families), in accordance with previous findings that NPHS1 and NPHS2 variants are amongst the most common underlying genetic causes in childhood SRNS in non-Iranian populations (17). This is also in line with findings reported by Basiratnia et al. (20) who identified disease-causing variants in NPHS2 in 15 out of 49 Iranian SRNS cases (31%) and 57% of familiar Iranian SRNS cases. Three of these 15 cases (20%) carried the NPHPS2 p.Pro118Leu variant homozygously while we detected this variant in 2 of 6 cases (30% of all our NPHS2 cases), indicating it represents a fairly common Iranian disease allele. We did not detect the previously described Iranian NPHS2 founder variant p.Arg238Ser reported by Basiratnia et al. (20). Interestingly, the proportion of childhood SRNS cases resulting from biallelic disease-causing NPHS2 variants seems fairly similar across many populations such as European populations [12–19% in sporadic cases (21, 22), 26% in familiar cases (23)], and 15% in Indian cases (24), while disease-causing NPHS2 variants are less frequently identified in Chinese, Japanese, or South African probands with SRNS (3–8%) (19, 25–29).
Although we were able to genetically solve 25% of cases by starting with Sanger sequencing of NPHS2, in the days of broad availability of NGS, performing NGS panel or ES as primary diagnostic step seems more efficient, given the overall high genetic variability in SRNS. Using ES in NPHS2 negative cases, we identified multiple biallelic disease-causing NPHS1 variants, two heterozygous disease-causing WT1 variants as well as biallelic disease-causing variants in COQ6, ARHGDIA, NUP205, and SGPL. Disease-causing variants in COQ6 represent a very rare cause of SRNS with until completion of this study less than 20 disease-causing alleles reported to date in the literature (30, 31) (Figure 2). Very recently, Drovandi et al. reported additional COQ6 cases as part of a large cohort of 116 individuals with primary coenzyme Q10 deficiency in which CoQ10 replacement had beneficial effects regarding disease progression (32, 33). Disease-causing variants in ARHGDIA likewise represent a rare cause of SRNS (Figure 2). ARHGDIA (Rho GDP-Dissociation Inhibitor Alpha) is expressed in podocytes playing an important role in maintaining Rho-GTPases in their inactive state and loss of function subsequently disturbs the actin-cytoskeleton arrangement, resulting in early-onset proteinuria and progressive loss of renal function in humans and mice (34–36). In line with findings in cases previously reported in the literature, our case presented with congenital nephrotic syndrome, confirming a crucial role of ARHGDIA for podocyte integrity. We further identified a homozygous small deletion encompassing exon 2 (the first coding exon) resulting in a start-loss in SGPL1 in a case with syndromic congenital SRNS. SGPL1 encodes sphingosine-1-phosphate lyase-1, a ubiquitously expressed enzyme located at the endoplasmic reticulum. Loss of function results in syndromic SRNS and adrenal insufficiency in humans with 20 disease alleles published to date (37, 38) (Figure 2). We also detected a homozygous NUP205 missense variant, previously reported as disease-causing, in a case with disease onset at 12 months of age. NUP205 encodes for a nuclear pore complex protein and a different homozygous disease-causing missense variant in NUP205 has been previously reported in a Turkish sib-pair affected by SRNS (39). Additionally, NUP205 loss of function has been previously associated with left-right patterning defects in humans (40) (Figure 2) as well as ciliogenesis defects in a frog knockdown model system (41). Our case did not exhibit any overt laterality disturbances. A summary of subcellular localisations and functions of proteins encoded for by genes in which variants have been identified in this cohort is shown in Figure 3.
Last, we identified a phenocopy in a case presenting clinically with SRNS where we identified a homozygous disease-causing NPHP1 frameshift variant. NPHP1 loss of function is a frequent cause of NPHP, with a deletion of NPHP1 representing the most frequent disease allele (42). Interestingly, Kirsty et al. have previously published a Filipino family with nephrotic syndrome but genetic diagnostics revealing disease-causing variants in NPHP4 associated with NPHP (43). While proteinuria occurs frequently in NPHP, usually it is not of the nephrotic range and instead of a glomerular protein pattern observed in SRNS, proteinuria in NPHP rather shows a tubular pattern. For our case, unfortunately no clinical reports stating what type of proteins were lost in the urine are available. It therefore remains unclear if the individual suffered from tubular or glomerular proteinuria. We cannot exclude that in addition to the genetically established NPHP, the individual independently also suffered from SRNS of another origin, including a non-monogenic form of SRNS.
