An overview on the RSV-mediated mechanisms in the onset of non-allergic asthma
 Sara Manti
Sara Manti Giovanni Piedimonte
Giovanni Piedimonte- 1Pediatric Pulmonology Unit, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 2Pediatric Unit, Department of Human Pathology of Adult and Childhood Gaetano Barresi, University of Messina, Messina, Italy
- 3Department of Pediatrics, Biochemistry and Molecular Biology, Tulane University, New Orleans, LA, United States
Respiratory syncytial virus (RSV) infection is recognized as an important risk factor for wheezing and asthma, since it commonly affects babies during lung development. While the role of RSV in the onset of atopic asthma is widely recognized, its impact on the onset of non-atopic asthma, mediated via other and independent causal pathways, has long been also suspected, but the association is less clear. Following RSV infection, the release of local pro-inflammatory molecules, the dysfunction of neural pathways, and the compromised epithelial integrity can become chronic and influence airway development, leading to bronchial hyperreactivity and asthma, regardless of atopic status. After a brief review of the RSV structure and its interaction with the immune system and neuronal pathways, this review summarizes the current evidence about the RSV-mediated pathogenic pathways in predisposing and inducing airway dysfunction and non-allergic asthma development.
Introduction
Respiratory syncytial virus (RSV) is the most common respiratory pathogen in infants and young children worldwide (1). Prospective epidemiologic studies support the association between RSV infection and short-term pulmonary morbidities during infancy, such as lower respiratory tract infections (LRTIs), and long-term pulmonary morbidities during childhood, such as airway hyperresponsiveness, wheezing, and asthma (2–9). Consistent literature findings evidence that RSV-caused asthma is closely related to the atopic constitution since a T helper (Th)2 dominance in immune response has been commonly reported. Studies support an intrisic “Th2-trophic” effects of the RSV as it: (1) stimulates the T cell responses to inhalant allergens, by triggering the local Th2 cytokine at the airway mucosa; (2) promotes eosinophils recruitment at lesional sites in the airway mucosa; (10, 11) and generates a Th2-polarized RSV-specific immunological memory, which, following a RSV reinfection, leads to intense infiltrates of eosinophils and Th2 cells secreting interleukin (IL)-4 in the lung tissue (12).
Whether the role of RSV in the onset of atopic asthma is widely recognized, its impact on the onset of non-atopic asthma, mediated via other and independent causal pathways, has long been suspected, but the association is less clear (2–9). The RSV-mediated persistent inflammation and airway hyperreactivity probably result from changes in the local and systemic immune response and alterations of the neural airway pathways that can occur in parallel and/or at different times (13–15). However, all these changes appear reversible, suggesting a transient respiratory dysfunction rather than chronic and irreversible damage, commonly featuring asthma (15). This review aims to summarize the current evidence about the RSV-mediated pathogenic pathways in predisposing and inducing airway dysfunction and non-allergic asthma development.
RSV structure and host cell interaction
Respiratory syncytial virus is a non-segmented, negative-sense, single-stranded RNA virus belonging to the Paramyxoviridae family whose genome is constituted by ten sequentially arranged genes encoding the following eleven proteins: three transmembrane surface glycoproteins [attachment G protein, fusion (F) protein, the small hydrophobic (SH) protein]; three genomic RNA-associated proteins forming the nucleocapsid [large (L) polymerase, N protein, phospo (P) protein]; two non-structural proteins (NS) (NS1 and NS2); two transcription and replication factors (M2-1, M2-2); and one unglycosylated matrix (M) protein (16, 17). The L, N, P, M-1, and M-2 proteins and the genomic RNA participate in creating the ribonucleoprotein (RNP) complex, and they are required for viral transcription and replication. The soluble form of G protein (Gs) and the NSs (NS1 and NS2) proteins downregulate the antiviral response (18).
While the proteins G and F are crucial for virus attachment and fusion, respectively; the SH protein, a pentameric ion channel, is involved in permeabilizing cell membrane and delaying apoptosis in infected cells (17).
Interestingly, the cytoskeleton plays a supporting role in the infectious cycle of RSV (19). The cytoskeleton is made up of the three following proteins: actin, intermediate filaments, and microtubules. Accounting for 5–10% protein, actin is the most abundant cytoskeletal protein, and it is present both in a globular monomeric form (G-actin) and filamentous form (F-actin) with a different polarity needed for intracellular transport. In response to varying stimuli, actin microfilaments undergo rapid cycles of polymerization/depolymerization to modulate shape changes, cell contraction and migration (20–25). Neural-Wiskott-Aldrich syndrome protein (N-WASP), a member of the WASP family, and ARP2/3 complex have a regulatory role in the actin polymerization (20–25). The role of actin rearrangement in the RSV infection has been confirmed by the evidence that by disrupting actin through cytochalasin D and latrunculin A, a significant decrease in the viral load of RSV occurred (21, 22). Actin is also involved in RSV endocytosis, replication, gene expression, and cell-to-cell spread. Following the virus entry, actin and actin-modulatory proteins facilitate the RSV transcription. Profilin, an actin modulatory protein, is also essential for RSV transcription. Via interaction with RSV M protein, actin mediates budding and virion particle transport (23–25). The crucial role of M protein in trafficking the viral particles emerges from the evidence that the absence of M protein leads to the accumulation of RNP complexes in the cytoplasm; thus, the viral filaments cannot be synthesized (26). The M-containing complex anchors the microtubule organizing center (MTOC) and interacts with the mature RNP, creating a M-RNPs which, in turn, complexes with the cytoplasmic tail of G and F to form the budding mature particles which will move to the plasma membrane (27). While the G protein is not required to generate progeny virus, the F protein is crucial, as the M protein associates and sorts into detergent-resistant membranes (DRMs) only when the F protein is present (27). Specifically, the formation of RSV filaments depends on the F-protein cytoplasmic tail (FCT), especially to a phenyalanine (Phe) residue at position 22, as a mutation in Phe22 causes the inability of RSV F to recruit viral proteins and form filaments (28). To avoid any pitfalls in the spreading of the viral particles, RSV modulates the cytoskeletal and actin rearrangement in creating filopodia, finger-like projections constituted by polymerized actin with free-barbed ends, at which can be added additional actin monomers (29). Cdc42, a GTPase, Rac, Phosphoinositide-3-kinase (PI3K), and Rho mediate the filopodia formation (24, 25, 29). The interaction between RhoA and F protein plays a key role in driving the cell-to-cell spread of RSV and creating syncytia, featuring the RSV infection. Higher expression of RhoA and phospho-myosin light chain (pMLC2) were observed following RSV infection, and the incubation with Rho kinase (ROCK), a regulator of actin activity, reversed this effect (30). Phosphoinositide-3-kinase, a family of cellular kinases, acts as a secondary messenger in modulating the phosphorylation of the serine/threonine kinase Akt. Several studies reported that the viral penetration into host cells depends on PI3K signaling activation, and the PI3K activity requires, in turn, the activation of RhO-family GTPases and actin cytoskeleton reorganization. The role of the PI3K signaling results from the evidence that an impaired activity or inhibition of PI3K affects RSV replication (31).
Lastly, the RSV acts by disrupting intermediate filaments to weak the cell, enhance the cell lysis, and favor the release of the viral progeny in the extracellular space (32).
RSV and immune-pulmonary pathways
RSV infection and innate immune
Respiratory syncytial virus infection elicits a strong systemic and airway immune response, involving neutrophils, natural killers, dendritic cells (DCs), macrophages and monocytes, eosinophils, T lymphocytes, and inducing the release of several pro-inflammatory cytokines and chemokines (33).
Primarily, RSV targets nasal epithelial cells that release pro-inflammatory mediators and recruit immune cells, such as monocytes-macrophages and DCs (33). Monocytes play two crucial roles: they induce the release of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, IL-1, IL-6, IL-8, IL-10, and IL-18; and promote a Th2-impaired immune response with a parallel decrease in lymphocyte maturation and interferon (IFN)-γ production, and a marked release of IL-4 and IL-13 (34). Recent findings suggest that the human innate lymphoid cells (ILCs), especially the type 2 ILCs (ILC2s), promote also the release of IL-4 and IL-13 with a further enhance of the Th2 immune response (33–35). Moreover, the Toll-like receptors (TLRs) present on the membrane of monocytes, trough the interaction with the RSV F protein, induce an enhanced binding of environmental lipopolysaccharides to airway epithelium, a mitogen-activated protein kinase (MAPK) activation, and a further pro-inflammatory cytokine production [(34, 35), Figure 1].
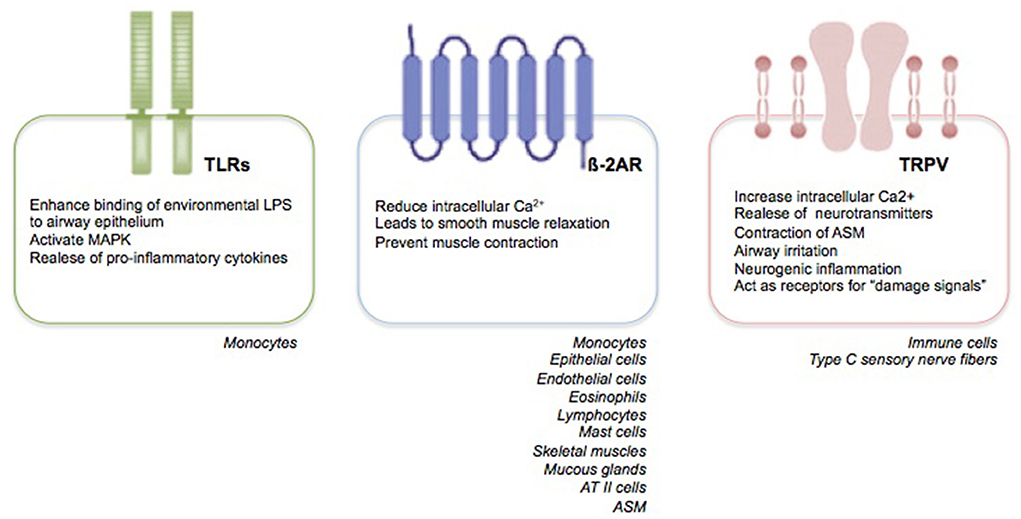
Figure 1. RSV receptors. Expression and function of RSV receptors.
The RSV G protein affects the innate immune response by binding the motif that resembles the CX3C chemokine fractalkine (Fkn). Mutation or deletion of the CX3C-Fkn motif is associated with higher production of IFNs and TNF-α in response to RSV infection (36).
The SH protein is able to activate the inflammasomes which recruit and activate caspase-1, which, in turn, processes pro-IL-1ß and pro-IL-18 to their active forms (37). Moreover, through inhibition of TNF-α signaling, SH protein delays cellular apoptosis (37).
Through NS1/2, RSV blocks the release and acitivity of type I and III IFNs (38). The crucial role of NS1 and NS2 is confirmed by the evidence that the NS genes' deletion significantly reduced RSV replication in IFN-competent cells but not in IFN-deficient cells (39). Moreover, NS1 and NS2 inhibited the IFN-signaling pathway and the formation of “NS-degradasome,” a complex with a protease/proteasomal activity involved in the suppression of IFN signaling (38).
