
Abstract
Aims/hypothesis
People with isolated impaired fasting glucose (IFG) have normal beta cell function. We hypothesised that an increased glucose threshold for beta cell secretion explains IFG.
Methods
We used graded glucose infusion to examine the relationship of insulin secretion rate (ISR) and glucagon secretion rate (GSR) with rising glucose. We studied 39 non-diabetic individuals (53 ± 2 years, BMI 30 ± 1 kg/m2), categorised by fasting glucose and glucose tolerance status. After an overnight fast, a variable insulin infusion was used to maintain glucose at ~4.44 mmol/l (07:00 to 08:30 hours). At 09:00 hours, graded glucose infusion commenced at 1 mg kg−1 min−1 and doubled every 60 min until 13:00 hours. GSR and ISR were calculated by nonparametric deconvolution from concentrations of glucagon and C-peptide, respectively.
Results
The relationship of ISR with glucose was linear and the threshold for insulin secretion in isolated IFG did not differ from that in people with normal fasting glucose and normal glucose tolerance. GSR exhibited a single-exponential relationship with glucose that could be characterised by G50, the change in glucose necessary to suppress GSR by 50%. G50 was increased in IFG compared with normal fasting glucose regardless of the presence of impaired or normal glucose tolerance.
Conclusions/interpretation
These data show that, in non-diabetic humans, alpha cell dysfunction contributes to the pathogenesis of IFG independently of defects in insulin secretion. We also describe a new index that quantifies the suppression of glucagon secretion by glucose.
Graphical abstract

Similar content being viewed by others



Introduction
The states of impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) are associated with a high, but heterogeneous, rate of progression to type 2 diabetes [1]. In Olmsted County, MN, USA, ‘only’ 40% of people with a fasting glucose ≥6.1 mmol/l progress to overt diabetes within 10 years [2]. This can be partially, but not completely, explained by lifestyle and heredity [3, 4]. In addition, a subset of people with isolated IFG with normal glucose tolerance (i.e. IFG/NGT) have normal beta cell function [5, 6], as quantified by the disposition index (DI) [7], in response to an oral challenge.
DI expresses insulin secretion as a function of insulin action [8] and is the standard measure of beta cell function. These data suggest that (at least in IFG/NGT) defects in the regulation of fasting glucose occur independently of defects in postprandial beta cell function [5]. This is in keeping with the results of genetic association studies that have identified loci that alter fasting glucose but not glucose tolerance and vice versa [9,10,11]. However, a recent longitudinal study in an Asian population [12] showed that the rate of conversion to type 2 diabetes is similar in IFG/NGT and in normal fasting glucose (NFG) with IGT (NFG/IGT), while it is highest in IFG with IGT (IFG/IGT). This suggests that progression to type 2 diabetes can occur in the absence of an abnormal beta cell response to an oral challenge.
The oral minimal model measures two components of beta cell responsivity (Φ) to an oral glucose challenge: dynamic (Φd), representing a response to rising glucose concentrations; and static (Φs), representing a response to constant glucose concentrations [6]. Neither component is abnormal in people with IFG/NGT. However, abnormalities of Φ are present in people with IGT [5]. This implies that changes in beta cell responses to glucose as quantified by Φ, and its components, cannot explain fasting hyperglycaemia.
If beta cell function is intact in people with isolated IFG, fasting insulin secretion would be expected to rise to restrain endogenous glucose production (EGP) and lower fasting glucose. Since this does not occur, one potential explanation is an increased threshold for glucose-stimulated insulin secretion as observed in people with mutations in glucokinase (GCK) [13]. Type 2 diabetes is characterised by an acquired defect in hepatic glucokinase activity [14, 15] but it is unknown if similar abnormalities occur in the beta cells of individuals with IFG/NGT. Our primary hypothesis is that IFG with or without abnormal glucose tolerance is associated with impaired beta cell sensing of glucose, i.e. an increased glucose threshold for insulin secretion.
Given that prediabetes predisposes to a disease (type 2 diabetes) characterised by defects in both beta cell and alpha cell function, there has been interest in the role of alpha cell dysfunction in the progression of prediabetes. This interest has been given added impetus by the observation that diabetes-associated genetic variation in TCF7L2 is associated with impaired postprandial glucagon suppression in non-diabetic individuals [16]. Elevated fasting glucagon has been associated with impaired insulin action [7, 17] and with progression of glucose intolerance [18]. Some [19, 20], but not all [6], prior experiments have failed to consistently document elevated fasting glucagon concentrations in people with IFG, compared with those with NFG. Nevertheless, in the presence of fasting hyperglycaemia, fasting glucagon concentrations indistinguishable from normal values imply a defect in the ability of glucose to suppress glucagon secretion by the alpha cell [21]. This formed the basis of our secondary hypothesis, namely that alpha cell dysfunction contributes to the pathogenesis of fasting hyperglycaemia.
To address these questions, we used a graded glucose infusion (GGI) [22] to measure insulin [23] and glucagon [24] secretion in response to rising glucose concentrations.