Overall, ES was very efficient in our cohort of consanguineous early-onset SRNS cases to establish a genetic diagnosis. Nevertheless, several cases remain unsolved, putatively due to variants outside of the coding regions, warranting genome sequencing (with RNA sequencing) or due to the presence of a polygenic or non-genetic mechanism.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Freiburg University ethics commission, votum 122/20 and samples processed under the Radboudumc Diagnostics Innovation programme (CMO2006-048) to establish a genetic diagnosis. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
MN, AR, EK, SS, TB, AS, AR, PN, SB, MK, and AA recruited the probands and/or were involved in their clinical care. MN, KMR, TM, RB, IS, SS, AK, RB, and MS conducted data analysis. MN and MS conceived the study. MS drafted the manuscript. MN and JH supervised the study. All authors read and approved the final version of the manuscript.
Funding
IS acknowledges funding from the Freiburg University Medical Faculty Hospital Berta-Ottenstein Clinical fellowship programme. MS acknowledges funding form the European Research Council (ERC): ERC starting grant TREATCilia (grant agreement no. 716344) and MS and AK received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 431984000—SFB 1453 (CRC Nephgen). MS, AK, and RB acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Excellence Initiative CIBSS—EXC-2189—Project ID 390939984.
Acknowledgments
The authors thank the families for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.974840/full#supplementary-material
Abbreviations
DNA, deoxyribonucleic acid; ES, exome sequencing; GATK, genome analysis toolkit; LOF, loss of function; OFC, occipitofrontal circumference; PCR, polymerase chain reaction; SD, standard deviation; SNV, single nucleotide variant; SRNS, steroid resistant nephrotic syndrome; SSNS, steroid sensitive nephrotic syndrome.
References
1. Sato M, Ishikura K, Ando T, Kikunaga K, Terano C, Hamada R, et al. Prognosis and acute complications at the first onset of idiopathic nephrotic syndrome in children: a nationwide survey in Japan (JP-SHINE study). Nephrol Dial Transplant. (2021) 36:475–81. doi: 10.1093/ndt/gfz185
2. The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J Pediatr. (1981). 98:561–4. doi: 10.1016/S0022-3476(81)80760-3
3. Machuca E, Benoit G, Nevo F, Tete MJ, Gribouval O, Pawtowski A, et al. Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. (2010) 21:1209–17. doi: 10.1681/ASN.2009121309
4. Trautmann A, Lipska-Zietkiewicz BS, Schaefer F. Exploring the clinical and genetic spectrum of steroid resistant nephrotic syndrome: the podonet registry. Front Pediatr. (2018) 6:200. doi: 10.3389/fped.2018.00200
5. Mao Y, Schneider R, van der Ven PFM, Assent M, Lohanadan K, Klambt V, et al. Recessive mutations in SYNPO2 as a candidate of monogenic nephrotic syndrome. Kidney Int Rep. (2021) 6:472–83. doi: 10.1016/j.ekir.2020.10.040
6. Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics. (2007) 119:e907–19. doi: 10.1542/peds.2006-2164
7. Srivastava T, Simon SD, Alon US. High incidence of focal segmental glomerulosclerosis in nephrotic syndrome of childhood. Pediatr Nephrol. (1999) 13:13–8. doi: 10.1007/s004670050555
8. Rheault MN, Gbadegesin RA. The genetics of nephrotic syndrome. J Pediatr Genet. (2016) 5:15–24. doi: 10.1055/s-0035-1557109
9. Frishberg Y, Rinat C, Megged O, Shapira E, Feinstein S, Raas-Rothschild A. Mutations in NPHS2 encoding podocin are a prevalent cause of steroid-resistant nephrotic syndrome among Israeli-Arab children. J Am Soc Nephrol. (2002) 13:400–5. doi: 10.1681/ASN.V132400
10. Lipska-Zietkiewicz BS, Ozaltin F, Holtta T, Bockenhauer D, Berody S, Levtchenko E, et al. Genetic aspects of congenital nephrotic syndrome: a consensus statement from the ERKNet-ESPN inherited glomerulopathy working group. Eur J Hum Genet. (2020) 28:1368–78. doi: 10.1038/s41431-020-0642-8
11. Otukesh H, Ghazanfari B, Fereshtehnejad SM, Bakhshayesh M, Hashemi M, Hoseini R, et al. NPHS2 mutations in children with steroid-resistant nephrotic syndrome. Iran J Kidney Dis. (2009) 3:99–102.