RSV infection and adaptive immune
After 4–7 days of infection, the adaptive immune response is activated; however, studies revealed an impairment in T cells response, DCs activity, and T–DC cells interaction, leading to an ineffective memory T cell response to counteract RSV infection (33, 34). Systemic T cell lymphopenia, especially in CD8+ and CD4+, has been reported in patients with early RSV infection compared to convalescence and in uninfected individuals (33, 34). Moreover, an inverse and significant correlation bewteen T cells count and RSV infection severity has also been documented (33, 34).
Given the G and F proteins, RSV elicits the adaptive immune response by inducing the synthesis of RSV-specific antibodies, immunoglobulin (Ig)A and IgG. The RSV-specific IgA are responsible for the defense of the mucosal surfaces, and, moreover, they downregulate the severity of the first infection as well as prevent the reinfection of the upper respiratory tract (40). The RSV-specific IgGs, produced after the first infection, are involved in the viral clearance (41). However, given to the high variability and glycosylation, RSV can change the G protein profile and escape the immune response. By binding the RSV-specific-IgGs, the soluble form of the G protein reduces the serum concentrations of RSV-IgG (42). Moreover, RSV F and G proteins inhibit mitogen-induced T-cell proliferation and decrease T cells functions (39). Lastly, RSV NS1 and NS2 proteins negatively impact the DC maturation into monocyte-derived DCs and affect their ability to interact with T-cell, resulting in a delay in the acquisition of T-cell memory, thus, in a weak adaptive immune response causing susceptibility to reinfection with RSV throughout life (38).
RSV infection and lung epithelium
From the upper respiratory tract, the virus moves to the lower airways, where it mainly targets ciliated cells and alveolar type II (ATII) cells.
In response to RSV infection, several changes occur at the airway epithelium. It produces cytokines and chemokines that modulate the influx of inflammatory cells into the infected lung tissue. Higher are the cytokines and chemokines levels in the respiratory tract secretions, and more severe is the RSV infection severity. Moreover, RSV, via the action of NS2 protein, induces epithelial cell shedding, which, in turn, accelerates the clearance of the virus-infected cells from airway mucosa but contributes to acute obstruction of the distal airways (43). Also, following the infection of basal cells, RSV promotes the IFN-mediated formation of epithelium with a profound loss of ciliated cells (44).
Intraepithelial DCs, alveolar type I (ATI) cells, basal epithelial cells of the bronchial epithelium, and airway smooth muscle (ASM) cells can also be infected by the virus (40). The RSV-infected ciliated cells release pro-inflammatory cytokines such as TNF-α, IL-33, and thymic stromal lymphopoietin (TSLP), which, in turn, promote a Th2-mediated inflammatory response and the recruitment of neutrophils and eosinophils (45). The RSV activates the neutrophils expressing specific activation markers, such as CD11b, CD18, and CD54, and induces the release of neutrophil elastase by neutrophils. Moreover, the neutrophil apoptosis and neutrophil extracellular trap (NET) are active during infection, and the peak of these activities coincides with the maximum in the viral load and clinical severity (46). Similarly, eosinophils are activated by the RSV and the high levels of leukotriene C4, eosinophil-derived neurotoxin (EDN), and eosinophil cationic protein (ECP) detected in the respiratory tract in RSV bronchiolitis support their role during the acute phase of RSV infection (47).
Respiratory syncytial virus may also modulate the human ASM (HASM) function by decreasing the synthesis of cyclic adenosine monophosphate (cAMP) and affecting the ß-2 adrenergic receptor (ß-2AR) functions (48). ß-adrenergic receptors (ß-AR) are transmembrane glycoprotein structures belonging to a major receptor family. They are coupled with guanine nucleotide (GTP) binding proteins (G proteins) and classified in the following three subtypes: ß-1, ß-2, and ß-3 (49). Looking specifically to the ß-2ARs, the latter are coded on chromosome 5 and expressed on epithelial and endothelial cells; eosinophils, lymphocytes, and mast cells; skeletal and uterine muscles; mucous glands, and, predominantly, ATII cells and ASM [(49), Figure 1]. The ß-2AR, existing both in activated and inactivated form, is composed of eight alpha helices; three of which are extracellular, and five intracellular. ß-2 adrenergic receptor is attached to the cellular membrane and transmits the signal intracellularly through heterotrimeric Gs proteins, consisting of alpha, beta, and gamma subunits (49). The catecholamines, such as epinephrine and norepinephrine, are responsible for ß-2ARs stimulation. Additionally, synthetic compounds, known as ß-2AR agonists, and classified in accordance to their duration effects into short-acting, long-acting, and ultra-long-acting drugs, have been tought to stimulate ß-2ARs selectively (50). Following the binding of the agonist ligand to the ß-2AR, the alpha subunit of the Gs protein stimulates the conversion of adenosine triphosphate (ATP) into cAMP, which, in turn, trough the catalytic subunit of protein kinase A enzyme, reduces the intracellular Ca2+ concentration, leads to smooth muscle relaxation, and prevents muscle contraction (50). Given their properties, the ß-2AR agonists are part of an effective therapeutic approach to relieve acute airway obstruction, such as during asthma exacerbation; however, they appear less effective when airway obstruction is caused by RSV infection (51). Despite evidence have shown the presence of fully functional ASM also in the early years of life (52, 53) as well as the efficiency of ß-2AR agonists also in newborns and young children (54–56), several clinical trials have failed to demonstrate a clinical benefit of ß-2AR agonists in infants suffering from RSV-mediated bronchiolits (51, 57).
Firstly, Moore et al. (48) investigated the potential influence of RSV on ß-2AR responsiveness by evaluating the isoproterenol (ISO)-cAMP formation, the ß-2AR density, and the Gi expression in HASM cells incubated with RSV. Their findings showed that RSV-infected HASM cells inhibited the ISO-induced cAMP production in a time- and dose-dependent manner and induced a reduction in the ß-2AR density (48). Supporting this evidence, authors showed that RSV could induce an airway insensitivity to ß-2AR-agonists, both directly and indirectly, by inducing heterologous keratinocyte cytokine (KC)/CXCR2-mediated desensitization of epithelial ß-2AR (58, 59). Interestingly, the ß-2AR desensitization occurs in the absence of internalization or degradation of the β2-AR as it results from receptor uncoupling due to phosphorylation by GRK2 (60). More recently, Harford et al. (61) investigated the density and activity of ß-2AR in primary HASM cells derived from pediatric lung tissue with RSV infection compared to non-infected control cells. Their findings showed that RSV induced simultaneously more effects, including a proteasome-mediated cleavage of ß-2AR, a ß-2AR ligand-independent activation of adenylyl-cyclase, and a decrease in cAMP release compared to the control cells, and, lastly, increased intracellular concentrations of Ca2+ resulting in ASM cells contraction (61). Lastly, another explanation for the lack of effectiveness of ß-2AR agonists in infants suffering from RSV infection could be due to the fact that RSV not only induces muscular constriction (bronchospasm) but also impacts the bronchiolar caliber, inducing lymphoid hyperplasia, edema, and mucous plugging, promoting an additional extrinsic compression. Thus, administering ß-2AR agonists did not affect the RSV-mediated airway obstruction (61).
RSV and neurological pathways
The RSV infection in early life causes airway hyperreactivity and inflammation also attributed to an inappropriate neural control of ASM (62, 63). The airway patency depends on the activity and interaction between adrenergic, cholinergic and non-adrenergic–non-cholinergic (NANC) pathways. The adrenergic system, poorly present in the smooth muscle, releases catecholamines and induces bronchorelaxation. The cholinergic pathway releases acetylcholine and induces bronchoconstriction. The NANC component is constituted by inhibitory (NANCi) and excitatory (NANCe) sub-systems. The first one regulates the relaxation of ASM mediated by neurotransmitter vasoactive intestinal peptide (VIP) and nitric oxide. The NANCe sub-system is constituted by unmyelinated (C-type) sensory nerve fibers, and it causes bronchoconstriction mediated by tachykinins such as neuropeptide substance P, neurokinins A and B (63). Substance P acts by binding NK-1, NK-2, and NK-3, three receptors with a rhodopsin-like structure, also expressed in the immune cells. Among these receptors, the NK-1 has a high affinity for substance P and mediates its pro-inflammatory and immunomodulatory effects, including an increased endothelial permeability; induction of T cells, B lymphocytes, monocytes, and macrophages proliferation and activities; chemotaxis-inducer effects; and degranulation of mast cells. The disruption of the NK-1 protects an immune-mediated lung injury, supporting the role of the P/NK-1 interaction in the RSV infection (59). Moreover, the cells expressing NK-1 receptor also show neural endopeptidase and kininase II, two peptidases that cleave the carboxyl-terminal dipeptide of substance P, thereby, inhibiting its actions. Piedimonte et al. (64, 65) showed the effectiveness of the corticosteroids in preventing the neurogenically-mediated vascular permeability of the airways, as they increase the peptidase activity, and it was completely reversed when both kininase II and neutral endopeptidase were simultaneously inhibited.
The upregulation of nerve growth factor (NGF) and its TrkA and p75NTR receptors has also been reported in a vivo model infected with RSV (66). Together with brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3), and neurotrophin 4/5 (NT-4/5), NGF is a neurotrophin (NT) involved in the neuronal development, survival, and function, such as synapse formation and plasticity (63, 67) (Table 1). The NT-mediated effects are the result of their interactions with the p75 neurotrophin receptor (p75NTR) and tropomyosin-related kinase (Trk) family, which may work separately as well as together. The p75NTR, belonging to the TNF receptor superfamily, is a low-affinity receptor for NGF and a receptor for the NTs precursor forms, and it mediates neurite outgrowth, migration, survival, cell cycle arrest, and apoptosis (63, 67). The Trk receptors, which include TrkA, TrkB, and TrkC, interact with all NTs and lead to an activation of several downstream signaling cascades, including PI3K/Akt (protein kinase B a.k.a. PKB) and phospholipase Cγ (PLC) pathways which, in turn, promote the neuronal development, axon and dendrite growth, membrane trafficking, glial differentiation, and interactions with adjacent neurons (67, 68). The crucial role of the NTs and their specific receptors in the RSV-mediated pathogenic mechanisms has been reported in several studies. Increased NGF protein levels and TrkA expression were detected in macrophages and airway epithelial cells in BAL of infants with acute RSV infection requiring ventilatory support (66). In addition, following RSV infection, NGF, by promoting overgrowth of neurites with higher substance P content, favored the sensory fiber responsiveness, acetylcholine and pro-inflammatory peptides release, and long-term remodeling of NANC in the airway (63, 66). Moreover, the NGF over-expression might further affect the ASM tone dysregulation via a decrease in catecholamine production, resulting from the adrenal medulla cell differentiation into nerve cells (68–71).
