
Abstract
Unlike bromine, the effect of iodine chemistry on the Arctic surface ozone budget is poorly constrained. We present ship-based measurements of halogen oxides in the high Arctic boundary layer from the sunlit period of March to October 2020 and show that iodine enhances springtime tropospheric ozone depletion. We find that chemical reactions between iodine and ozone are the second highest contributor to ozone loss over the study period, after ozone photolysis-initiated loss and ahead of bromine.
Similar content being viewed by others
Main
While the importance of bromine chemistry for the Arctic surface ozone budget is well documented, the effect of iodine chemistry has received less attention1,2. We present observations over the sunlit period in the Arctic that demonstrate active iodine chemistry and show that it causes substantial ozone destruction. Iodine enhances springtime tropospheric ozone depletion and is the second most important ozone loss pathway, after ozone photolysis-initiated loss and ahead of bromine over the whole sunlit period.
Halogen compounds can cause catalytic destruction of ozone1. Past observations have shown that up to 45% of the ozone loss in the tropical marine boundary layer (BL) can be attributed to halogen-mediated depletion3. Episodic increases of reactive bromine cause ozone depletion in the Arctic BL2. In the Antarctic, elevated levels of iodine compounds have been observed throughout the sunlit period, affecting the oxidizing capacity of the atmosphere and forming ultrafine aerosol particles4. However, the relative importance of iodine chemistry in the Arctic remains unclear owing to the limited reports on the presence of active iodine chemistry. Iodine oxide (IO) has been reported in the sub-Arctic BL, where it was associated with air masses passing over polynyas5. Iodine in particulate matter6,7 and a snowpack source of molecular iodine8 have also been reported. Owing to a lack of widespread ground- or satellite-based observations, there is high uncertainty in the role of iodine chemistry as compared with bromine chemistry in the Arctic9.
We present a dataset of gas-phase halogens and ozone in the BL obtained from various locations in the remote high Arctic. In combination with a photochemical model, the data demonstrate a substantial role of iodine in Arctic BL ozone loss. Observations were made as part of the ship-based Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition10, where halogen oxides were measured during the sunlit period from March to October 2020. In situ measurements of particulate iodine, iodic acid (HIO3) and other related trace gases were also made. The cruise track and a detailed description of the expedition are given in Supplementary Fig. 1.
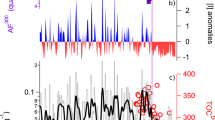
Ozone shows drastic loss during the boreal spring (March, April and May), when near-complete depletion is regularly observed (Fig. 1a,b), highlighting that ozone depletion in the central Arctic is widespread. For the remaining sunlit period, ozone rises to higher levels (10 and 40 ppbv). The drastic ozone depletion in spring coincides with ‘bromine explosion’ episodes, when bromine oxide (BrO) reaches values as high as 14.8 ± 0.8 pptv (Fig. 1c,d). Elevated BrO has been observed previously across the Arctic, including in regions with fresh sea-ice11. During boreal spring, the cruise drifted through areas where the influence of first-year sea-ice was higher (Supplementary Fig. 2), thus such enhancements were expected. Atmospheric transport calculations show that air masses observed during bromine explosion events were strongly influenced by contact with snow-covered sea-ice (Supplementary Fig. 3). For the other months, BrO mole fractions remain below the instrumental detection limit (2σ ranging between 0.5 and 3 pptv). Ship-based observations are in good agreement with the satellite-based TROPOspheric Monitoring Instrument (TROPOMI) retrievals12 (Supplementary Fig. 4). The observed seasonal variability in BrO is also seen in situ in the measured bromine-containing trace gas, where the signal is visible only in spring when ozone depletion occurs (Supplementary Fig. 5). Therefore, the combined in situ and satellite observations confirm that lower tropospheric bromine chemistry remains active during spring in the high Arctic.