12. Najafi M, Kordi-Tamandani DM, Behjati F, Sadeghi-Bojd S, Bakey Z, Karimiani EG, et al. Mimicry and well known genetic friends: molecular diagnosis in an Iranian cohort of suspected Bartter syndrome and proposition of an algorithm for clinical differential diagnosis. Orphanet J Rare Dis. (2019) 14:41. doi: 10.1186/s13023-018-0981-5
13. Lipska-Zietkiewicz BS. Genetic steroid-resistant nephrotic syndrome overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. GeneReviews((R)). Seattle, WA: University of Washington, Seattle (1993).
14. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. (2018) 39:1517–24. doi: 10.1002/humu.23626
15. Nagano C, Yamamura T, Horinouchi T, Aoto Y, Ishiko S, Sakakibara N, et al. Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Sci Rep. (2020) 10:270. doi: 10.1038/s41598-019-57149-5
16. Tan W, Lovric S, Ashraf S, Rao J, Schapiro D, Airik M, et al. Analysis of 24 genes reveals a monogenic cause in 11.1% of cases with steroid-resistant nephrotic syndrome at a single center. Pediatr Nephrol. (2018) 33:305–14. doi: 10.1007/s00467-017-3801-6
17. Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2018) 13:53–62. doi: 10.2215/CJN.04120417
18. Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2015) 26:1279–89. doi: 10.1681/ASN.2014050489
19. Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2017) 32:1181–92. doi: 10.1007/s00467-017-3590-y
20. Basiratnia M, Yavarian M, Torabinezhad S, Erjaee A. NPHS2 gene in steroid-resistant nephrotic syndrome: prevalence, clinical course, and mutational spectrum in South-West Iranian children. Iran J Kidney Dis. (2013) 7:357–62.
21. Ruf RG, Fuchshuber A, Karle SM, Lemainque A, Huck K, Wienker T, et al. Identification of the first gene locus (SSNS1) for steroid-sensitive nephrotic syndrome on chromosome 2p. J Am Soc Nephrol. (2003) 14:1897–900. doi: 10.1097/01.ASN.0000070070.03811.02
22. Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. (2004) 15:722–32. doi: 10.1097/01.ASN.0000113552.59155.72
23. Caridi G, Trivelli A, Sanna-Cherchi S, Perfumo F, Ghiggeri GM. Familial forms of nephrotic syndrome. Pediatr Nephrol. (2010) 25:241–52. doi: 10.1007/s00467-008-1051-3
24. Vasudevan A, Siji A, Raghavendra A, Sridhar TS, Phadke KD. NPHS2 mutations in Indian children with sporadic early steroid resistant nephrotic syndrome. Indian Pediatr. (2012) 49:231–3. doi: 10.1007/s13312-012-0057-x
25. Yu Z, Ding J, Huang J, Yao Y, Xiao H, Zhang J, et al. Mutations in NPHS2 in sporadic steroid-resistant nephrotic syndrome in Chinese children. Nephrol Dial Transplant. (2005) 20:902–8. doi: 10.1093/ndt/gfh769
26. Maruyama K, Iijima K, Ikeda M, Kitamura A, Tsukaguchi H, Yoshiya K, et al. NPHS2 mutations in sporadic steroid-resistant nephrotic syndrome in Japanese children. Pediatr Nephrol. (2003) 18:412–6. doi: 10.1007/s00467-003-1120-6
27. Ogino D, Hashimoto T, Hattori M, Sugawara N, Akioka Y, Tamiya G, et al. Analysis of the genes responsible for steroid-resistant nephrotic syndrome and/or focal segmental glomerulosclerosis in Japanese patients by whole-exome sequencing analysis. J Hum Genet. (2016) 61:137–41. doi: 10.1038/jhg.2015.122
28. Sako M, Nakanishi K, Obana M, Yata N, Hoshii S, Takahashi S, et al. Analysis of NPHS1, NPHS2, ACTN4, and WT1 in Japanese patients with congenital nephrotic syndrome. Kidney Int. (2005) 67:1248–55. doi: 10.1111/j.1523-1755.2005.00202.x
29. Chernin G, Heeringa SF, Gbadegesin R, Liu J, Hinkes BG, Vlangos CN, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2008) 23:1455–60. doi: 10.1007/s00467-008-0861-7
30. Park E, Lee C, Kim NKD, Ahn YH, Park YS, Lee JH, et al. Genetic study in korean pediatric patients with steroid-resistant nephrotic syndrome or focal segmental glomerulosclerosis. J Clin Med. (2020) 9:2013. doi: 10.3390/jcm9062013