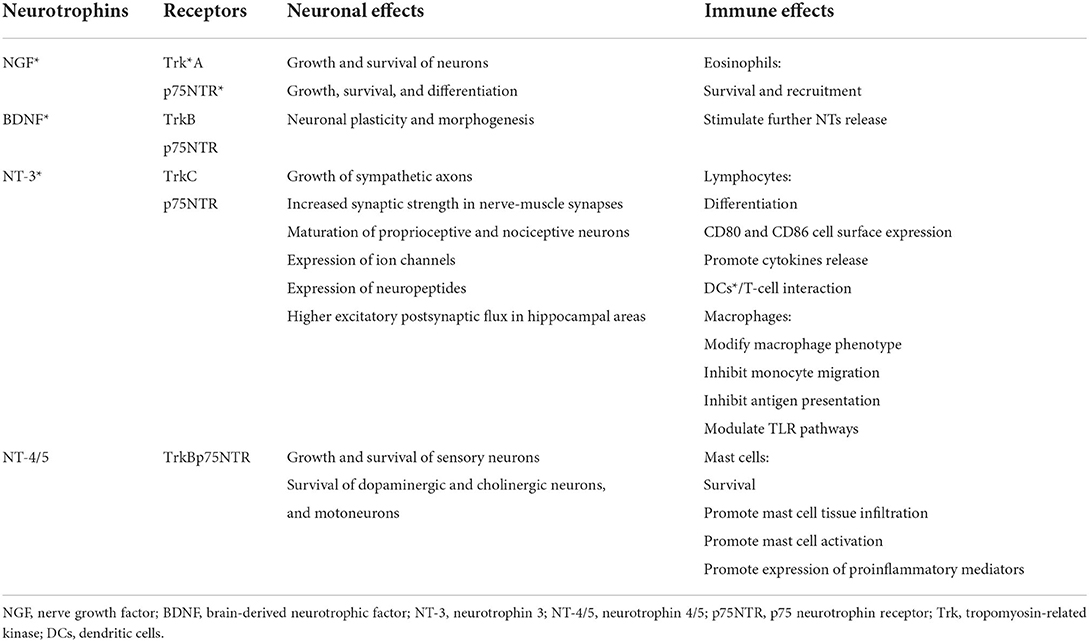
Table 1. The neurotrophins: their receptors and neuronal and immune effects.
The slow-conducting non-myelinated C-fibers represent up to 75% of vagal bronchopulmonary afferents. They innervate the airways from upper (nose, larynx, trachea) to the lower tract, including the parenchyma and alveolar wall. They express the transient receptor potential (TRP) ion channel family, consisting of 28 ion channels and classified, in accordance with their structure and activation mechanisms, into six subgroups, including: ankyrin (TRPA, 1 channel), canonical (TRPC, 7 channels), melastatin (TRPM, 8 channels), mucolipin (PRTML, 3 channels), polycystin (TRPP, 3 channels), and vanilloid (TRPV, 6 channels) families (72). Specifically, the TRPV family (TRPV1–6) are non-selective cationic ligand-gated channels with high permeability to Ca2+. They are commonly expressed by non-neuronal cells, including immune cells and type C sensory nerve fibers of the respiratory tract, and neuronal cells [(73–75), Figure 1]. TRPV family is triggered by exogenous mediators, such as high temperature, osmolarity, exposure to air pollutants, cigarette smoke, allergens and viral agents, capsaicin (CPS); and endogenous stimuli, such as bioactive pro-inflammatory lipids [thromboxanes, prostaglandins E2 (PGE2), leukotrienes, and arachidonic acid derivatives] (73, 74). Following exogenous and endogenous stimuli, TRPVs allow extracellular Ca2+ entrance into neuronal cells, which, in turn, leads to the release of neurotransmitters and result in contraction of ASM, contributing to the onset of the airway mechanisms of defense, such as mucociliary clearance, reflex bronchoconstriction, airway irritation, neurogenic inflammation, and cough reflex (76). Moreover, TRPVs act as receptors for “damage signals” able to transfer the signal neuronal fibers to the immune cells, thus, inducing and perpetuating a “pro-inflammatory status,” also attributed to the release of IL-6 and neuropeptides substance P (77).
Recently, the TRPV family has gained great interest from researchers, which focused their attention, especially on the functions of TRPV1 and TRPV4, mainly expressed in the respiratory tract. Because TRPV1 channels are commonly co-localized with sensory neuropeptides, including calcitonin gene-related peptide (CGRP) and tachykinins, the TRPV1 activation causes a “neurogenic inflammatory reaction” featured by bronchoconstriction, inflammatory cell chemotaxis, and airway mucosal oedema (78). TRPV4 is expressed in mammalian tissues, including lung (human bronchial epithelial cells and alveolar wall), brain, sensory neurons, sympathetic nerves, salivary gland, sweat glands, inner ear, heart, kidney, intestine, skin, endothelium, and fat tissue (79). The TRPV4 activation induces both the activation of K+ and Ca2+ channels, resulting in a further ASM contraction (80). Taking into account their functions, it well appears how a prolonged and intense stimulation of TRPV1 and TRPV4 plays a crucial role in the pathogenesis of some airway diseases, such as chronic cough and asthma, as well as viral-mediated airway damage, since TRPV1 and TRPV4 are both involved into host–pathogen contacts including the binding, entry and replication of the viruses (81, 82). In this regard, in a vivo model, authors firstly reported that the RSV induced a neurogenic inflammation mediated by TRPV1 and capsaicin, resulting from the upregulation of NK-1 both in the airway epithelium and vascular epithelium (83). Later, the same authors reported that, during the RSV infection, the stimulation of the TRPV1-expressing sensory nerves was also involved in the overexpression of NK-1 receptors in CD4+ T cells and also showed chemotactic effects on this subset of lymphocytes (84). Furthermore, inoculation of in vivo model with RSV, induced over-expression of NGF, phenotypic switch in tachykininergic innervation of the airways, and a long-lasting airway inflammation (67, 85, 86). In line with these findings, in a vitro model, Omar et al. (81) reported an up-regulation in TRPV1 expression after 12 h post-RSV infection. Interestingly, this effect was independent of replicating virus as the virus-induced soluble factors were sufficient to increase channel expression. Moreover, the inhibition of RSV infection by using capsazepine induced a down-regulation of TRPV1, suggesting that these receptors have key role in virus-induced airway damage (81). Similarly, Jing et al. reported a down-regulation in the TRPV1 signaling pathway and in airway inflammation and mucus hypersecretion when qingfei oral liquid was administered to RSV-infected asthmatic mice models (87). More recently, this evidence was confirmed in human bronchial epithelium from children with asthma both at baseline and after RSV infection (88). Specifically, children with asthma were intrinsically reporting higher basal TRPV1 protein expression when compared to children without asthma; moreover, a further increase in TRPV1 expression was also noted in asthmatic children during RSV infection since the virus promoted higher intracellular Ca2+ levels as well as NGF overexpression (88). In addition to the asthma status, the patient's age is also a factor affecting the TRVP1 expression during RSV infection (89). By comparing the TRPV1-mediated Ca2+ changes in human bronchial epithelial cells from children and adults with and without asthma, at baseline and after RSV infection, authors noted that TRPV1 expression, localization, and activity were higher in asthmatic children but not in adults, supporting the evidence that RSV entry and/or replication is more efficient in the bronchial epithelium from children but not in adults, a population in which RSV did not affect the TRPV1 function regardless of the asthma status (89).
RSV and non-allergic asthma: In vivo and human models
Since it commonly affects babies during lung development, the RSV is widely recognized as an important risk factor for wheezing and asthma. Although the role of RSV in the onset of atopic asthma is widely recognized, its impact on the onset of non-atopic asthma, mediated via other and independent causal pathways, has long been also suspected but the association is less clear. Following RSV infection, the release of local proinflammatory molecules, the dysfunction of neural pathways, and the compromised epithelial integrity can become chronic and influence airway development, leading to bronchial hyperreactivity and asthma.
In vivo models
Although human studies are essential to assess or refute data from experimental models, many of RSV's behaviors have been reproduced and replicated in the murine models, as they are highly susceptible to RSV infection, permissive to viral replication, and are strictly reflecting the virus and specific T and B cells interactions. In infected animals, RSV infection causes pulmonary damage similar to that observed in humans, featured by degeneration of nasal epithelial mucosa, peribronchiolitis, interstitial pneumonitis, and perivasculitis (10, 11). Accordingly, several studies in animal model systems, firstly, highlighted the key role of Th2-polarized response in the immunopathogenesis of RSV-induced airway inflammation (10, 11). Later, an “asthma-promoting” effect of the RSV infection, regardless of atopic status, has also been postulated (90). Studies conducted on Balb/C model showed that a population of IFN-γ-secreting CD8+ T cells, potentially attenuating the pathogenic Th2 host response to the RSV G-protein, was involved in the RSV-induced airways inflammation (91).
Authors postulated that the age at first infection determined the type of cytokine production and, consequently, the disease patterns during reinfection (12). To investigate the rechallenge, mice were infected at 1 day or 4 or 8 weeks of age and reinfected at 12 weeks. While neonatal priming produced a more severe inflammatory cell recruitment, such as Th2 and eosinophils, delayed priming led to an increased IFN-γ production and a less severe disease in later life. These results showed the crucial importance of the age at the first infection in determining the outcome of reinfection and suggested that the environment of the neonatal lung is a major determinant of cytokine production and disease patterns in later life (12). In vitro studies have also shown that IFN-γ activates eosinophils, prolongs their survival and promotes the synthesis of leukotrienes from these cells. Considering this evidence, Wedde-Beer et al. postulated that leukotrienes, originating from the interaction between P-containing nerves and mast cells, may be important mediators of RSV-induced airway inflammation (92). Accordingly, rats were inoculated at 2 or 12 weeks of age with RSV or virus-free medium and treated with montelukast or its vehicle starting 1 day before inoculation. The authors reported greater microvascular permeability in the intrapulmonary airways of RSV-infected rats not receiving montelukast treatment compared to the control group. Moreover, a significant increase in 5-lipoxygenase-encoding mRNA and cysteinyl leukotrienes levels and mast cells was detected in the lung tissues of RSV-infected rats, suggesting that the increase in vascular permeability might be promoted by mast cells-derived leukotrienes (92).
The potential effect of the RSV-induced upregulation of NGF must also be considered as a pathogenic mechanism in the onset of non-atopic asthma. Because NGF is released from airway epithelial cells, it increases the release of substance P and other tachykinins from adult sensory neurons and induces sensory hyperinnervation in the airways of transgenic mice. Substance P, in turn, activates mast cells releasing leukotrienes, which further sensitize C-type neurons to release neurotransmitters, thereby reactivating the mast cells and creating a vicious circle contributing to an exaggerated inflammation of the lower respiratory tract (92). In a mouse model, the expression of NGF, trkA, and p75 declined with age, but the RSV increased their levels both in weanling and adult rats. Confirming the role of the NGF and its receptors as major determinants of neurogenic inflammation in RSV infection, authors also reported that exogenous NGF upregulated NK1 receptor expression in the lungs and, on the contrary, the anti-NGF antibody inhibited NK1 receptor and, thereby, the neurogenic inflammation in RSV-infected lungs (86). The evidence that NGF expression was inversely correlated with the age, supported the hypothesis that this neurotrophin is critical for the neuronal plasticity; thereby, variations in its expression can result in permanent changes in sensorineural lung pathways further contributing in airway hyperresponsiveness and asthma susceptibility (93). These findings were confirmed by Piedimonte et al. (79) that showed an increase in NGF and neurotrophin receptor expression in early life during RSV infection and a RSV-related abnormal remodeling of neuronal networks in the respiratory tract, resulting in bronchial hyperreactivity and airway obstruction. Interestingly, the neurogenic inflammation and the bronchial hyperreactivity were long lasting in mice up to 60 days after intranasal inoculation of RSV (94). Confirming these findings, animal data revealed that the RSV-induced airway hyperreactivity was long-lasting for weeks after the inoculation of the virus (95–97). This protracted inflammatory response may be due to the persistence of viral genomic in mouse lung tissue, which has been demonstrated to last for at least 67 days following RSV inoculation (95–98).