Methods
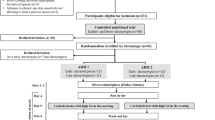
Screening
After approval from the Mayo Clinic Institutional Review Board, we recruited individuals through a combination of intramural and extramural advertising as well as from a database of prior participants whose fasting glucose and glucose tolerance status was known. To be eligible, individuals had no history of chronic illness or upper gastrointestinal surgery. Additionally, they were not taking medications that could affect glucose metabolism. Potentially eligible individuals who showed interest in participating were invited to the Clinical Research and Trials Unit (CRTU) for a screening visit. After written, informed consent was obtained, participants underwent a 2 h 75 g OGTT to characterise their glucose tolerance status, as previously described [5]. All participants were instructed to follow a weight-maintenance diet containing 55% carbohydrate, 30% fat and 15% protein for at least 3 days prior to the study. Body composition was measured at the time of screening using dual-energy x-ray absorptiometry (Lunar, Madison, WI, USA).
Experimental design
Participants were admitted to the CRTU at 17:00 hours on the day before the study. After consuming a standard 10 kcal/kg caffeine-free meal, they fasted overnight. The following morning at 06:00 hours a dorsal hand vein was cannulated and placed in a heated Plexiglas box maintained at 55°C to allow sampling of arterialised venous blood. The contralateral forearm vein was cannulated for glucose and hormone infusions. At 07:00 hours (−120 min) insulin (0.1 U/ml Humulin R; Eli Lilly, Indianapolis, IN, USA) was infused at a variable rate to lower and maintain glucose concentrations at 4.44 mmol/l (80 mg/dl) [25]. The insulin infusion was discontinued at 08:30 hours (−30 min). At 09:00 hours (0 min) a GGI using 50% dextrose commenced at 1 mg kg−1 min−1 and then increased to 2 (10:00 hours), 4 (11:00 hours) and 8 mg kg−1 min−1 (12:00 hours) every 60 min [22]. The experiment ended at 13:00 hours when all infusions were stopped. Participants consumed a meal and left the CRTU when it was safe to do so.
Analytic techniques
All blood was immediately placed on ice after collection, centrifuged at 4°C, separated and stored at −80°C until assay. Plasma glucose concentrations were measured using a Yellow Springs Glucose Analyzer (Yellow Springs Instruments, Yellow Springs, OH, USA). Plasma insulin concentrations were measured using a chemiluminescence assay (Access Assay; Beckman, Chaska, MN, USA). Plasma C-peptide was measured using a two-site immunoenzymatic sandwich assay (Roche Diagnostics, Indianapolis, IN, USA). Glucagon was measured using a two-site ELISA (Mercodia, Winston Salem, NC, USA) in accordance with the manufacturer’s instructions.
Calculations
The insulin secretion rate (ISR) was calculated from C-peptide concentrations using nonparametric deconvolution and population-based measures of C-peptide kinetics [26]. Linear regression was used to describe the increase of ISR in response to the observed glucose concentrations for each individual. We characterised individual responses using the ΔISR/ΔGlucose gradient and the intercept of this slope at a glucose of 4.44 mmol/l. We then utilised the individualised linear relationships, together with a non-negativity constraint, to predict the response of ISR over a wider range of 3.3–15 mmol/l. The lower limit was chosen to avoid the presumed glucose concentration at which insulin secretion is suppressed [27].
Likewise, glucagon secretion rate (GSR) was calculated from the measured glucagon concentrations observed during the experiments using nonparametric deconvolution and the population model of glucagon kinetics we described recently [24]. The exponential relationship of GSR to glucose concentrations in each individual was then used to derive the increment in glucose concentration necessary to suppress GSR by 50% (G50) in each participant. Please refer to the electronic supplementary material (ESM) Methods for more details.
The oral minimal model [8] was used to calculate insulin secretion and action at the time of screening as previously described [28, 29]
Statistical analysis
All continuous data are summarised as means ± SEM. AUC and area above basal (AAB) were calculated using the trapezoidal rule. One-way ANOVA and a Tukey’s post hoc test were used to determine between-group differences (parametric data). For nonparametric data, a Kruskal–Wallis test followed by a Dunn’s post hoc test was used. When necessary, multivariate analysis adjusting for the effects of age, sex and weight was performed. BlueSky Statistics software v.7.10 (BlueSky Statistics, Chicago, IL, USA) and Prism 5 v.5.0f (GraphPad Software, San Diego, CA, USA) were utilised for the statistical analysis. Residuals for the conditional logistic regression of a particular variable with the covariates were used to confirm or refute the contribution of that variable to variation in the tested index. A p value <0.05 was considered statistically significant. Our power calculation for the glucose threshold of insulin secretion was based on observed (mean ± SD) glucose concentrations of 4.8 ± 0.5 mmol/l [16]. Assuming similar variability, 15 participants with IFG would give us the ability to detect a 0.36 mmol/l (7%) difference in glucose threshold compared with people with NFG.