31. Wang N, Zheng Y, Zhang L, Tian X, Fang Y, Qi M, et al. A family segregating lethal primary coenzyme Q10 deficiency due to two novel COQ6 variants. Front Genet. (2021) 12:811833. doi: 10.3389/fgene.2021.811833
32. Justine Perrin R, Rousset-Rouviere C, Garaix F, Cano A, Conrath J, Boyer O, et al. COQ6 mutation in patients with nephrotic syndrome, sensorineural deafness, and optic atrophy. JIMD Rep. (2020) 54:37–44. doi: 10.1002/jmd2.12068
33. Drovandi S, Lipska-Zietkiewicz BS, Ozaltin F, Emma F, Gulhan B, Boyer O, et al. Variation of the clinical spectrum and genotype-phenotype associations in Coenzyme Q10 deficiency associated glomerulopathy. Kidney Int. (2022) 2022:S0085-2538(22)00338-6. doi: 10.1016/j.kint.2022.02.040
34. Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest. (2013) 123:3243–53. doi: 10.1172/JCI69134
35. Narayan A, Karunakar P, Krishnamurthy S, Deepthi B, Jose D. Homozygous ARHGDIA gene mutation in an 11-month-old infant with steroid-resistant nephrotic syndrome. Indian J Pediatr. (2022) 89:411. doi: 10.1007/s12098-021-04053-4
36. Gupta IR, Baldwin C, Auguste D, Ha KC, El Andalousi J, Fahiminiya S, et al. ARHGDIA: a novel gene implicated in nephrotic syndrome. J Med Genet. (2013) 50:330–8. doi: 10.1136/jmedgenet-2012-101442
37. Lovric S, Goncalves S, Gee HY, Oskouian B, Srinivas H, Choi WI, et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest. (2017) 127:912–28. doi: 10.1172/JCI89626
38. Maharaj A, Theodorou D, Banerjee II, Metherell LA, Prasad R, et al. A sphingosine-1-phosphate lyase mutation associated with congenital nephrotic syndrome and multiple endocrinopathy. Front Pediatr. (2020) 8:151. doi: 10.3389/fped.2020.00151
39. Braun DA, Sadowski CE, Kohl S, Lovric S, Astrinidis SA, Pabst WL, et al. Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet. (2016) 48:457–65. doi: 10.1038/ng.3512
40. Chen W, Zhang Y, Yang S, Shi Z, Zeng W, Lu Z, et al. Bi-Allelic mutations in NUP205 and NUP210 are associated with abnormal cardiac left-right patterning. Circ Genom Precis Med. (2019) 12:e002492. doi: 10.1161/CIRCGEN.119.002492
41. Marquez J, Bhattacharya D, Lusk CP, Khokha MK. Nucleoporin NUP205 plays a critical role in cilia and congenital disease. Dev Biol. (2021) 469:46–53. doi: 10.1016/j.ydbio.2020.10.001
42. Stokman M, Lilien M, Knoers N. Nephronophthisis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., editors. GeneReviews((R)). Seattle, WA: University of Washington, Seattle (1993).
Keywords: nephrotic syndrome, Iran, SRNS, ARHGDIA, NUP205, COQ6, SGPL1
Citation: Najafi M, Riedhammer KM, Rad A, Torbati PN, Berutti R, Schüle I, Schroda S, Meitinger T, Ćomić J, Bojd SS, Baranzehi T, Shojaei A, Azarfar A, Khazaei MR, Köttgen A, Backofen R, Karimiani EG, Hoefele J and Schmidts M (2022) High detection rate for disease-causing variants in a cohort of 30 Iranian pediatric steroid resistant nephrotic syndrome cases. Front. Pediatr. 10:974840. doi: 10.3389/fped.2022.974840
Received: 21 June 2022; Accepted: 11 August 2022;
Published: 22 September 2022.
Edited by:
Nina Mann, Boston Children‘s Hospital and Harvard Medical School, United StatesReviewed by:
Daw-Yang Hwang, National Health Research Institutes, TaiwanBixia Zheng, Nanjing Children's Hospital, China
Copyright © 2022 Najafi, Riedhammer, Rad, Torbati, Berutti, Schüle, Schroda, Meitinger, Ćomić, Bojd, Baranzehi, Shojaei, Azarfar, Khazaei, Köttgen, Backofen, Karimiani, Hoefele and Schmidts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam Schmidts, miriam.schmidts@uniklinik-freiburg.de
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
§ORCID: Korbinian M. Riedhammer orcid.org/0000-0002-7503-5801