New data support the Th17 cell differentiation during RSV infection and Treg airway accumulation during RSV clearance (99). In a mice model, authors found that RSV infection, through activation Notch-1/DLL3, increased the mRNA expression of IL-17A and IL-17A/Foxp3 and Treg levels in the hilar lymph nodes and mesenteric lymph nodes. In contrast, the mRNA expression of IL-4 and other Th2 cytokines was unchanged (100, 101).
Recently, it has been reported that RSV infection contributes to the onset of asthma in later life by inducing High Mobility Group Box-1 (HMGB1), as a result of necroptosis, a programmed cell death of airway epithelial cells (102, 103). The inhibition of necroptosis decreased the severity of bronchiolitis by reducing viral load, prevented the airway epithelial cells remodeling, and the asthma development in later life (102, 103).
In addition to the immune dysfunction and neurogenic inflammation, non-atopic asthma would be the result of an impaired relationship between epithelium and mesenchymal structures. An exaggerated release and responsiveness to trophic factors and a subsequent abnormal growth of smooth muscle, nerves, and blood vessels are considered crucial events in remodeling the airways. The barrier dysfunction could enhance the sampling of luminal antigens by intraepithelial DCs, and facilitate translocation of inhaled particles, allergens, bacterial and viral pathogens through the lung, resulting in an inappropriate immune response and airway inflammation (104, 105).
The integrity of the airway epithelial barrier is regulated by several mechanisms, including the assembly of the epithelial apical junctional complex (AJC), which are composed of apically located tight junctions (TJs) and underlying adherens junctions (AJs), also containing adhesion, scaffolding, signaling, and cytoskeletal proteins (104). The TJs limit the passage of ions and uncharged solutes thanks to their adhesive properties. Three major types of transmembrane proteins are described: (1) members of the claudin family, (2) the TJ-associated MARVEL proteins (TAMP) family, which includes occludin, tricellulin, and Marvel D3, and (3) immunoglobulin-like proteins, which include junctional adhesion molecule A (JAM-A) and coxsackievirus and adenovirus receptor (CAR) proteins (104, 105). The AJs are involved in starting and maintaining of epithelial cell–cell contacts, and enabling TJ assembly. The E-cadherin and nectins are the main transmembrane adhesion proteins involved in forming epithelial AJs, cell–cell adhesion, cell signaling, proliferation, and differentiation. The cytoplasmic side of TJs is organized by multifunctional scaffolding proteins of the zonula occludens (ZO) family, whereas β-catenin, α-catenin, and p120 catenin form a complex with the cytoplasmic domain of E-cadherin (106). C57BL/6 mice, intranasally inoculated with RSV, showed a significant peribronchial inflammation compared with non-infected controls and UV-inactivated RSV-inoculated animals. Moreover, RSV infection increased the permeability of the airway epithelial barrier, decreased the expression of several TJ proteins, and prevented the accumulation of cleaved extracellular fragments of E-cadherin in BAL and tracheal epithelial cells (107). Another study found that RSV infection led to decreased mRNA expression of claudin-1 and occludin in lung samples of wild-type BALB/c mice (108). These studies supported the evidence that RSV induces a AJCs disorganization and dysfunction, and it was likely a result of cortical F-actin cytoskeletal remodeling, which is, in turn, is regulated by Protein kinase D (PKD)-mediated phosphorylation of cortactin, an actin-binding protein that regulates F-actin dynamics between polymerization and depolymerization steps during plasma membrane remodeling (109).
Globally, an abnormal remodeling of the airways substructures, especially involving the mucosal neural network, occurs when the airways are infected during critical developmental windows in early life. However, because these changes seem to be the result of a transient derangement in airway development, these modifications would return to baseline condition when the virus is cleaved, thereby, justifying the evidence that some children with previous RSV infection do not develop asthma. On the other hand, RSV-associated long-term consequences have been reported in neonatal mice, such as persistent mucus production, subepithelial fibrosis, and increased collagen deposition, contributing to promoting and maintaining airway remodeling also in later life (109–111). Moreover, 30 days after inoculation, neither evidence of active RSV infection nor immunostaining and no viral nucleic sequences were detected in a mouse model. In contrast, the substance P in the lung and the capsaicin-induced plasma extravasation were significantly higher in the infected mice compared with pathogen-free rat, supporting the evidence that the sensory innervation of airways remain susceptible to the proinflammatory effects even if RSV infection is disappeared (112). In conclusion, RSV seems to exert a dual influence: in short-term postsynaptic manner, deriving from up-regulation of the substance P, and in long-term presynaptic manner, by remodeling sensory innervation (112).
In humans
Similarly to other viruses, RSV shows a close relantionship with the development of wheeze and asthma in later life, regardless atopic status (2, 113). Several birth cohort studies reported that one-third of children with RSV developed recurrent wheezing and asthma, but not allergic sensitization (90, 114, 115). In line with these findings, the Tucson respiratory study reported that children with a previous RSV infection were commonly experiencing wheezing and lower forced expiratory volume at first second (FEV1) by the age 6 years (90). Moreover, the risk decreased significantly with the age and it was not significant by age 13 (90). There was not any significant relationship with atopic status development; as sensitization did not appear as a risk factor for infants suffering from RSV-caused wheezing (90, 116). By enrolling 95,310 children by Carroll et al. (117) and Wu et al. (9), the Tennessee Asthma Bronchiolitis Study (TABS) suggested a causal relationship between severe RSV infection and the onset of asthma by age 5.5 years. They also reported that during the winter months was recorded the greatest number of bronchiolitis-related hospitalizations. Moreover, the authors observed that children born 4 months prior to the annual peak of bronchiolitis-related hospitalizations were 29% more likely to develop asthma compared with infants born 1 year from this time point. This trend was similar troughout the 5-year study although the peak of bronchiolitis-related hospitalizations shifted by up to 6 weeks. Furthermore, if on one side the authors did not state if children were infected with a specific virus, including RSV, on the other hand, up to 70% of severe bronchiolitis were due to RSV infection (118). In accordance with several longitudinal studies, these changes appears to be transient rather than persisting over the time. Pullan et al. (119) showed that children younger than 1 year of age and with severe RSV infections experienced wheezing primarily during the first 4 years of life, while, by age 10 years, no significant difference in incidence are reported compared to the control group. Similar results were observed in the Tucson Children's Respiratory Study of 1,246 children as children, who experienced a severe RSV infection by up to 3 years of life, were at a significantly increased risk of wheezing at ages 6 and 11 years. However, by age 13 years no significant differences were recorded compared to the control group (120). These data were not confirmed by the longitudinal study by Sigurs et al. (121). Authors described a significant increase in asthma by age 13 years; probably, the differential findings reported in the studies above reported are due to several factors involved in the RSV-asthma relationship, including genetic variability among investigated cohorts as well as the difference in pathogenicity of circulating RSV strains (121).
In two controlled, randomized, double-blinded trials performed in preterm infants receiving palivizumab to prevent RSV bronchiolitis, authors reported that the palivizumab administration was protective against recurrent wheeze up to 3 year of life; supporting the evidence that an early-life RSV bronchiolitis can have a continuing causal impact beyond infant infection (122, 123). Confirming these findings, authors demonstrated that RSV prophylaxis in non-atopic children decreased the risk of recurrent wheezing up to 80% and, interestingly, it did not have effects on infants with a positive family history for atopy (124). Later, a Finnish study reported a significant association between RSV infection and self-reported asthma in adolescents aged from 15 to 18 years (125). In a 7 year follow-up study enrolling 127 steroid-naive children, authors reported that the first severe RSV/rhinovirus–negative wheezing episode was a risk factor for developing non-allergic asthma, together with the age of first episode of wheezing <12 months, and exposure to tobacco smoking (113). Despite this increasing body of evidence supporting a relationship between RSV infection and non-atopic asthma, it remains unclear whether the virus contributes really to the onset of asthma or is simple trigger in subjects with asthma susceptibility (126). Authors hypothesized that probably the association between RSV infection and asthma could be also due to a shared genetic predisposition, as reported in a large population of 8,280 twin pairs in Denmark (126). Furthermore, genetic polymorphisms in the gene encoding IkBa, a negative regulator of NF-kB, have been recently associated with RSV and asthma (127). The gain-of-function polymorphisms in the promoter region of IL-8, a chemokine secreted by epithelial cells and macrophages, lead to an increase in RSV infection severity and in developing wheezing and asthma (128–130). Polymorphisms in other chemokines, such CX3CL1, CX3CR1, and CCL5, seem further predispose to asthma onset (131, 132). Mainly, CX3CR1, expressed in human airway epithelial, binds RSV G protein and mediates the viral entry in the host cells; thus, CX3CR1 mutations and/or complete or partial deletion of RSV G protein results in a less efficient viral entry and a decreased virus replication (133).
Gain-of-function polymorphisms in IL-4 and IL-13 cytokine genes as well as deficit in IFN-γ expression have been also associated with developing wheezing during at the age 6 years (134, 135).
Several mechanisms causing RSV-mediated long-term pulmonary effects have also been investigated. The effects of maternal RSV infection on postnatal offspring immunity and neurotrophins release, ASM contractility, and development of wheezing and asthma are still under debate. However, the recent discovery that viral antigens can impact postnatal immunity in vivo model and negatively affect the perinatal period in humans showed the possibility that a challenge in immunity can ocurr after postnatal virus (136, 137). In utero exposure to RSV caused a chronic airway dysfunction by influencing cell-mediated host immunity, local NGF expression, neurotransmitters release and neuronal hyperreactivity, and ASM contractility during RSV-mediated LRTIs in early-life (66, 138–140). Neurotrophin levels and immunoreactivity for neurotrophin-receptor were found to be strongly increased in the BAL fluid of mechanically ventilated infants with severe RSV infection (66). Children with severe RSV-bronchiolitis are reporting an increased bronchial responsiveness and altered epithelial immune response to the viral agents compared with children without bronchiolitis (141). It seems that in subjects with chronic obstructive pulmonary disease, the RSV can persist contributing to the pathogenesis of stable disease (142). Smyth et al. (143) studied the behavior of mediators of lymphocyte activity [IL-4, CD25, and soluble intercellular adhesion molecule 1 (sICAM-1)] in 94 children affected by RSV-mediated bronchiolitis. Authors reported that the serum CD25 levels were elevated during acute infection and they remained raised during convalescence up to 150 days after infection, also in absence of acute infection (143). This finding was in contrast to the rapid decline in serum CD25 levels recorded in children after acute infection mediated by other viral agents, such as measles or dengue fever (143). Thereby, RSV could mediate a persistent inflammatory response in the airway that continues for longer than is generally believed. Similarly, Pala et al. (144) compared the production of cytokines of children 7 years after acute RSV bronchiolitis to healthy children. The post-bronchiolitic children showed a significant response in cells producing IL-4, suggesting the role of this cytokine in the RSV-mediated long-term airway effects (144). Moreover, since RSV induced a strong release of IL-10, IL-11, and prostaglandin E2 (PGE2), molecules well-known for their immunosuppressive effects, authors hypothesized that these mediators might be responsible for the delayed protective RSV specific immune response, further contributing to RSV-mediated long-term damage in the airway (145). More recently, Bertrand et al. (146) reported that the cytokine (IL-3, IL-4, IL-10 and IL-13) and chemokine (IL-1β, IL-6, TNF-β, MCP-1/CCL2, MIP-1α/CCL3, and IL-8/CXCL8) levels in the BAL and nasopharyngeal aspirates of children with RSV-mediated bronchiolitis were significantly higher compared to control group, and, moreover, a direct correlation between IL-3 and IL-12p40 levels and development of wheezing later in life was also observed.