a–f, Time series (a,c,e) and geographical distibutions (b,d,f) of the ozone (a,b), BrO (c,d) and IO (e,f) mixing ratios as observed during the MOSAiC expedition. BrO was above the detection limit (DL) only during the spring, coinciding with drastic ozone depletion. IO mixing ratios were larger during the spring, but IO was also detected during the sunlit period throughout the expedition, showing its widespread nature (a zoom of only spring is shown in Supplementary Fig. 8). The error bars for the BrO and IO data include the sensitivity of the sum of squares with respect to variations of the fitted parameters around the minimum and the noise on the measurements, along with errors in the result due to the path length retrieval using the oxygen dimer.
In contrast, observations of IO show the presence of active iodine chemistry on most days during the whole sunlit period (that is, boreal spring, summer and autumn; Fig. 1e,f). While day-to-day variation due to changing meteorological conditions is observed, elevated levels of IO (usually between 0.2 and 1 pptv) were regularly observed throughout the expedition. In spring, levels further enhanced up to 2.9 ± 0.3 pptv were also measured. Nonetheless, these values are lower than the spring peak in BrO during MOSAiC and are also lower than the highest IO mixing ratios (~20 pptv) reported in the Antarctic4. Previous observations of integrated vertical columns during spring suggested the presence of IO in the Arctic13, and the fact that we observed IO throughout the sunlit period along the MOSAiC cruise track shows that iodine chemistry is widespread across the Arctic. The absolute values are lower than those in the Antarctic, which probably explains the lack of satellite-based detection14. In addition to measurements of IO in the gas phase, we conducted in situ observations of gas-phase concentrations of HIO3, which show the same seasonal cycle as IO. HIO3 peaks in the spring (with a seasonal mean of 0.09 ± 0.14 pptv) compared with lower values during summer (0.009 ± 0.016 pptv). The enhanced spring values reflect a seasonal cycle similar to the one in the Antarctic4, which was attributed to biological activity underneath the sea-ice producing iodine compounds that diffuse through brine channels15. Another possible source is direct photochemical snowpack emission of I2, with higher emissions expected through the spring with increasing radiation8. Past model estimates have shown that observed snowpack I2 emissions can explain up to 1.6 pptv of IO8 but would not explain the peak IO mixing ratios observed during MOSAiC. Background levels for the rest of the sunlit period (that is, boreal summer and autumn) can also be explained by the deposition of ozone to seawater followed by a reaction with seawater iodide leading to emissions of I2 and HOI16. During this period, the observed air masses were predominantly exposed to the open ocean (Supplementary Fig. 3). This indicates that, when larger areas of the Arctic become ice free in the future, a continuously increasing oceanic flux of iodine compounds to the polar atmosphere can be expected. The reported increasing trend in ozone concentrations in the Arctic lower troposphere17 will also increase the net sea–air flux of iodine compounds, which in turn will suppress the background Arctic ozone levels due to the buffering effect of iodine. Indeed, ice-core observations of snowpack-deposited iodine reflect this feedback cycle18 and suggest that future increases in emissions of iodine compounds in the Arctic will play a greater role in modulating ozone.
Elevated BrO mixing ratios were observed during periods with depleted ozone. However, the highest IO values were observed on the days with the lowest ozone. This suggests that, in addition to bromine, iodine also plays a role in determining the degree of ozone loss (Supplementary Fig. 9a,b). Iodine is a more potent ozone destruction agent than bromine owing to its faster chemistry19. Hitherto, ozone depletion in the Arctic has been mostly attributed to bromine, with a small contribution from chlorine, although it has been suggested that iodine could also play a role8. Our observations show that iodine chemistry played a crucial role during at least some periods of extremely low ozone. Model simulations (Supplementary Fig. 9c and Supplementary Table 2) show that, during spring, the observed IO concentrations can increase ozone destruction by a factor of 2–3 fold, depending on the bromine loading (Supplementary Fig. 9d).