Results
Participant characteristics by fasting glucose and by glucose tolerance status
A total of 39 participants were studied. At the time of screening, 14 participants were classified as NFG/NGT. Participants with IFG and/or IGT were heavier. This trend was not explained by differences in lean body mass (Table 1). As previously observed, participants with NFG/IGT and those with IFG/IGT exhibited defects in insulin secretion and action as quantified by the oral minimal model [6]. See also ESM Fig. 1.
Glucose, insulin, C-peptide and glucagon concentrations in groups classified by fasting glucose and by glucose tolerance status
By design, at the time when the insulin infusion was discontinued (−30 min), glucose concentrations did not differ between groups. However, in people with IFG/NGT, glucose concentrations over the subsequent 30 min (−30 to 0 min) were significantly increased compared with those with NFG/NGT. In response to GGI, glucose concentrations were higher in those with IFG/NGT and IFG/IGT (Fig. 1b and c, respectively), but by the end of the study did not differ between groups. See also ESM Table 1.

Glucose (a–c), insulin (d–f), C-peptide (g–i) and glucagon (j–l) concentrations during the GGI, for participants with NFG/IGT (black circles), those with IFG/NGT (black squares) and those with IFG/IGT (black triangles), when compared with those with NFG/NGT (white circles)
Higher insulin concentrations at the time when insulin infusion was discontinued (−30 min) were observed in the IFG/NGT and the IFG/IGT groups. Insulin concentrations in response to GGI did not differ between these two groups (Fig. 1e and f, respectively). See also ESM Table 1.
Fasting C-peptide concentrations were significantly increased in people with NFG/IGT, compared with those with NFG/NGT. In response to GGI, C-peptide concentrations were increased in all three groups with prediabetes compared with NFG/NGT (Fig. 1g–l). See also ESM Table 1.
Fasting glucagon concentrations as well as the concentrations observed during the GGI did not differ between groups (Fig. 1j–l). See also ESM Table 1.
ISR over time and its relationship with glucose concentrations in groups classified by fasting glucose and by glucose tolerance status
The ISR over time did not differ between groups whether considered as absolute values (Fig. 2a–c) or as integrated AUC (data not shown). See also ESM Fig. 2 for data from representative individuals.

The mean (± SEM) ISR over time observed during the GGI for participants with NFG/IGT (a), those with IFG/NGT (b) and those with IFG/IGT (c), compared with those with NFG/NGT. The corresponding panels (d–f) show the relationship of ISR (mean ± SEM) and glucose concentrations. In addition, the symbols (black circle [NFG/IGT], black square [IFG/NGT], black triangle [IFG/IGT] and white circle [NFG/NGT]) in (d–f) show the relationship between mean fasting ISR and glucose on the day of the screening visit
The model for the relationship of ISR with glucose predicts that ISR is completely suppressed at 3.0 ± 0.3 vs 2.5 ± 0.4 vs 3.3 ± 0.3 vs 2.7 ± 0.2 mmol/l (NFG/NGT vs NFG/IGT vs IFG/NGT vs IFG/IGT, respectively, p=0.50; Fig. 2d–f). AUC was compared for glucose values ≤5.55 mmol/l, values >5.55 and ≤7.77 mmol/l, and values >7.77 mmol/l. These thresholds were chosen using the criteria for diagnosis of IFG and of IGT.
This analysis showed that for glucose concentrations >5.55 mmol/l, people with IFG and/or IGT exhibited a similar relationship to that observed in NFG/NGT. On the other hand, for glucose concentrations ≤5.55 mmol/l, people with NFG/IGT secreted more insulin (0.35 ± 0.03 vs 0.55 ± 0.06 vs 0.43 ± 0.12 vs 0.44 ± 0.05 nmol/min per mmol/l, NFG/NGT vs NFG/IGT vs IFG/NGT vs IFG/IGT, respectively, p=0.04; Fig. 2d–f).
At the time of the screening OGTT, the relationship of fasting ISR with fasting glucose exhibited a similar relationship to that observed during the GGI study day (as shown in Fig. 2d–f).
Indices of insulin secretion derived from the GGI in groups classified by fasting glucose status and by glucose tolerance status and the relationship of these indices with fasting and post-challenge glucose concentrations
The ΔISR/ΔGlucose gradient observed during GGI did not differ across the groups defined by fasting glucose status and by glucose tolerance status (Fig. 3a).

(a, d) The mean (± SEM) gradient of the relationship between ISR and glucose (ΔISR/ΔGlucose) and its intercept (a and d, respectively) for participants with NFG/NGT (white circles), NFG/IGT (black circles), IFG/NGT (black squares) and IFG/IGT (black triangles) during the GGI. There was no correlation of either variable with fasting (b and e, respectively) and peak (c and f, respectively) glucose concentrations at the time of the screening OGTT. R2 and p values represent results from a multivariate analysis adjusting for the effects of age, sex and weight. The inset panels represent residuals after adjusting for these covariates
To avoid categorical errors in the classification of participants with/without IFG and/or IGT, we examined the relationship of individual fasting glucose concentrations (and post-challenge values) observed at the time of screening with these indices. After adjusting for the effects of age, sex and weight, the ΔISR/ΔGlucose gradient did not correlate with fasting glucose concentrations observed at the time of screening (Fig. 3b) or with peak post-OGTT glucose (Fig. 3c) or with 120 min glucose concentrations (data not shown).