Other possible mechanism might include the “hitchhike effect” yet described in patients suffering from chronic bacterial colonization. Specifically, it seems that the neutrophilic airway inflammation following a chronic colonization could contribute to development asthma in the pediatric population (147).
Probably, the development of RSV-induced asthma requires a “two-hit” model (148) featured by the co-existence of at least two among individual (genetic, immune response in the lung environment), developmental (lung remodeling), and environmental (exposure to inhalants) factors (Figure 2). Whether only one factor is present, the patient will not develop chronic airway inflammation and asthma; conversely, whether two factors are coexisting, the patient will report long-term symptoms and/or asthma (148). This might explain why not all children with severe RSV bronchiolitis develop asthma as well as why other children with early wheezing show resolution of their illness by their adolescence (148).
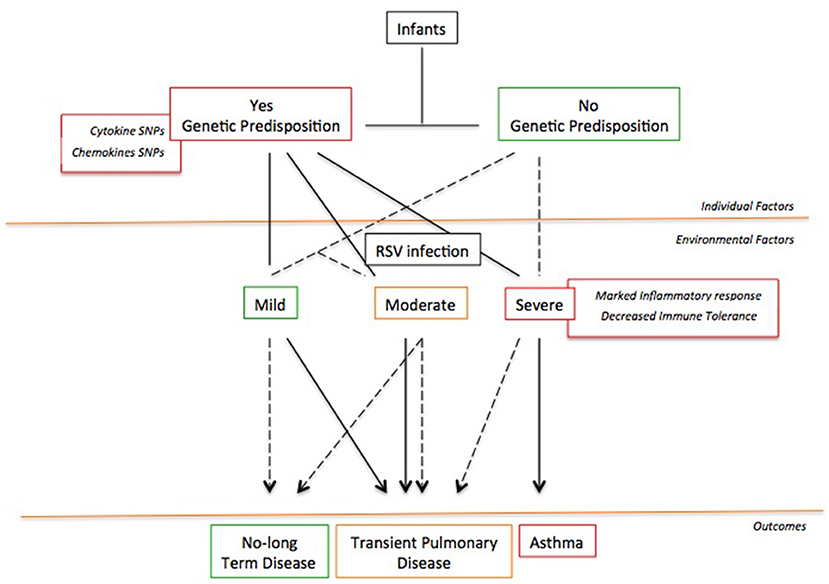
Figure 2. The “Two-hit” hypothesis. The co-existence of at least two or more risk factors favors the asthma development following a severe RSV infection.
Future prospectives
Compared with other environmental factors, RSV infection remains the main cause of LRTIs in infants, and wheezing and asthma during childhood. Despite several experimental and human studies have demonstrated a close relationship between RSV infection and the subsequent lon-term airway effects, to date, the exact mecahnism underlying the RSV infection and asthma development remains to be elucidated. Respiratory syncytial virus shows an intricate relationship with the local and systemic immune response of the host. Consistent literature findings evidence that RSV-caused asthma is closely related to atopic constitution, since a Th2 dominance in immune response has been commonly reported. Additionally, other important cellular mediators of RSV-mediated inflammation and immune responses are endothelial cells, lymphocytes, macrophages, and mast cells. Moreover, despite its simple structure, RSV shows a complex relationship also with neuronal pathway of the airways. Respiratory syncytial virus makes the airways abnormally susceptible to the RSV-caused proinflammatory effects by upregulating NK-1 receptor gene expression and, thereby, increasing the synthesis of the substance P and the density of its receptors on target immune cells, including lymphocytes, macrophages, mast cells, and endothelial cells. Thus, RSV can establish crucial interactions between neuronal airway system and immune response that result in long-term airway dysfunction, predisposing to the onset and maintaninance of chronic persistent airway hyperreactivity and inflammation (149–151). Additionally, the RSV-immune system-neuronal pathway interactions are influenced not only by the viral factors but also by host factors, such as genetic susceptibility, that modify the efficiency of the response to the virus, viral replication, and virus-mediated injury to the airways. All this may justify the differences in severity infection, damage extention, and duration and magnitude of the RSV effects in later life among the pediatric population. Thereby, due to the pathophysiologic mechanisms involved in the RSV infection, it is likely that future treatment strategies should be focused on modulating the interactions between the virus, host immune response and neuronal pathways. Currently, RSV-mediated treatment is limited to supportive care; and no vaccine is licensed to prevent RSV infection. The only prevention strategy available is palivizumab, which is indicated only in a cluster of preterm newborns or those with comorbidities. On the other hand, vaccine development has encountered several challenges, such as the immaturity of the immune response in infants; thus, most newborns remain unprotected against RSV. It appears that the two feasible strategies for protecting all infants against this virus are maternal immunization and immunization of infants with long-acting monoclonal antibodies (mAbs). The latter seems to provide consistent protection against RSV for at least 5 months, covering the duration of the RSV season, offering great flexibility in the timing of administration, and regardless of the gestational age, presence of comorbidities, and maturity of the immune system. Accordingly, using long-acting mAbs appears to be the only available strategy for protecting all newborns entering their first RSV. Moreover, using long-acting mAbs could also postpone the risk of RSV-mediated bronchiolitis throughout life. As reported above, the age at the first infection represents one of the risk factors for developing non-allergic asthma. Earlier the newborn meets the virus, more severe it will be the airway damage. The early RSV-related abnormal remodeling of neuronal networks in the respiratory tract will lead to bronchial hyperreactivity and airway obstruction; thus, the baby will experience early wheezing during the first 4 years of life (12, 85, 113). On the contrary, if the child meets the virus late, it is reasonable to hypothesize that the RSV-mediated damage to the airways will be less severe. The baby will develop a more mature immune response and will have an advanced lung maturity; therefore, the onset of wheezing will be late, or it will not occur (12, 85, 113).
Lastly, a global understanding of the principal mediators and the risk factors for onset of wheezing and for its progression to asthma is critical for prevention strategies after an initial RSV infection as well as for the development of effective therapeutic strategies for viral-induced wheezing/asthma, especially in the pediatric population.
Author contributions
SM and GP reviewed the literature on the subject, drafted the final version of the manuscript, finally approved the version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Both authors made substantial contribution to the conception of the work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Piedimonte G. RSV infections: state of the art. Cleve Clin J Med. (2015) 82(11 Suppl 1):S13–8. doi: 10.3949/ccjm.82.s1.03
2. Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. (2010) 65:1045–52. doi: 10.1136/thx.2009.121582
3. Fjaerli HO, Farstad T, Rød G, Ufert GK, Gulbrandsen P, Nakstad B. Acute bronchiolitis in infancy as risk factor for wheezing and reduced pulmonary function by seven years in Akershus County, Norway. BMC Pediatr. (2005) 5:31. doi: 10.1186/1471-2431-5-31
4. Cassimos DC, Tsalkidis A, Tripsianis GA, Stogiannidou A, Anthracopoulos M, Ktenidou-Kartali S, et al. Asthma, lung function and sensitization in school children with a history of bronchiolitis. Pediatr Int. (2008) 50:51–6. doi: 10.1111/j.1442-200X.2007.02509.x
5. Zomer-Kooijker K, van der Ent CK, Ermers MJ, Uiterwaal CS, Rovers MM, Bont LJ, et al. Increased risk of wheeze and decreased lung function after respiratory syncytial virus infection. PLoS ONE. (2014) 9:e87162. doi: 10.1371/journal.pone.0087162
6. Riikonen R, Lauhkonen E, Törmänen S, Backman K, Koponen P, Helminen M, et al. Prospective study confirms that bronchiolitis in early infancy increases the risk of reduced lung function at 10–13 years of age. Acta Paediatr. (2019) 108:124–30. doi: 10.1111/apa.14412
7. Wu P, Hartert TV. Evidence for a causal relationship between respiratory syncytial virus infection and asthma. Expert Rev Anti Infect Ther. (2011) 9:731–45. doi: 10.1586/eri.11.92
8. Stensballe LG, Simonsen JB, Thomsen SF, Larsen AM, Lysdal SH, Aaby P, et al. The causal direction in the association between respiratory syncytial virus hospitalization and asthma. J Allergy Clin Immunol. (2009) 123:131.e1–7.e1. doi: 10.1016/j.jaci.2008.10.042
9. Wu P, Dupont WD, Griffin MR, Carroll KN, Mitchel EF, Gebretsadik T, et al. Evidence of a causal role of winter virus infection during infancy in early childhood asthma. Am J Respir Crit Care Med. (2008) 178:1123–9. doi: 10.1164/rccm.200804-579OC
10. Openshaw P, Murphy EE, Hosken NA, Maino V, Davis K, Murphy K, et al. Heterogeneity of intracellular cytokine synthesis at the single-cell level in polarized T helper 1 and T helper 2 populations. J Exp Med. (1995) 182:1357–67. doi: 10.1084/jem.182.5.1357
11. Varga SM, Wang X, Welsh RM, Braciale TJ. Immunopathology in RSV infection is mediated by a discrete oligoclonal subset of antigen-specific CD4(+) T cells. Immunity. (2001) 15:637–46. doi: 10.1016/S1074-7613(01)00209-6
12. Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med. (2002) 196:1381–6. doi: 10.1084/jem.20020943
13. Lemanske RF Jr. Immunologic mechanisms in RSV-related allergy and asthma. In: Hiatt PW, editor. RSV and Asthma: Is There a Link? New York: American Thoracic Society (1998), p. 11–6.
14. Larsen GL. RSV infection and airway neural control in animal models. In: Hiatt PW, editor. RSV and Asthma: Is There a Link? New York: American Thoracic Society (1998), p. 17–20.