A substantial effect of iodine, which has not been considered to date, is its cumulative effect on ozone destruction over the sunlit period. While bromine plays a major role in spring, BrO is not observed above the detection limit during the rest of the year. However, IO is also observed during summer and autumn, that is, throughout the sunlit period, albeit at lower levels than during spring. Given that iodine was found to be persistent during MOSAiC, it is expected to affect large areas of the Arctic region. Model simulations (Fig. 2 and Supplementary Table 2) show the relative contribution of individual reaction pathways to the chemical loss of ozone on days when halogen observations were available. During spring, most of the ozone depletion is driven by halogens. The maximum individual daily contribution of bromine to ozone loss was ~70% (3 April 2020), while the maximum single-day iodine contributed loss was about 75% (26 March 2020). Together, iodine and bromine chemistry contribute a maximum of 92% of the ozone loss on 26 March 2020. When averaged over spring, iodine and bromine contribute about 56% of the ozone loss (bromine 26%, iodine 29%). This shows that, even during spring, the integrated relative loss of ozone due to iodine is comparable to that of bromine. During the other sunlit seasons, increased solar radiation results in the dominant role of ozone photolysis loss (followed by the reaction of O1D with water vapour), but iodine chemistry still accounts for as much as 25% of the monthly ozone loss during August. As BrO was not observed above the detection limit, the relative contribution of bromine is expected to be smaller. When averaged over the whole sunlit period, photolysis-driven loss accounts for 60 ± 20%, iodine chemistry contributes 16 ± 10% and bromine chemistry accounts for 14 ± 9% of the total ozone loss. This makes iodine chemistry the second most important ozone loss pathway in the Arctic atmosphere. Our study confirms, via direct field observations, that iodine chemistry is critical for understanding the ozone budget in the Arctic. This changes the decades of paradigm on the drivers of Arctic photochemical ozone loss, where the cumulative effect of iodine on the Arctic ozone budget was not considered, although reported emissions of I2 have demonstrated a possible role8.

a, The relative loss for each day of the expedition. b–d, The monthly median value (horizontal line within the box), quartiles (box) and range excluding outliers (with error bars indicating 1.5 times the quartiles) and outliers (diamonds, data beyond 1.5 times the quartiles) for the three most important loss processes: Ox (ozone photolysis followed by reaction of O1D with H2O) (b), bromine (c) and iodine (d). The mean and standard deviation for the measurement period are also indicated for the sunlit period. The results show that iodine-catalysed reactions represent a significant photochemical loss pathway for ozone, during the spring and cumulatively throughout the year.
These results show the importance of iodine chemistry in regulating the pan-Arctic oxidizing capacity through ozone destruction. Iodine was as important as bromine during spring when the depletion of ozone was observed and represents the second most important chemical loss pathway over the entire sunlit period. The presence of iodine compounds along the whole cruise track, together with past observations of molecular iodine8 and aerosol-based iodine6,20, suggests that this effect is not localized but rather pan-Arctic. Given the increase in the iodine atmospheric loading due to enhanced anthropogenic ozone-induced ocean iodine emissions18, as well as the thinning and shrinking of Arctic sea-ice expected in the near future probably leading to increases in iodine emissions, these results indicate that iodine chemistry could play an increasingly important role in the future and must be considered for accurate quantification of the ozone budget in the Arctic.
Methods
Observations of ozone and other trace gases
Ozone ambient air concentrations were monitored using commercial instruments in three different sea-laboratory containers: a Thermo Fisher Scientific instrument (model 49i) in the Atmospheric Radiation Measurement (ARM) container, a Thermo Environmental Instruments analyser (model 49c) in the University of Colorado (CU) container and a 2B Technologies (model 205) in the so-called Swiss container. The individual ozone time series were cleaned for local anthropogenic pollution spikes (ship exhaust or other ongoing activities, for example, use of skidoos) and were cross-calibrated using the ARM instrument as a calibration reference. These cross-calibrated individual datasets were used to generate the hourly-averaged merged dataset used in this study.
CH4 dry air mole fractions were measured aboard Polarstern in two different containers and on sea-ice at Met City with cavity ring-down spectrometers: a Picarro model G2311-f at Met City and in the CU container and a Picarro model G2401 in the Swiss container. Mole fractions were adjusted after cross-calibration against whole-air discrete samples collected for post-cruise analysis by the NOAA Global Monitoring Laboratory Carbon Cycle Cooperative Global Air Sampling Network (https://gml.noaa.gov/ccgg/). These cross-calibrated individual datasets were used to generate the hourly-averaged merged dataset used in this study.