Since the relationship of ISR to glucose for every individual is described both by its gradient and by its intercept, we also examined the latter variable across the groups defined by fasting glucose status (Fig. 3d) and by glucose tolerance status. In addition, we examined the relationship of individual fasting glucose concentrations (Fig. 3e) and post-challenge values (Fig. 3f) observed at the time of screening after adjusting for the effects of age, sex and weight. A similar lack of correlation was observed.
The relationship of GSR and glucose concentrations in groups classified by fasting glucose status and by glucose tolerance status
Fasting GSR did not differ across the four groups. Similarly, rates of glucagon secretion did not differ over time in the NFG/IGT, IFG/NGT and IFG/IGT groups compared with the NFG/NGT group (Fig. 4a–c). In all groups, GSR exhibited an inverse exponential relationship with increasing glucose concentrations. Although nadir glucagon secretion did not differ among the groups (Fig. 4d, e), participants with IFG/NGT (Fig. 4e) and IFG/IGT (Fig. 4f) required higher glucose concentrations to suppress GSR to the values observed in NFG/NGT. See also ESM Fig. 2 for data from representative individuals.

The mean (± SEM) GSR over time observed during the GGI for participants with NFG/IGT (a), those with IFG/NGT (b) and those with IFG/IGT (c), respectively, compared with those with NFG/NGT. The corresponding panels (d–f) show the relationship of GSR (mean ± SEM) and glucose concentrations. In addition, the symbols (black circle [NFG/IGT], black square [IFG/NGT], black triangle [IFG/IGT] and white circle [NFG/NGT]) in panels (d–f) show the relationship between mean fasting GSR and glucose on the day of the screening visit
At the time of the screening OGTT, the relationship of GSR with fasting glucose exhibited a similar relationship to that observed during the GGI study day (as shown in Fig. 4d–f).
The G50 in groups classified by fasting glucose status and by glucose tolerance status and its relationship with fasting and post-challenge glucose concentrations
The inverse exponential relationship of glucagon secretion with glucose concentrations enables calculation of the increase in glucose concentration necessary to suppress GSR by 50%. This index (G50) allows comparison of alpha cell responsivity to glucose (Fig. 5a).

(a) The mean G50 (± SEM) for participants with NFG/NGT (white circles), NFG/IGT (black circles), IFG/NGT (black squares) and IFG/IGT (black triangles) during the GGI. *p<0.05 for a post hoc Tukey’s test vs the NFG/NGT group after a one-way ANOVA test. G50 correlated with fasting (b) and peak (c) glucose concentrations at the time of the screening OGTT. R2 and p values represent results from a multivariate analysis adjusting for the effects of age, sex and weight. The inset panels represent residuals after adjusting for these covariates
In this cohort, G50 differed significantly (p<0.01) across the four groups characterised by fasting glucose, glucose tolerance or peak post-OGTT glucose concentrations. People with IFG/NGT (2.4 ± 0.2 mmol/l) and IFG/IGT (2.7 ± 0.6 mmol/l), but not those with NFG/IGT (1.6 ± 0.3 mmol/l), exhibited higher values than those observed in people with NFG/NGT (1.4 ± 0.2 mmol/l; Fig. 5a).
To avoid categorical errors in the classification of participants with/without IFG and/or IGT, we examined the relationship of individual fasting glucose concentrations (and post-challenge values) observed at the time of screening with G50. After adjusting for the effects of age, sex and weight, G50 correlated (R2=0.15, p=0.02) with fasting glucose concentrations observed at the time of screening (Fig. 5b). G50 also correlated with peak post-OGTT glucose (Fig. 5c) and with 120 min glucose concentrations (data not shown) observed at the time of screening.
Discussion
Prediabetes is a heterogenous condition with variable progression to type 2 diabetes. Conventionally, a 2 h 75 g OGTT has been used to classify prediabetes based on fasting and 120 min glucose concentrations (ESM Fig. 1, Table 1) [30]. Of the resulting three subgroups of prediabetes, the group with isolated IFG (i.e. IFG/NGT) has courted the most controversy [31]. Prior work suggested that these individuals exhibit normal beta cell function in response to an oral challenge although they still are at increased risk of progression to type 2 diabetes [12]. In addition, we previously demonstrated that beta cell function quantified by DI did not correlate with fasting glucose concentrations in this population [5].
A possible explanation for these observations is that people with IFG/NGT have an altered set-point for insulin secretion but a normal response to a subsequent glucose challenge. Indeed, there is considerable genetic evidence to suggest that fasting glucose concentrations are regulated independently of postprandial glucose concentrations [9, 32, 33]. To test our primary hypothesis, we used an experimental design where glucose concentrations were lowered to similar values in all participants. We then compared the relationship of ISR with changing glucose concentrations, during a GGI (Fig. 2).