15. Colten HR, Krause JE. Pulmonary inflammation–a balancing act. N Engl J Med. (1997) 336:1094–6. doi: 10.1056/NEJM199704103361511
16. Troy NM, Bosco A. Respiratory viral infections and host responses; insights from genomics. Respir Res. (2016) 17:156. doi: 10.1186/s12931-016-0474-9
17. Fuentes S, Tran KC, Luthra P, Teng MN, He B. Function of the respiratory syncytial virus small hydrophobic protein. J Virol. (2007) 81:8361–6. doi: 10.1128/JVI.02717-06
18. Bueno SM, González PA, Pacheco R, Leiva ED, Cautivo KM, Tobar HE, et al. Host immunity during RSV pathogenesis. Int Immunopharmacol. (2008) 8:1320–9. doi: 10.1016/j.intimp.2008.03.012
19. Shaikh FY, Utley TJ, Craven RE, Rogers MC, Lapierre LA, Goldenring JR et al. Respiratory syncytial virus assembles into structured filamentous virion particles independently of host cytoskeleton and related proteins. PLoS ONE. (2012) 7:e40826. doi: 10.1371/journal.pone.0040826
20. Ulloa L, Serra R, Asenjo A, Villanueva N. Interactions between cellular actin and human respiratory syncytial virus (HRSV). Virus Res. (1998) 53:13–25. doi: 10.1016/S0168-1702(97)00121-4
21. Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog. (2013) 9:e1003309. doi: 10.1371/journal.ppat.1003309
22. Mehedi M, Collins PL, Buchholz UJ. A novel host factor for human respiratory syncytial virus. Commun Integr Biol. (2017) 10:e1319025. doi: 10.1080/19420889.2017.1319025
23. Shahriari S, Wei KJ, Ghildyal R. Respiratory syncytial virus Matrix (M) protein interacts with actin in vitro and in cell culture. Viruses. (2018) 10:535. doi: 10.3390/v10100535
24. Jeffree CE, Brown G, Aitken J, Su-Yin DY, Tan BH, Sugrue RJ. Ultrastructural analysis of the interaction between F-actin and respiratory syncytial virus during virus assembly. Virology. (2007) 369:309–23. doi: 10.1016/j.virol.2007.08.007
25. Gower TL, Pastey MK, Peeples ME, Collins PL, McCurdy LH, Hart TK, et al. RhoA signaling is required for respiratory syncytial virus-induced syncytium formation and filamentous virion morphology. J Virol. (2005) 79:5326–36. doi: 10.1128/JVI.79.9.5326-5336.2005
26. Mitra R, Baviskar P, Duncan-Decocq RR, Patel D, Oomens AG. The human respiratory syncytial virus matrix protein is required for maturation of viral filaments. J Virol. (2012) 86:4432–43. doi: 10.1128/JVI.06744-11
27. Henderson G, Murray J, Yeo RP. Sorting of the respiratory syncytial virus matrix protein into detergent-resistant structures is dependent on cell-surface expression of the glycoproteins. Virology. (2002) 300:244–54. doi: 10.1006/viro.2002.1540
28. Shaikh FY, Cox RG, Lifland AW, Hotard AL, Williams JV, Moore ML, et al. A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles. mBio. (2012) 3:e00270-11. doi: 10.1128/mBio.00270-11
29. Carlier MF, Ducruix A, Pantaloni D. Signalling to actin: the Cdc42-N-WASP-Arp2/3 connection. Chem Biol. (1999) 6:R235–40. doi: 10.1016/S1074-5521(99)80107-0
30. Linfield DT, Gao N, Raduka A, Harford TJ, Piedimonte G, Rezaee F. RSV attenuates epithelial cell restitution by inhibiting actin cytoskeleton-dependent cell migration. Am J Physiol Lung Cell Mol Physiol. (2021) 321:L189–203. doi: 10.1152/ajplung.00118.2021
31. Chen LF, Xu WB, Li YY, Chen NH, Luo D, Song QY, et al. Inhibitory effect of PIK-24 on respiratory syncytial virus entry by blocking phosphatidylinositol-3 kinase signaling. Antimicrob Agents Chemother. (2020) 64:e00608-20. doi: 10.1128/AAC.00608-20
32. Ferreira LR, Moussatché N, Moura Neto V. Rearrangement of intermediate filament network of BHK-21 cells infected with vaccinia virus. Arch Virol. (1994) 138:273–85. doi: 10.1007/BF01379131
33. Russell CD, Unger SA, Walton M, Schwarze J. The human immune response to respiratory syncytial virus infection. Clin Microbiol Rev. (2017) 30:481–502. doi: 10.1128/CMR.00090-16
34. Bergeron HC, Tripp RA. Immunopathology of RSV: an updated review. Viruses. (2021) 13:2478. doi: 10.3390/v13122478
35. Gasteiger G, D'Osualdo A, Schubert DA, Weber A, Bruscia EM, Hartl D. Cellular innate immunity: an old game with new players. J Innate Immunity. (2017) 9:111–25. doi: 10.1159/000453397
36. Liang B, Kabatova B, Kabat J, Dorward DW, Liu X, Surman S, et al. Effects of alterations to the CX3C motif and secreted form of human respiratory syncytial virus (RSV) G protein on immune responses to a parainfluenza virus vector expressing the RSV G protein. J Virol. (2019) 93:e02043-18. doi: 10.1128/JVI.02043-18
37. Gan SW, Tan E, Lin X, Yu D, Wang J, Tan GM, et al. The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J Biol Chem. (2012) 287:24671–89. doi: 10.1074/jbc.M111.332791
38. Sedeyn K, Schepens B, Saelens X. Respiratory syncytial virus nonstructural proteins 1 and 2: exceptional disrupters of innate immune responses. PLoS Pathog. (2019) 15:e1007984. doi: 10.1371/journal.ppat.1007984
39. Schlender J, Zimmer G, Herrler G, Conzelmann KK. Respiratory syncytial virus (RSV) fusion protein subunit F2, not attachment protein G determines the specificity of RSV infection. J Virol. (2003) 77:4609–16. doi: 10.1128/JVI.77.8.4609-4616.2003
40. Weltzin R, Hsu SA, Mittler ES, Georgakopoulos K, Monath TP. Intranasal monoclonal immunoglobulin A against respiratory syncytial virus protects against upper and lower respiratory tract infections in mice. Antimicrob Agents Chemother. (1994) 38:2785–91. doi: 10.1128/AAC.38.12.2785
41. Prince GA, Hemming VG, Horswood RL, Baron PA, Chanock RM. Effectiveness of topically administered neutralizing antibodies in experimental immunotherapy of respiratory syncytial virus infection in cotton rats. J Virol. (1987) 61:1851–4. doi: 10.1128/jvi.61.6.1851-1854.1987
42. Bukreyev A, Yang L, Fricke J, Cheng L, Ward JM, Murphy BR, et al. The secreted form of respiratory syncytial virus G glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on Fc receptor-bearing leukocytes. J Virol. (2008) 82:12191–204. doi: 10.1128/JVI.01604-08
43. Liesman RM, Buchholz UJ, Luongo CL, Yang L, Proia AD, DeVincenzo JP, et al. RSV-encoded NS2 promotes epithelial cell shedding and distal airway obstruction. J Clin Invest. (2014) 124:2219–33. doi: 10.1172/JCI72948
44. Persson BD, Jaffe AB, Fearns R, Danahay H. Respiratory syncytial virus can infect basal cells and alter human airway epithelial differentiation. PLoS ONE. (2014) 9:e102368. doi: 10.1371/journal.pone.0102368
45. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
46. Emboriadou M, Hatzistilianou M, Magnisali C, Sakelaropoulou A, Exintari M, Conti P, et al. Human neutrophil elastase in RSV bronchiolitis. Ann Clin Lab Sci. (2007) 37:79–84.
47. Harrison AM, Bonville CA, Rosenberg HF, Domachowske JB. Respiratory syncytical virus-induced chemokine expression in the lower airways: eosinophil recruitment and degranulation. Am J Respir Crit Care Med. (1999) 159:1918–24. doi: 10.1164/ajrccm.159.6.9805083
48. Moore PE, Cunningham G, Calder MM, DeMatteo AD Jr, Peeples ME, Summar ML, et al. Respiratory syncytial virus infection reduces beta2-adrenergic responses in human airway smooth muscle. Am J Respir Cell Mol Biol. (2006) 35:559–64. doi: 10.1165/rcmb.2005-0282OC
49. Johnson M. The beta-adrenoceptor. Am J Respir Crit Care Med. (1998) 158(5 Pt 3):S146–53. doi: 10.1164/ajrccm.158.supplement_2.13tac110
50. Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. (2006) 117:18–25. doi: 10.1016/j.jaci.2005.11.012
51. Ralston SL, Lieberthal AS, Meissner HC, Alverson BK, Baley JE, Gadomski AM, et al. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics. (2014) 134:e1474–502. doi: 10.1542/peds.2014-2742
52. Danopoulos S, Bhattacharya S, Mariani TJ, Al Alam D. Transcriptional characterisation of human lung cells identifies novel mesenchymal lineage markers. Eur Respir J. (2020) 55:1900746. doi: 10.1183/13993003.00746-2019
53. Roesler AM, Wicher SA, Ravix J, Britt RD Jr, Manlove L, Teske J, et al. Calcium sensing receptor in developing human airway smooth muscle. J Cell Physiol. (2019) 234:14187–97. doi: 10.1002/jcp.28115
54. Busi LE, Restuccia S, Tourres R, Sly PD. Assessing bronchodilator response in preschool children using spirometry. Thorax. (2017) 72:367–72. doi: 10.1136/thoraxjnl-2015-207961
55. Castro-Rodriguez JA, Rodrigo GJ. Beta-agonists through metered-dose inhaler with valved holding chamber versus nebulizer for acute exacerbation of wheezing or asthma in children under 5 years of age: a systematic review with meta-analysis. J Pediatr. (2004) 145:172–7. doi: 10.1016/j.jpeds.2004.04.007
56. Goldstein AB, Castile RG, Davis SD, Filbrun DA, Flucke RL, McCoy KS, et al. Bronchodilator responsiveness in normal infants and young children. Am J Respir Crit Care Med. (2001) 164:447–54. doi: 10.1164/ajrccm.164.3.2005080
57. Cai Z, Lin Y, Liang J. Efficacy of salbutamol in the treatment of infants with bronchiolitis: a meta-analysis of 13 studies. Medicine. (2020) 99:e18657. doi: 10.1097/MD.0000000000018657
58. Davis IC, Xu A, Gao Z, Hickman-Davis JM, Factor P, Sullender WM, et al. Respiratory syncytial virus induces insensitivity to beta-adrenergic agonists in mouse lung epithelium in vivo. Am J Physiol. Lung Cell Mol Physiol. (2007) 293:L281–9. doi: 10.1152/ajplung.00458.2006
59. Traylor ZP, Yu EN, Davis IC. Respiratory syncytial virus induces airway insensitivity to beta-agonists in BALB/c mice. Am J Physiol. Lung Cell Mol Physiol. (2010) 298:L437–45. doi: 10.1152/ajplung.00363.2009
60. Davis IC, Sullender WM, Hickman-Davis JM, Lindsey JR, Matalon S. Nucleotide-mediated inhibition of alveolar fluid clearance in BALB/c mice after respiratory syncytial virus infection. Am J Physiol. Lung Cell Mol Physiol. (2004) 286:L112–20. doi: 10.1152/ajplung.00218.2003
61. Harford TJ, Rezaee F, Gupta MK, Bokun V, Naga Prasad SV, Piedimonte G. Respiratory syncytial virus induces β2-adrenergic receptor dysfunction in human airway smooth muscle cells. Sci Signal. (2021) 14:eabc1983. doi: 10.1126/scisignal.abc1983
62. Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Modern Pathol. (2007) 20:108–19. doi: 10.1038/modpathol.3800725