CO ambient air mole fractions were measured in the ARM and Swiss containers by using an off-axis integrated cavity output spectroscopy instrument (Los Gatos Research model 098-0014) and a Picarro model G2401, respectively. Mole fractions were also adjusted after cross-calibration against whole-air discrete samples collected for post-cruise analysis, and these cross-calibrated individual datasets were used to generate the hourly-averaged merged dataset used in this study.
NOx (sum of NO and NO2) ambient air mole fractions were measured by chemiluminescence22 during the global-scale airborne Atmospheric Tomography mission (ATom; https://espo.nasa.gov/atom/) onboard the NASA-DC8 aircraft22,23. For our analysis, we used data collected in the Arctic (70–90° N) marine boundary layer (<1 km altitude).
IO and BrO observations
Observations of IO and BrO were made using the multi-axis differential optical absorption spectroscopy technique24,25,26. This technique uses scattered sunlight along different elevation angles to compute the concentration and vertical profiles of spectroscopically active chemical species in the atmosphere. The instrument, which has been used to measure halogen oxides in remote environments in the past27, consists of a telescope connected to a spectrometer using an optical fibre. The collected spectra are then analysed using the QDOAS software28 to identify the absorption due to the oxygen dimer (O4) in two different wavelength regions and the absorptions due to BrO and IO. This analysis results in the differential slant column densities (DSCDs) relative to the zenith for both species. Further details of the spectral analysis and the different molecular absorption cross-sections are given in the Supplementary Information. The calculation of mixing ratios is a two-step process where the O4 DSCDs are used to estimate the light paths and subsequently derive the BrO and IO concentrations. The AC-2 radiative transfer model29 is used to estimate the light path using the measured O4 DSCDs, and these light paths are then used to calculate the trace gas mixing ratios in the boundary layer.
Iodine concentrations in aerosols
Iodine concentrations in aerosols were measured in the Swiss container using an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (AMS)30 equipped with a 1 µm aerodynamics lens allowing the detection of non-refractive components of particulate matter. The particulate non-refractory iodide concentrations were retrieved by fitting the I+ ion in high-resolution analysis using PIKA. Here I+ (m/z = 126.9) was the dominant iodine-containing fragment ion, consistent with previous studies31. We used a relative ionization efficiency of 0.73 for iodide based on AMS calibration using iodide salts performed in ref. 32. The iodine concentration was corrected for composition-dependent collection efficiency33. No pollution mask was applied for the iodide data because the changes in iodide concentration were independent of pollution markers such as C4H9 (m/z = 57). Data during maintenance periods (of the filter and for calibrations) were discarded.
HIO3 and Br− observations
A chemical ionization atmospheric-pressure interface time-of-flight (CI-APi-TOF) mass spectrometer, described in detail in ref. 34, was used to detect traces of iodic acid (HIO3) and the signal of Br−. In brief, the CI-APi-TOF sampled from a new particle formation-dedicated inlet that was designed to minimize diffusional losses with other measurements (reaching a combined inlet flow of 70 lpm). The new particle formation inlet was ~1.3 m long with a diameter of 10.2 cm, and the CI-APi-TOF sampled the core flow utilizing a short, ¾ inch inlet tube of the chemical ionization inlet. The instrument was calibrated using sulphuric acid, and we obtained a calibration factor of 6 × 109 including diffusional losses in the used new particle formation inlet. To calculate HIO3 concentrations, we used the sum of two distinct HIO3 peaks in the spectra, that is, IO3− (exact mass 174.8898 Th) and HNO3IO3− (exact mass 237.8854), normalized the signals using the sum of charger ions (NO3−, HNO3NO3− and (HNO3)2NO3−) and multiplied the result by the calibration factor. We use the same calibration factor as for sulphuric acid, which charges at the kinetic limit because the proton affinity of IO3− is significantly lower than that of the charger ions, thus collision limit charging can be assumed35.
The Br− signal is a trace of, for example, neutral HBr or a Br− fragment from other Br−-containing molecule that charges with the aforementioned charger ions. The signal reported here is a Br− signal (exact mass 78.9189 Th, identification based on both the exact mass and isotopic pattern) normalized with the charger ions only.