We did not observe a significant difference of the ΔISR/ΔGlucose gradient across the spectrum of prediabetes when compared with NFG/NGT (Fig. 3), at least in the range of glucose concentrations observed during the study. These observations are congruent with the data obtained in response to OGTT (Table 1), where, as before, beta cell responsivity to glucose (Φ) is unchanged across categories of prediabetes [6]. However, when expressed as a function of prevailing insulin sensitivity (Si ) as a DI, beta cell function decreases with worsening glucose tolerance (Table 1). Future development of a novel model enabling expression of insulin secretion as a function of insulin action during GGI may allow discrimination of differences in islet function across subtypes of prediabetes, as is the case for OGTT.
During the GGI, we did not observe glucose concentrations below values where insulin secretion would be completely suppressed. In addition, we and others [22] did not observe an inflection point at which the ΔISR/ΔGlucose relationship changed over the range of observed glucose concentrations. More germane to our hypothesis, ISRs at glucose concentrations within the normal fasting range did not differ from those observed in NFG/NGT in people with IFG/NGT and IFG/IGT. This leads us to reject our primary hypothesis. Indeed beta cell responsivity to (fasting) glucose at the time of screening (Φbasal; Table 1) also did not differ between these two groups. It is also notable that the relationship of fasting ISR to fasting glucose concentrations observed at the time of screening was congruent with that observed during the GGI (Fig. 2).
Intriguingly, both Φbasal from the OGTT and the ISR observed during the GGI in this range of glucose concentrations were increased in people with NFG/IGT. Impaired insulin action is the primary defect observed in such individuals [34] and several authors have proposed that the consequent chronic beta cell stimulation may impair insulin secretion [35, 36]. NEFA are elevated after an overnight fast in individuals with impaired insulin action [37] and the rise in NEFA stimulates beta cell function [38, 39]. However, differences in NEFA concentrations are unlikely to explain our observation in NFG/IGT given the exposure to insulin (which would suppress NEFA) prior to the GGI. Both the gradient and intercept (Fig. 3) for the relationship of ISR with glucose concentrations did not correlate with fasting and post-challenge glucose concentrations observed at the time of screening.
Since defects in beta cell glucose sensing alone do not explain IFG, we examined whether alpha cell dysfunction contributes to the dysregulation of fasting glucose. Prior work using an OGTT or a mixed meal challenge has failed to show significant differences in absolute fasting glucagon concentrations across the subgroups of prediabetes [6]. This also appeared to be the case in our experiments (ESM Fig. 1, Table 1). However, there is genetic and observational evidence to suggest that alpha cell dysfunction contributes to progression to type 2 diabetes [16, 18].
When GSRs were deconvolved from glucagon concentrations observed during the GGI, we demonstrated a single-exponential relationship of glucagon secretion with glucose concentrations (Fig. 4). This relationship can be quantified by the change in glucose necessary to suppress the GSR by 50% (G50). In fact, people with IFG with or without IGT exhibited higher values of G50 compared with people with NFG, implying that alpha cell insensitivity to glucose contributes to IFG (Fig. 5). Indeed, when fasting GSRs are plotted against fasting glucose concentrations observed at the time of screening, these points lie on the regression line for glucagon secretion and glucose concentrations observed during the GGI (Fig. 4). This observation demonstrates that alpha cell dysfunction, as characterised during the GGI, can explain fasting hyperglycaemia in people with IFG. Impaired suppression of glucagon secretion by glucose, as quantified by increased G50, also correlated with increased post-challenge glucose concentrations at the time of screening (Fig. 5). This is consistent with the previously observed effects of impaired glucagon suppression on postprandial glucose concentrations even when insulin secretion is relatively preserved [40]. There was no correlation of G50 with surrogates of beta cell function (data not shown).
This experiment has some limitations that need to be addressed. The first is the relatively limited numbers of individuals in the subcategories of prediabetes. Similarly, given the variability in fasting and post-OGTT glucose concentrations, there is the possibility of categorical classification errors [41, 42]. To overcome these limitations, we have analysed the relationship of the study variables with fasting and post-challenge glucose concentrations observed at the time of screening, as continuous variables. To ensure comparable glucose concentrations across all participants on the day of the GGI, we infused insulin (−120 to −30 min). Although exogenous insulin can alter beta cell function, which might affect insulin secretion [43], the short t1/2 of insulin would ensure the absence of exogenous insulin by the start of glucose infusion (0 min). C-peptide-based measures of insulin secretion ensure that our results are not influenced by the presence of exogenous insulin. It is notable that the relationships of ISR and of GSR with glucose concentrations in the fasting state were congruent between the screening and study days (Figs 2, 4). This also suggests that exogenous insulin infusion did not alter islet function during the GGI.