63. Piedimonte G. Neural mechanisms of respiratory syncytial virus-induced inflammation and prevention of respiratory syncytial virus sequelae. Am J Respir Crit Care Med. (2001) 163(3 Pt 2):S18–21. doi: 10.1164/ajrccm.163.supplement_1.2011113
64. Piedimonte G, McDonald DM, Nadel JA. Glucocorticoids inhibit neurogenic plasma extravasation and prevent virus-potentiated extravasation in the rat trachea. J Clin Invest. (1990) 86:1409–15. doi: 10.1172/JCI114855
65. Piedimonte G, McDonald DM, Nadel JA. Neutral endopeptidase and kininase II mediate glucocorticoid inhibition of neurogenic inflammation in the rat trachea. J Clin Invest. (1991) 88:40–4. doi: 10.1172/JCI115302
66. Tortorolo L, Langer A, Polidori G, Vento G, Stampachiacchere B, Aloe L, et al. Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am J Respir Crit Care Med. (2005) 172:233–7. doi: 10.1164/rccm.200412-1693OC
67. Manti S, Brown P, Perez MK, Piedimonte G. The role of neurotrophins in inflammation and allergy. Vit Horm. (2017) 104:313–41. doi: 10.1016/bs.vh.2016.10.010
68. Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. (2006) 361:1545–64. doi: 10.1098/rstb.2006.1894
69. Li QG, Wu XR, Li XZ, Yu J, Xia Y, Wang AP, et al. Neural-endocrine mechanisms of respiratory syncytial virus-associated asthma in a rat model. Genet Mol Res. (2012) 11:2780–9. doi: 10.4238/2012.August.24.3
70. Rossi GA, Colin AA. Respiratory syncytial virus-host interaction in the pathogenesis of bronchiolitis and its impact on respiratory morbidity in later life. Pediatr Allergy Immunol. (2017) 28:320–31. doi: 10.1111/pai.12716
71. Rezaee F, Gibson LF, Piktel D, Othumpangat S, Piedimonte G. Respiratory syncytial virus infection in human bone marrow stromal cells. Am J Respir Cell Mol Biol. (2011) 45:277–86. doi: 10.1165/rcmb.2010-0121OC
72. Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calc. (2005) 38:233–52. doi: 10.1016/j.ceca.2005.06.028
73. Groneberg DA, Niimi A, Dinh QT, Cosio B, Hew M, Fischer A, et al. Increased expression of transient receptor potential vanilloid-1 in airway nerves of chronic cough. Am J Respir Crit Care Med. (2004) 170:1276–80. doi: 10.1164/rccm.200402-174OC
74. McLeod RL, Fernandez X, Correll CC, Phelps TP, Jia Y, Wang X, et al. TRPV1 antagonists attenuate antigen-provoked cough in ovalbumin sensitized guinea pigs. Cough Lond Engl. (2006) 2:10. doi: 10.1186/1745-9974-2-10
75. Nicotera AG, Spanò M, Decio A, Valentini G, Saia M, Di Rosa. G. Epileptic phenotype and cannabidiol efficacy in a Williams-Beuren syndrome patient with atypical deletion: a case report. Front Neurol. (2021) 12:659543. doi: 10.3389/fneur.2021.659543
76. Du Q, Liao Q, Chen C, Yang X, Xie R, Xu J. The role of transient receptor potential vanilloid 1 in common diseases of the digestive tract and the cardiovascular and respiratory system. Front Physiol. (2019) 10:1064. doi: 10.3389/fphys.2019.01064
77. Tränkner D, Hahne N, Sugino K, Hoon MA, Zuker C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc Natl Acad Sci USA. (2014) 111:11515–20. doi: 10.1073/pnas.1411032111
78. Watanabe N, Horie S, Michael GJ, Keir S, Spina D, Page CP, et al. Immunohistochemical co-localization of transient receptor potential vanilloid (TRPV)1 and sensory neuropeptides in the guinea-pig respiratory system. Neuroscience. (2006) 141:1533–43. doi: 10.1016/j.neuroscience.2006.04.073
79. Jia Y, Lee LY. Role of TRPV receptors in respiratory diseases. Biochim Biophys Acta. (2007) 1772:915–27. doi: 10.1016/j.bbadis.2007.01.013
80. Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res. (2006) 99:988–95. doi: 10.1161/01.RES.0000247065.11756.19
81. Omar S, Clarke R, Abdullah H, Brady C, Corry J, Winter H, et al. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE. (2017) 12:e0171681. doi: 10.1371/journal.pone.0171681
82. Kitagawa Y, Tamai I, Hamada Y, Usui K, Wada M, Sakata M, et al. Orally administered selective TRPV1 antagonist, JTS-653, attenuates chronic pain refractory to non-steroidal anti-inflammatory drugs in rats and mice including post-herpetic pain. J Pharmacol Sci. (2013) 122:128–137. doi: 10.1254/jphs.12276FP
83. Piedimonte G, Rodriguez MM, King KA, McLean S, Jiang X. Respiratory syncytial virus upregulates expression of the substance P receptor in rat lungs. Am J Physiol. (1999) 277:L831–40. doi: 10.1152/ajplung.1999.277.4.L831
84. Auais A, Adkins B, Napchan G, Piedimonte G. Immunomodulatory effects of sensory nerves during respiratory syncytial virus infection in rats. Am J Physiol Lung Cell Mol Physiol. (2003) 285:L105–13. doi: 10.1152/ajplung.00004.2003
85. Piedimonte G. Contribution of neuroimmune mechanisms to airway inflammation and remodeling during and after respiratory syncytial virus infection. Pediatr Infect Dis J. (2003) 22(2 Suppl):S66–75. doi: 10.1097/01.inf.0000053888.67311.1d
86. Hu C, Wedde-Beer K, Auais A, Rodriguez MM, Piedimonte G. Nerve growth factor and nerve growth factor receptors in respiratory syncytial virus-infected lungs. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L494–502. doi: 10.1152/ajplung.00414.2001
87. Jing X, Yan W, Zeng H, Cheng W. Qingfei oral liquid alleviates airway hyperresponsiveness and mucus hypersecretion via TRPV1 signaling in RSV-infected asthmatic mice. Biomed Pharmacother. (2020) 128:110340. doi: 10.1016/j.biopha.2020.110340
88. Harford TJ, Grove L, Rezaee F, Scheraga R, Olman MA, Piedimonte G. RSV infection potentiates TRPV1-mediated calcium transport in bronchial epithelium of asthmatic children. Am J Physiol. Lung Cell Mol Physiol. (2021) 320:L1074–84. doi: 10.1152/ajplung.00531.2020
89. Harford TJ, Rezaee F, Scheraga RG, Olman MA, Piedimonte G. Asthma predisposition and respiratory syncytial virus infection modulate transient receptor potential vanilloid 1 function in children's airways. J Allergy Clin Immunol. (2018) 141:414.e4–6.e4. doi: 10.1016/j.jaci.2017.07.015
90. Stein RT, Sherrill D, Morgan WJ, Holberg CJ, Halonen M, Taussig LM, et al. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet. (1999) 354:541–5. doi: 10.1016/S0140-6736(98)10321-5
91. Hussell T, Baldwin CJ, O'Garra A, Openshaw PJ. CD8+ T cells control Th2-driven pathology during pulmonary respiratory syncytial virus infection. Eur J Immunol. (1997) 27:3341–9. doi: 10.1002/eji.1830271233
92. Wedde-Beer K, Hu C, Rodriguez MM, Piedimonte G. Leukotrienes mediate neurogenic inflammation in lungs of young rats infected with respiratory syncytial virus. Am J Physiol. Lung Cell Mol Physiol. (2002) 282:L1143–50. doi: 10.1152/ajplung.00323.2001
93. Borchers AT, Chang C, Gershwin ME, Gershwin LJ. Respiratory syncytial virus–a comprehensive review. Clin Rev Allergy Immunol. (2013) 45:331–79. doi: 10.1007/s12016-013-8368-9
94. Zang N, Li S, Li W, Xie X, Ren L, Long X, et al. Resveratrol suppresses persistent airway inflammation and hyperresponsivess might partially via nerve growth factor in respiratory syncytial virus-infected mice. Int Immunopharmacol. (2015) 28:121–8. doi: 10.1016/j.intimp.2015.05.031
95. Chávez-Bueno S, Mejías A, Gómez AM, Olsen KD, Ríos AM, Fonseca-Aten M, et al. Respiratory syncytial virus-induced acute and chronic airway disease is independent of genetic background: an experimental murine model. Virol J. (2005) 2:46. doi: 10.1186/1743-422X-2-46
96. Jafri HS, Chavez-Bueno S, Mejias A, Gomez AM, Rios AM, Nassi SS, et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis. (2004) 189:1856–65. doi: 10.1086/386372
97. Estripeaut D, Torres JP, Somers CS, Tagliabue C, Khokhar S, Bhoj VG, et al. Respiratory syncytial virus persistence in the lungs correlates with airway hyperreactivity in the mouse model. J Infect Dis. (2008) 198:1435–43. doi: 10.1086/592714
98. Schwarze J, O'Donnell DR, Rohwedder A, Openshaw PJ. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am J Respir Crit Care Med. (2004) 169:801–5. doi: 10.1164/rccm.200308-1203OC
99. Lotz MT, Peebles RS Jr. Mechanisms of respiratory syncytial virus modulation of airway immune responses. Curr Allergy Asthma Rep. (2012) 12:380–7. doi: 10.1007/s11882-012-0278-z
100. Shi T, Li N, He Y, Feng J, Mei Z, Du Y, et al. Th17/Treg cell imbalance plays an important role in respiratory syncytial virus infection compromising asthma tolerance in mice. Microb Pathog. (2021) 156:104867. doi: 10.1016/j.micpath.2021.104867
101. Qin L, Qiu K, Hu C, Wang L, Wu G, Tan Y. Respiratory syncytial virus promoted the differentiation of Th17 cells in airway microenvironment through activation of Notch-1/Delta3. J Med Microbiol. (2019) 68:649–56. doi: 10.1099/jmm.0.000959
102. Simpson J, Loh Z, Ullah MA, Lynch JP, Werder RB, Collinson N, et al. Respiratory syncytial virus infection promotes necroptosis and HMGB1 release by airway epithelial cells. Am J Respir Critical Care Med. (2020) 201:1358–71. doi: 10.1164/rccm.201906-1149OC
103. Manti S, Harford TJ, Salpietro C, Rezaee F, Piedimonte G. Induction of high-mobility group Box-1 in vitro and in vivo by respiratory syncytial virus. Pediatr Res. (2018) 83:1049–56. doi: 10.1038/pr.2018.6
104. Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. (2014) 134:509–20. doi: 10.1016/j.jaci.2014.05.049
105. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. (2008) 178:1271–81. doi: 10.1164/rccm.200801-136OC
106. Saito M, Tucker DK, Kohlhorst D, Niessen CM, Kowalczyk AP. Classical and desmosomal cadherins at a glance. J Cell Sci. (2012) 125(Pt 11):2547–52. doi: 10.1242/jcs.066654
107. Smallcombe CC, Linfield DT, Harford TJ, Bokun V, Ivanov AI, Piedimonte G, et al. Disruption of the airway epithelial barrier in a murine model of respiratory syncytial virus infection. Am J Physiol. Lung Cell Mol Physiol. (2019) 316:L358–68. doi: 10.1152/ajplung.00345.2018
108. Kast JI, McFarlane AJ, Głobińska A, Sokolowska M, Wawrzyniak P, Sanak M, et al. Respiratory syncytial virus infection influences tight junction integrity. Clin Exp Immunol. (2017) 190:351–9. doi: 10.1111/cei.13042
109. Weed SA, Parsons JT. Cortactin: coupling membrane dynamics to cortical actin assembly. Oncogene. (2001) 20:6418–34. doi: 10.1038/sj.onc.1204783