Satellite-based BrO observations
Satellite BrO columns from the TROPOMI instrument on Sentinel-5 Precursor were retrieved following the approach described in ref. 36 for the slant columns. In brief, the differential optical absorption spectroscopy method was applied to TROPOMI radiances in the fitting range of 334.6–358 nm. As interfering species, O3, NO2, HCHO, OClO and O4 were included in the fit, as well as a correction term for the Ring effect, a liner intensity offset and a polynomial of degree five. We used daily row-specific background spectra created using TROPOMI data over the equatorial Pacific taken on the same day. The resulting offset in the absolute values was accounted for by assuming a constant vertical BrO column of 3.5 × 1013 molecules cm−2 over the background region and adding this to the fitted differential slant columns. The slant columns were converted to tropospheric columns using the stratospheric correction approach from ref. 37 using operational stratospheric ozone and NO2 columns from Sentinel-5 Precursor and tropopause heights derived from the National Centers for Environmental Prediction Reanalysis 1 product38. For the conversion to vertical columns, an airmass factor appropriate for a surface BrO layer of 400 m thickness over a bright surface was applied.
Modelling
The impact of both halogens on the atmospheric oxidation capacity was estimated using the Tropospheric Halogen Chemistry Model chemical box model39,40. Further details regarding the model setup along with a list of reactions included in the model are given in the Supplementary Information.
Data availability
The data that support the findings of this study are available online in the following repositories. The halogen data is available at Mahajan, Anoop (2022), ‘Substantial contribution of iodine to Arctic ozone destruction - data’, Mendeley Data, V1, https://doi.org/10.17632/bn7ytz4mfz.1. Merged datasets for ozone (https://doi.pangaea.de/10.1594/PANGAEA.944393), methane (https://doi.pangaea.de/10.1594/PANGAEA.944291) and carbon monoxide (https://doi.pangaea.de/10.1594/PANGAEA.944389) are available. The satellite-retrieved BrO is available at (https://www.iup.uni-bremen.de/doas/mosaic.htm). The backtrajectory data along with the sea-ice influence are available at https://srvx1.img.univie.ac.at/webdata/mosaic/mosaic.html.
Code availability
The model code is available at Mahajan, Anoop (2022), ‘Substantial contribution of iodine to Arctic ozone destruction - data’, Mendeley Data, V1, https://doi.org/10.17632/bn7ytz4mfz.1.
References
Saiz-Lopez, A. & von Glasow, R. Reactive halogen chemistry in the troposphere. Chem. Soc. Rev. https://doi.org/10.1039/c2cs35208g (2012).
Barrie, L. A., Bottenheim, J. W., Schnell, R. C., Crutzen, P. J. & Rasmussen, R. A. Ozone destruction and photochemical reactions at polar sunrise in the lower Arctic atmosphere. Nature 334, 138–141 (1988).
Read, K. A. et al. Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean. Nature 453, 1232–1235 (2008).
Saiz-Lopez, A. et al. Boundary layer halogens in coastal Antarctica. Sci. (80-.) 317, 348–351 (2007).
Mahajan, A. S. et al. Evidence of reactive iodine chemistry in the Arctic boundary layer. J. Geophys. Res. 115, D20303 (2010).
Baccarini, A. et al. Frequent new particle formation over the high Arctic pack ice by enhanced iodine emissions. Nat. Commun. 11, 1–11 (2020).
Allan, J. D. et al. Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA. Atmos. Chem. Phys. 15, 5599–5609 (2015).
Raso, A. R. W. et al. Active molecular iodine photochemistry in the Arctic. Proc. Natl Acad. Sci. USA 114, 10053–10058 (2017).
Halfacre, J. W. & Simpson, W. R. in Chemistry in the Cryosphere, volume 3 (eds. Shepson P. B. & Domine, F.) pp 411–452 (World Scientific, 2021).
Shupe, M. D, et al. Overview of the MOSAiC expedition: Atmosphere. Elem. Sci. Anth. 10:1. https://doi.org/10.1525/elementa.2021.00060 (2022).