Strengths of the experiment include the ability to compare beta cell and alpha cell function in response to an oral and intravenous challenge, and the relatively large overall sample size. The ability to deconvolve glucagon secretion from glucagon concentrations allows description of the exponential relationship between glucagon secretion and glucose concentrations. Furthermore, we propose a novel index that can quantify the suppression of the alpha cell by glucose in an individual. This index (G50) is increased in individuals with IFG and indeed appears to be the only identifiable abnormality of islet function in people with isolated IFG (IFG/NGT). An important corollary of this work is to emphasise the heterogeneity of prediabetes and the need to adapt therapy to the underlying pathogenic mechanisms. In a setting where alpha cell function is a significant contributor to hyperglycaemia, it may be reasonable to avoid pharmacotherapy that significantly increases fasting glucagon secretion [44].
In summary, we conclude that contrary to our hypothesis, defects in insulin secretion at low to normal glucose concentrations do not play a role in the pathogenesis of isolated IFG. Somewhat paradoxically, insulin secretion at lower glucose concentrations was enhanced in people with NFG/IGT. Whether this is a marker of early beta cell stress or of progression to type 2 diabetes remains to be ascertained. More importantly, we provide additional evidence for alpha cell dysfunction contributing to the pathogenesis of prediabetes, particularly in isolated IFG. In addition, we describe a novel index, G50, which is a marker of alpha cell responsiveness to glucose and may be useful in better characterising the heterogenous pathophysiology of prediabetes.
Data availability
The de-identified datasets generated during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Φ:
-
Beta cell responsivity
- Φd :
-
Dynamic beta cell responsivity
- Φs :
-
Static beta cell responsivity
- CRTU:
-
Clinical Research and Trials Unit
- DI:
-
Disposition index
- G50 :
-
Increment in glucose concentration necessary to suppress GSR by 50%
- GGI:
-
Graded glucose infusion
- GSR:
-
Glucagon secretion rate
- IFG:
-
Impaired fasting glucose
- IFG/IGT:
-
Impaired fasting glucose with impaired glucose tolerance
- IGT:
-
Impaired glucose tolerance
- IFG/NGT:
-
Impaired fasting glucose with normal glucose tolerance
- ISR:
-
Insulin secretion rate
- NFG:
-
Normal fasting glucose
- NFG/IGT:
-
Normal fasting glucose with impaired glucose tolerance
- S i :
-
Insulin sensitivity
References
Knowler WC, Barrett-Connor E, Fowler SE et al (2002) Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 346:393–403. https://doi.org/10.1056/NEJMoa012512
Dinneen SF, Maldonado D 3rd, Leibson CL et al (1998) Effects of changing diagnostic criteria on the risk of developing diabetes. Diabetes Care 21:1408–1413. https://doi.org/10.2337/diacare.21.9.1408
Imamura F, Mukamal KJ, Meigs JB et al (2013) Risk factors for type 2 diabetes mellitus preceded by beta-cell dysfunction, insulin resistance, or both in older adults: the Cardiovascular Health Study. Am J Epidemiol 177:1418–1429. https://doi.org/10.1093/aje/kws440
Nguyen NT, Nguyen X-MT, Lane J, Wang P (2011) Relationship between obesity and diabetes in a US adult population: findings from the National Health and Nutrition Examination Survey, 1999-2006. Obes Surg 21:351–355. https://doi.org/10.1007/s11695-010-0335-4
Sathananthan A, Man CD, Zinsmeister AR et al (2012) A concerted decline in insulin secretion and action occurs across the spectrum of fasting and postchallenge glucose concentrations. Clin Endocrinol 76:212–219. https://doi.org/10.1111/j.1365-2265.2011.04159.x
Bock G, Dalla Man C, Campioni M et al (2006) Pathogenesis of pre-diabetes: mechanisms of fasting and postprandial hyperglycemia in people with impaired fasting glucose and/or impaired glucose tolerance. Diabetes 55:3536–3549. https://doi.org/10.2337/db06-0319
Sharma A, Varghese RT, Shah M et al (2018) Impaired Insulin Action is Associated with Increased Glucagon Concentrations in Non-Diabetic Humans. J Clin Endocrinol Metab 103:314–319. https://doi.org/10.1210/jc.2017-01197
Cobelli C, Dalla Man C, Toffolo G, Basu R, Vella A, Rizza R (2014) The oral minimal model method. Diabetes 63:1203–1213. https://doi.org/10.2337/db13-1198
Smushkin G, Vella A (2010) Genetics of type 2 diabetes. Curr Opin Clin Nutr Metab Care 13:471–477. https://doi.org/10.1097/MCO.0b013e32833a558d
Stolerman ES, Florez JC (2009) Genomics of type 2 diabetes mellitus: implications for the clinician. Nat Rev 5:429–436. https://doi.org/10.1038/nrendo.2009.129
Saxena R, Hivert MF, Langenberg C et al (2010) Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 42:142–148. https://doi.org/10.1038/ng.521