110. Becnel D, You D, Erskin J, Dimina DM, Cormier SA. A role for airway remodeling during respiratory syncytial virus infection. Respir Res. (2005) 6:122. doi: 10.1186/1465-9921-6-122
111. Srinivasa BT, Restori KH, Shan J, Cyr L, Xing L, Lee S, et al. STAT6 inhibitory peptide given during RSV infection of neonatal mice reduces exacerbated airway responses upon adult reinfection. J Leuk Biol. (2017) 101:519–529. doi: 10.1189/jlb.4A0215-062RR
112. Piedimonte G, Hegele RG, Auais A. Persistent airway inflammation after resolution of respiratory syncytial virus infection in rats. Pediatr Res. (2004) 55:657–65. doi: 10.1203/01.PDR.0000112244.72924.26
113. Lukkarinen M, Koistinen A, Turunen R, Lehtinen P, Vuorinen T, Jartti T. Rhinovirus-induced first wheezing episode predicts atopic but not nonatopic asthma at school age. J Allergy Clin Immunol. (2017) 140:988–95. doi: 10.1016/j.jaci.2016.12.991
114. Castro-Rodríguez JA, Holberg CJ, Wright AL, Martinez FD. A clinical index to define risk of asthma in young children with recurrent wheezing. Am J Respir Crit Care Med. (2000) 162(4 Pt 1):1403–6. doi: 10.1164/ajrccm.162.4.9912111
115. Henderson J, Hilliard TN, Sherriff A, Stalker D, Al Shammari N, Thomas HM. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: a longitudinal birth cohort study. Pediatr Allergy Immunol. (2005) 16:386–92. doi: 10.1111/j.1399-3038.2005.00298.x
116. Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B, Björkstén B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics. (1995) 95:500–5. doi: 10.1542/peds.95.4.500
117. Carroll KN, Wu P, Gebretsadik T, Griffin MR, Dupont WD, Mitchel EF, et al. Season of infant bronchiolitis and estimates of subsequent risk and burden of early childhood asthma. J Allergy Clin Immunol. (2009) 123:964–6. doi: 10.1016/j.jaci.2008.12.011
118. Papadopoulos NG, Moustaki M, Tsolia M, Bossios A, Astra E, Prezerakou A, et al. Association of rhinovirus infection with increased disease severity in acute bronchiolitis. Am J Respir Crit Care Med. (2002) 165:1285–9. doi: 10.1164/rccm.200112-118BC
119. Pullan CR, Hey EN. Wheezing, asthma, and pulmonary dysfunction 10 years after infection with respiratory syncytial virus in infancy. Brit Med J. (1982) 284:1665–9. doi: 10.1136/bmj.284.6330.1665
120. Taussig LM, Wright AL, Morgan WJ, Harrison HR, Ray CG. The Tucson Children's Respiratory Study Design I and implementation of a prospective study of acute and chronic respiratory illness in children. Am J Epidemiol. (1989) 129:1219–31. doi: 10.1093/oxfordjournals.aje.a115242
121. Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. (2005) 171:137–41. doi: 10.1164/rccm.200406-730OC
122. Blanken MO, Rovers MM, Molenaar JM, Winkler-Seinstra PL, Meijer A, Kimpen JL, et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med. (2013) 368:1791–9. doi: 10.1056/NEJMoa1211917
123. Yoshihara S, Kusuda S, Mochizuki H, Okada K, Nishima S, Simões EA, et al. Effect of palivizumab prophylaxis on subsequent recurrent wheezing in preterm infants. Pediatrics. (2013) 132:811–8. doi: 10.1542/peds.2013-0982
124. Simões EA, Carbonell-Estrany X, Rieger CH, Mitchell I, Fredrick L, Groothuis JR, et al. The effect of respiratory syncytial virus on subsequent recurrent wheezing in atopic and nonatopic children. J Allergy Clin Immunol. (2010) 126:256–62. doi: 10.1016/j.jaci.2010.05.026
125. Ruotsalainen M, Hyvärinen MK, Piippo-Savolainen E, Korppi M. Adolescent asthma after rhinovirus and respiratory syncytial virus bronchiolitis. Pediatr Pulmonol. (2013) 48:633–9. doi: 10.1002/ppul.22692
126. Thomsen SF, van der Sluis S, Stensballe LG, Posthuma D, Skytthe A, Kyvik KO, et al. Exploring the association between severe respiratory syncytial virus infection and asthma: a registry-based twin study. Am J Respir Crit Care Med. (2009) 179:1091–7. doi: 10.1164/rccm.200809-1471OC
127. Ali S, Hirschfeld AF, Mayer ML, Fortuno ES III, Corbett N, Kaplan M, et al. Functional genetic variation in NFKBIA and susceptibility to childhood asthma, bronchiolitis, bronchopulmonary dysplasia. J Immunol. (2013) 190:3949–58. doi: 10.4049/jimmunol.1201015
128. Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax. (2000) 55:1023–7. doi: 10.1136/thorax.55.12.1023
129. Goetghebuer T, Isles K, Moore C, Thomson A, Kwiatkowski D, Hull J. Genetic predisposition to wheeze following respiratory syncytial virus bronchiolitis. Clin Exp Allergy. (2004) 34:801–3. doi: 10.1111/j.1365-2222.2004.1947.x
130. Puthothu B, Krueger M, Heinze J, Forster J, Heinzmann A. Impact of IL8 and IL8-receptor alpha polymorphisms on the genetics of bronchial asthma and severe RSV infections. Clin Mol Allergy. (2006) 4:2. doi: 10.1186/1476-7961-4-2
131. Tremblay K, Lemire M, Provost V, Pastinen T, Renaud Y, Sandford AJ, et al. Association study between the CX3CR1 gene and asthma. Genes Immun. (2006) 7:632–9. doi: 10.1038/sj.gene.6364340
132. Tian M, Liu F, Wen GY, Shi SY, Chen RH, Zhao DY. Effect of variation in RANTES promoter on serum RANTES levels and risk of recurrent wheezing after RSV bronchiolitis in children from Han, Southern China. Eur J Pediatr. (2009) 168:963–7. doi: 10.1007/s00431-008-0870-3
133. Jeong KI, Piepenhagen PA, Kishko M, DiNapoli JM, Groppo RP, Zhang L, et al. CX3CR1 is expressed in differentiated human ciliated airway cells and co-localizes with respiratory syncytial virus on cilia in a G protein-dependent manner. PLoS ONE. (2015) 10:e0130517. doi: 10.1371/journal.pone.0130517
134. Ermers MJ, Hoebee B, Hodemaekers HM, Kimman TG, Kimpen JL, Bont L. IL-13 genetic polymorphism identifies children with late wheezing after respiratory syncytial virus infection. J Allergy Clin Immunol. (2007) 119:1086–91. doi: 10.1016/j.jaci.2006.12.655
135. Stern DA, Guerra S, Halonen M, Wright AL, Martinez FD. Low IFN-gamma production in the first year of life as a predictor of wheeze during childhood. J Allergy clin Immunol. (2007) 120:835–41. doi: 10.1016/j.jaci.2007.05.050
136. Baker CA, Swainson L, Lin DL, Wong S, Hartigan-O'Connor DJ, Lifson JD, et al. Exposure to SIV in utero results in reduced viral loads and altered responsiveness to postnatal challenge. Sci Transl Med. (2015) 7:300ra125. doi: 10.1126/scitranslmed.aac5547
137. Manti S, Esper F, Alejandro-Rodriguez M, Leonardi S, Betta P, Cuppari C, et al. Respiratory syncytial virus seropositivity at birth is associated with adverse neonatal respiratory outcomes. Pediatr Pulmonol. (2020) 55:3074–9. doi: 10.1002/ppul.25001
138. Brown PM, Harford TJ, Agrawal V, Yen-Lieberman B, Rezaee F, Piedimonte G. Prenatal exposure to respiratory syncytial virus alters postnatal immunity and airway smooth muscle contractility during early-life reinfections. PLoS ONE. (2017) 12:e0168786. doi: 10.1371/journal.pone.0168786
139. Bonini S, Lambiase A, Bonini S, Angelucci F, Magrini L, Manni L, et al. Circulating nerve growth factor levels are increased in humans with allergic diseases and asthma. Proc Natl Acad Sci USA. (1996) 93:10955–60. doi: 10.1073/pnas.93.20.10955
140. Piedimonte G, Walton C, Samsell L. Vertical transmission of respiratory syncytial virus modulates pre- and postnatal innervation and reactivity of rat airways. PLoS ONE. (2013) 8:e61309. doi: 10.1371/journal.pone.0061309
141. Edwards MR, Bartlett NW, Hussell T, Openshaw P, Johnston SL. The microbiology of asthma. Nat Rev Microbiol. (2012) 10:459–471. doi: 10.1038/nrmicro2801
142. Sikkel MB, Quint JK, Mallia P, Wedzicha JA, Johnston SL. Respiratory syncytial virus persistence in chronic obstructive pulmonary disease. Pediatric Infect Dis J. (2008) 27(10 Suppl):S63–70. doi: 10.1097/INF.0b013e3181684d67
143. Smyth RL, Fletcher JN, Thomas HM, Hart CA. Immunological responses to respiratory syncytial virus infection in infancy. Arch Dis childhood. (1997) 76:210–4. doi: 10.1136/adc.76.3.210
144. Pala P, Bjarnason R, Sigurbergsson F, Metcalfe C, Sigurs N, Openshaw PJ. Enhanced IL-4 responses in children with a history of respiratory syncytial virus bronchiolitis in infancy. Eur Respir J. (2002) 20:376–82. doi: 10.1183/09031936.02.00249902
145. Bartz H, Büning-Pfaue F, Türkel O, Schauer U. Respiratory syncytial virus induces prostaglandin E2, IL-10 and IL-11 generation in antigen presenting cells. Clin Exp Immunol. (2002) 129:438–45. doi: 10.1046/j.1365-2249.2002.01927.x
146. Bertrand P, Lay MK, Piedimonte G, Brockmann PE, Palavecino CE, Hernández J, et al. Elevated IL-3 and IL-12p40 levels in the lower airway of infants with RSV-induced bronchiolitis correlate with recurrent wheezing. Cytokine. (2015) 76:417–23. doi: 10.1016/j.cyto.2015.07.017
147. Drews AC, Pizzichini MM, Pizzichini E, Pereira MU, Pitrez PM, Jones MH, et al. Neutrophilic airway inflammation is a main feature of induced sputum in nonatopic asthmatic children. Allergy. (2009) 64:1597–601. doi: 10.1111/j.1398-9995.2009.02057.x
148. Knudson CJ, Varga SM. The relationship between respiratory syncytial virus and asthma. Vet Pathol. (2015) 52:97–106. doi: 10.1177/0300985814520639
149. Bozic CR, Lu B, Höpken UE, Gerard C, Gerard NP. Neurogenic amplification of immune complex inflammation. Science. (1996) 273:1722–5. doi: 10.1126/science.273.5282.1722
150. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol. (2002) 76:5654–66. doi: 10.1128/JVI.76.11.5654-5666.2002
Keywords: respiratory syncytial virus, wheezing, asthma, experimental studies, human studies, immune system, neurogenic inflammation
Citation: Manti S and Piedimonte G (2022) An overview on the RSV-mediated mechanisms in the onset of non-allergic asthma. Front. Pediatr. 10:998296. doi: 10.3389/fped.2022.998296
Received: 19 July 2022; Accepted: 19 August 2022;
Published: 20 September 2022.
Edited by:
Mauricio Tomas Caballero, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Michael David Shields, Queen's University Belfast, United KingdomRaymond Pickles, University of North Carolina at Chapel Hill, United States
Copyright © 2022 Manti and Piedimonte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Piedimonte, gpiedimonte@tulane.edu