McElroy, C. T., McLinden, C. A. & McConnell, J. C. Evidence for bromine monoxide in the free troposphere during the Arctic polar sunrise. Nature 397, 338–341 (1999).
Seo, S., Richter, A., Blechschmidt, A. M., Bougoudis, I. & Philip Burrows, J. First high-resolution BrO column retrievals from TROPOMI. Atmos. Meas. Tech. 12, 2913–2932 (2019).
Wittrock, F., Müller, R., Richter, A., Bovensmann, H. & Burrows, J. P. Measurements of iodine monoxide (IO) above Spitsbergen. Geophys. Res. Lett. 27, 1471–1474 (2000).
Schönhardt, A. et al. Simultaneous satellite observations of IO and BrO over Antarctica. Atmos. Chem. Phys. 12, 6565–6580 (2012).
Saiz-Lopez, A., Blaszczak-Boxe, C. S. & Carpenter, L. J. A mechanism for biologically-induced iodine emissions from sea-ice. Atmos. Chem. Phys. 15, 9731–9746 (2015).
Carpenter, L. J. et al. Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 6, 108–111 (2013).
Sharma, S. et al. A factor and trends analysis of multidecadal lower tropospheric observations of Arctic aerosol composition, black carbon, ozone, and mercury at Alert, Canada. J. Geophys. Res. Atmos. 124, 14133–14161 (2019).
Cuevas, C. A. et al. Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century. Nat. Commun. 9, 1452 (2018).
Saiz-Lopez, A. et al. Atmospheric chemistry of iodine. Chem. Rev. 112, 1773–1804 (2012).
Sturges, W. T. & Barrie, L. A. Chlorine, bromine and iodine in Arctic aerosols. Atmos. Environ. 22, 1179–1194 (1988).
Nixdorf, U. et al. MOSAiC extended acknowledgement. Zenodo https://doi.org/10.5281/ZENODO.5541624 (2021).
Bourgeois, I. et al. Comparison of airborne measurements of NO, NO2, HONO, NOy, and CO during FIREX-AQ. Atmos. Meas. Tech 15, 4901–4930 (2022).
Wofsy, S. C. et al. ATom: merged atmospheric chemistry, trace gases, and aerosols. ORNL DAAC https://doi.org/10.3334/ORNLDAAC/1581 (2018).
Lohberger, F., Hönninger, G. & Platt, U. Ground-based imaging differential optical absorption spectroscopy of atmospheric gases. Appl. Opt. 43, 4711–4717 (2004).
Platt, U. & Stutz, J. Differential Optical Absorption Spectroscopy: Principles and Applications (Springer, 2008).
Plane, J. M. C. & Saiz-Lopez, A. in Analytical Techniques for Atmospheric Measurement (ed. Heard, D. E.) pp 147–188 (Wiley-Blackwell, 2006).
Prados-Roman, C. et al. Iodine oxide in the global marine boundary layer. Atmos. Chem. Phys. 15, 583–593 (2015).
Fayt, C. & Van Roozendael, M. QDOAS 1.00 software user manual. Royal Belgian Institute for Space Aeronomy http://uv-vis.aeronomie.be/software/QDOAS/ (2013).
Benavent Oltra, N. Desarrollo de un instrumento MAX-DOAS para medidas urbanas de calidad del aire y de gases traza en la atmósfera polar (Universidad Politécnica de Madrid Escuela, 2020).
DeCarlo, P. F. et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 78, 8281–8289 (2006).
Allan, J. D. et al. Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA. Atmos. Chem. Phys. 15, 5599–5609 (2015).
Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Nault, B. A. & Schroder, J. C. A. Tom: L2 particulate iodine from high-resolution aerosol mass spectrometer (HR-AMS). ORNL DAAC https://doi.org/10.3334/ORNLDAAC/1773 (2020).
Middlebrook, A. M., Bahreini, R., Jimenez, J. L. & Canagaratna, M. R. Evaluation of composition-dependent collection efficiencies for the aerodyne aerosol mass spectrometer using field data. Aerosol Sci. Technol. 46, 258–271 (2012).
Jokinen, T. et al. Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF. Atmos. Chem. Phys. 12, 4117–4125 (2012).