Han SJ, Kim HJ, Kim DJ, Lee KW, Cho NH (2017) Incidence and predictors of type 2 diabetes among Koreans: A 12-year follow up of the Korean Genome and Epidemiology Study. Diabetes Res Clin Pract 123:173–180. https://doi.org/10.1016/j.diabres.2016.10.004
Rudland VL, Hinchcliffe M, Pinner J et al (2016) Identifying Glucokinase Monogenic Diabetes in a Multiethnic Gestational Diabetes Mellitus Cohort: New Pregnancy Screening Criteria and Utility of HbA1c. Diabetes Care 39:50–52. https://doi.org/10.2337/dc15-1001
Basu A, Basu R, Shah P et al (2001) Type 2 diabetes impairs splanchnic uptake of glucose but does not alter intestinal glucose absorption during enteral glucose feeding: additional evidence for a defect in hepatic glucokinase activity. Diabetes 50:1351–1362. https://doi.org/10.2337/diabetes.50.6.1351
Basu A, Basu R, Shah P et al (2000) Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism: evidence for a defect in hepatic glucokinase activity. Diabetes 49:272–283. https://doi.org/10.2337/diabetes.49.2.272
Shah M, Varghese RT, Miles JM et al (2016) TCF7L2 Genotype and alpha-Cell Function in Humans Without Diabetes. Diabetes 65:371–380. https://doi.org/10.2337/db15-1233
Faerch K, Vistisen D, Pacini G et al (2016) Insulin Resistance Is Accompanied by Increased Fasting Glucagon and Delayed Glucagon Suppression in Individuals With Normal and Impaired Glucose Regulation. Diabetes 65:3473–3481. https://doi.org/10.2337/db16-0240
Adams JD, Dalla Man C, Laurenti MC et al (2020) Fasting glucagon concentrations are associated with longitudinal decline of beta-cell function in non-diabetic humans. Metabolism 105:154175. https://doi.org/10.1016/j.metabol.2020.154175
Faerch K, Vaag A, Holst JJ, Glumer C, Pedersen O, Borch-Johnsen K (2008) Impaired fasting glycaemia vs impaired glucose tolerance: similar impairment of pancreatic alpha and beta cell function but differential roles of incretin hormones and insulin action. Diabetologia 51:853–861. https://doi.org/10.1007/s00125-008-0951-x
Abdul-Ghani MA, Jenkinson CP, Richardson DK, Tripathy D, DeFronzo RA (2006) Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: results from the Veterans Administration Genetic Epidemiology Study. Diabetes 55:1430–1435. https://doi.org/10.2337/db05-1200
Bock G, Dalla Man C, Campioni M et al (2007) Effects of nonglucose nutrients on insulin secretion and action in people with pre-diabetes. Diabetes 56:1113–1119. https://doi.org/10.2337/db06-1272
Shankar SS, Shankar RR, Mixson LA et al (2016) Linearity of beta-cell response across the metabolic spectrum and to pharmacology: insights from a graded glucose infusion-based investigation series. Am J Physiol Endocrinol Metab 310:E865–E873. https://doi.org/10.1152/ajpendo.00527.2015
Toffolo G, Breda E, Cavaghan MK, Ehrmann DA, Polonsky KS, Cobelli C (2001) Quantitative indexes of beta-cell function during graded up&down glucose infusion from C-peptide minimal models. Am J Physiol Endocrinol Metab 280:E2–E10. https://doi.org/10.1152/ajpendo.2001.280.1.E2
Laurenti MC, Vella A, Adams JD, Schembri Wismayer DJ, Egan AM, Dalla Man C (2021) Assessment of individual and standardized glucagon kinetics in healthy humans. Am J Physiol Endocrinol Metab 320:E71–E77. https://doi.org/10.1152/ajpendo.00488.2020
Vella A, Shah P, Basu R, Basu A, Holst JJ, Rizza RA (2000) Effect of glucagon-like peptide 1(7-36) amide on glucose effectiveness and insulin action in people with type 2 diabetes. Diabetes 49:611–617. https://doi.org/10.2337/diabetes.49.4.611
Van Cauter E, Mestrez F, Sturis J, Polonsky KS (1992) Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 41:368–377. https://doi.org/10.2337/diab.41.3.368
Mitrakou A, Ryan C, Veneman T et al (1991) Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am J Phys 260:E67–E74. https://doi.org/10.1152/ajpendo.1991.260.1.E67
Dalla Man C, Campioni M, Polonsky K et al (2005) Two-hour seven-sample oral glucose tolerance test and meal protocol: minimal model assessment of beta-cell responsivity and insulin sensitivity in nondiabetic individuals. Diabetes 54(11):3265–3273. https://doi.org/10.2337/diabetes.54.11.3265
Shankar SS, Vella A, Raymond RH et al (2016) Standardized Mixed-Meal Tolerance and Arginine Stimulation Tests Provide Reproducible and Complementary Measures of beta-Cell Function: Results From the Foundation for the National Institutes of Health Biomarkers Consortium Investigative Series. Diabetes Care 39(9):1602–1613. https://doi.org/10.2337/dc15-0931
Colagiuri S (2021) Definition and Classification of Diabetes and Prediabetes and Emerging Data on Phenotypes. Endocrinol Metab Clin N Am 50:319–336. https://doi.org/10.1016/j.ecl.2021.06.004
Kim SH, Reaven GM (2008) Isolated impaired fasting glucose and peripheral insulin sensitivity: not a simple relationship. Diabetes Care 31:347–352. https://doi.org/10.2337/dc07-1574
Byrne MM, Sturis J, Clement K et al (1994) Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 93:1120–1130. https://doi.org/10.1172/JCI117064
Matschinsky FM, Glaser B, Magnuson MA (1998) Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes 47:307–315. https://doi.org/10.2337/diabetes.47.3.307
Varghese RT, Dalla Man C, Sharma A et al (2016) Mechanisms Underlying the Pathogenesis of Isolated Impaired Glucose Tolerance in Humans. J Clin Endocrinol Metab 101:4816–4824. https://doi.org/10.1210/jc.2016-1998
Leahy JL (1990) Natural history of beta-cell dysfunction in NIDDM. Diabetes Care 13:992–1010. https://doi.org/10.2337/diacare.13.9.992
Polonsky KS, Sturis J, Bell GI (1996) Seminars in Medicine of the Beth Israel Hospital, Boston. Non-insulin-dependent diabetes mellitus - a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med 334:777–783. https://doi.org/10.1056/NEJM199603213341207
Lu J, Varghese RT, Zhou L, Vella A, Jensen MD (2017) Glucose tolerance and free fatty acid metabolism in adults with variations in TCF7L2 rs7903146. Metabolism 68:55–63. https://doi.org/10.1016/j.metabol.2016.11.018
Broussard JL, Kolka CM, Castro AVB et al (2015) Elevated nocturnal NEFA are an early signal for hyperinsulinaemic compensation during diet-induced insulin resistance in dogs. Diabetologia 58:2663–2670. https://doi.org/10.1007/s00125-015-3721-6
Miller MR, Pereira RI, Langefeld CD et al (2012) Levels of free fatty acids (FFA) are associated with insulin resistance but do not explain the relationship between adiposity and insulin resistance in Hispanic Americans: the IRAS Family Study. J Clin Endocrinol Metab 97:3285–3291. https://doi.org/10.1210/jc.2012-1318
Shah P, Basu A, Basu R, Rizza R (1999) Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Phys 277:E283–E290. https://doi.org/10.1152/ajpendo.1999.277.2.E283
Bergman M, Abdul-Ghani M, DeFronzo RA et al (2020) Review of methods for detecting glycemic disorders. Diabetes Res Clin Pract 165:108233. https://doi.org/10.1016/j.diabres.2020.108233
Balion CM, Raina PS, Gerstein HC et al (2007) Reproducibility of impaired glucose tolerance (IGT) and impaired fasting glucose (IFG) classification: a systematic review. Clin Chem Lab Med 45:1180–1185. https://doi.org/10.1515/CCLM.2007.505
Lopez X, Cypess A, Manning R, O'Shea S, Kulkarni RN, Goldfine AB (2011) Exogenous insulin enhances glucose-stimulated insulin response in healthy humans independent of changes in free fatty acids. J Clin Endocrinol Metab 96:3811–3821. https://doi.org/10.1210/jc.2011-0627
Ferrannini E, Muscelli E, Frascerra S et al (2014) Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Investig 124:499–508. https://doi.org/10.1172/JCI72227
Acknowledgements
The authors wish to acknowledge the excellent editorial assistance of M. M. Davis, Endocrine Research Unit, Mayo Clinic, Rochester, MN, USA and the discussions that informed the study design with R. A. Rizza, Mayo Clinic College of Medicine, Rochester, MN, USA.
Authors’ relationships and activities
AV is the recipient of an investigator-initiated grant from Novo Nordisk and has consulted for vTv Therapeutics, Zeeland Pharmaceuticals, Crinetics and Rezolute. The other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
JDK researched data and ran the studies, contributed to the discussion and reviewed/edited the manuscript; MCL undertook mathematical modelling of insulin and glucagon secretion, contributed to the discussion and reviewed/edited the manuscript; AME researched data and ran the studies, contributed to the discussion and reviewed/edited the manuscript; DSW assisted with data management and organisation as well as with the data analysis, contributed to the discussion and reviewed/edited the manuscript; KRB supervised the statistical analyses, contributed to the discussion and reviewed/edited the manuscript; CC and CDM supervised the mathematical modelling, contributed to the discussion and reviewed/edited the manuscript; AV designed the study, oversaw its conduct, researched data and wrote the first draft of the manuscript. AV is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. The order of first authors was determined by the time that each joined the project. All authors approved the final version of the manuscript.
Funding
The authors acknowledge the support of the Mayo Clinic General Clinical Research Center (DK TR000135). AV is supported by DK78646, DK116231 and DK126206. JDK received grant support from the Endocrine Fellows Foundation. CDM was supported by MIUR (Italian Ministry for Education) under the initiative ‘Departments of Excellence’ (Law 232/2016).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM
(PDF 362 kb)
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kohlenberg, J.D., Laurenti, M.C., Egan, A.M. et al. Differential contribution of alpha and beta cell dysfunction to impaired fasting glucose and impaired glucose tolerance. Diabetologia 66, 201–212 (2023). https://doi.org/10.1007/s00125-022-05794-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05794-3