Sipilä, M. et al. Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3. Nature 537, 532–534 (2016).
Seo, S., Richter, A., Blechschmidt, A. M., Bougoudis, I. & Philip Burrows, J. First high-resolution BrO column retrievals from TROPOMI. Atmos. Meas. Tech. 12, 2913–2932 (2019).
Theys, N. et al. Global observations of tropospheric BrO columns using GOME-2 satellite data. Atmos. Chem. Phys. 11, 1791–1811 (2011).
Kalnay, E. et al. The NCEP/NCAR 40-year reanalysis project. Bull. Am. Meteorol. Soc 437, 472–472 (1996).
Mahajan, A. S. Reactive Halogen Species in the Marine Boundary Layer (University of Leeds, 2009).
Saiz-Lopez, A. et al. On the vertical distribution of boundary layer halogens over coastal Antarctica: implications for O3, HOx, NOx and the Hg lifetime. Atmos. Chem. Phys. 8, 887–900 (2008).
Acknowledgements
This study received funding from the European Research Council Executive Agency under the European Union’s Horizon 2020 Research and Innovation Program (project ERC‐2016‐COG 726349 CLIMAHAL and ERC-2016-STG 714621 GASPARCON) and the European Commission via the EMME-CARE project and was supported by the Consejo Superior de Investigaciones Científicas of Spain. This work was supported by the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement no. 856612 and the Academy of Finland (project no. 334514). The Indian Institute of Tropical Meteorology is funded by the Ministry of Earth Sciences, Government of India. Ozone, CO, CH4 and AMS measurements were funded by the Swiss National Science Foundation (grant 200021_188478), the Swiss Polar Institute and U.S. National Science Foundation grants 1914781 and 1807163. J.S. holds the Ingvar Kamprad chair for extreme environments research, sponsored by Ferring Pharmaceuticals. Data reported in this manuscript were produced as part of the international MOSAiC expedition with tag MOSAiC20192020, with activities supported by Polarstern expedition AWI-PS122_00. We thank all those who contributed to MOSAiC and made this endeavour possible21. We thank all the individuals who helped operate the MAX-DOAS instrument during the expedition. H.S. was funded by the European ERA-PLANET projects iGOSP and iCUPE (consortium agreement no. 689443 for both projects). We thank FORMAS and the Swedish Polar Research Secretariat for support. We gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (project no. 268020496 – TRR 172) within the Transregional Collaborative Research Center ‘ArctiC Amplification: Climate Relevant Atmospheric and SurfaCe Processes, and Feedback Mechanisms (AC)3’ in subproject C03. We thank I. Bourgeois (NOAA/CIRES) for providing the ATom NOx data. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
A.S.-L. devised the research; C.A.C., J.A.S., N.B., J.S., H.A., A.D., P.S.P., S.B., K.A., T.J., L.L. J.Q., I.B., T.L., B.B., D. Howard, S.A., L.B., D. Helmig, J.H., H.-W.J. and K.P. performed the measurements during the cruise. A.S.M., N.B. and D.G.-N. conducted the MAX-DOAS data analysis. A.M.B., B.Z. and A.R. analysed and retrieved the satellite data. Q.L. and A.S.M. set up the model and Q.L. conducted the modelling. A.S.M., N.B., A.S.-L., Q.L., C.A.C., J.S., H.A., T.J., L.L.J.Q., R.P.F., H.S., L.D. and K.R.D. analysed the data; S.B., M.D. and A.S. performed FLEXPART analyses. A.S.M., N.B. and A.S.-L. wrote the paper with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Geoscience thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Xujia Jiang.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary description of data, model, instruments and further information of the campaign, Figs. S1–S9′ and Tables S1–S3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Benavent, N., Mahajan, A.S., Li, Q. et al. Substantial contribution of iodine to Arctic ozone destruction. Nat. Geosci. 15, 770–773 (2022). https://doi.org/10.1038/s41561-022-01018-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41561-022-01018-w
This article is cited by
-
Widespread detection of chlorine oxyacids in the Arctic atmosphere
Nature Communications (2023)
