
Abstract
Promotion of hepatic glycogen synthesis and inhibition of hepatic glucose production are effective strategies for controlling hyperglycemia in type 2 diabetes mellitus (T2DM), but agents with both properties were limited. Herein we report coronarin A, a natural compound isolated from rhizomes of Hedychium gardnerianum, which simultaneously stimulates glycogen synthesis and suppresses gluconeogenesis in rat primary hepatocytes. We showed that coronarin A (3, 10 μM) dose-dependently stimulated glycogen synthesis accompanied by increased Akt and GSK3β phosphorylation in rat primary hepatocytes. Pretreatment with Akt inhibitor MK-2206 (2 μM) or PI3K inhibitor LY294002 (10 μM) blocked coronarin A-induced glycogen synthesis. Meanwhile, coronarin A (10 μM) significantly suppressed gluconeogenesis accompanied by increased phosphorylation of MEK, ERK1/2, β-catenin and increased the gene expression of TCF7L2 in rat primary hepatocytes. Pretreatment with β-catenin inhibitor IWR-1-endo (10 μM) or ERK inhibitor SCH772984 (1 μM) abolished the coronarin A-suppressed gluconeogenesis. More importantly, we revealed that coronarin A activated PI3K/Akt/GSK3β and ERK/Wnt/β-catenin signaling via regulation of a key upstream molecule IRS1. Coronarin A (10, 30 μM) decreased the phosphorylation of mTOR and S6K1, the downstream target of mTORC1, which further inhibited the serine phosphorylation of IRS1, and subsequently increased the tyrosine phosphorylation of IRS1. In type 2 diabetic ob/ob mice, chronic administration of coronarin A significantly reduced the non-fasting and fasting blood glucose levels and improved glucose tolerance, accompanied by the inhibited hepatic mTOR/S6K1 signaling and activated IRS1 along with enhanced PI3K/Akt/GSK3β and ERK/Wnt/β-catenin pathways. These results demonstrate the anti-hyperglycemic effect of coronarin A with a novel mechanism by inhibiting mTORC1/S6K1 to increase IRS1 activity, and highlighted coronarin A as a valuable lead compound for the treatment of T2DM.
Similar content being viewed by others

Introduction
Type 2 diabetes mellitus (T2DM) is characterized with hyperglycemia due to insulin resistance and impaired insulin secretion. The liver plays crucial role in maintaining blood glucose homeostasis by coordinating the glucose storage, utilization and production [1, 2]. In diabetic subjects, the ability of liver to store glycogen was typically diminished, and the hepatic gluconeogenesis was aberrantly activated, which jointly broke the whole body glucose homeostasis and enhanced both of postprandial and fasting blood glucose [3, 4]. Thus, targeting on promotion of hepatic glycogen synthesis and inhibition of hepatic gluconeogenesis could be more effective in suppression of diabetic symptoms [5]. Nevertheless, the agents which could stimulate glycogen synthesis and simultaneously inhibit gluconeogenesis were limited.
Insulin activates phosphoinositide 3-kinase (PI3K)/Akt signaling cascade and subsequently stimulates the phosphorylation and inactivation of GSK3β [6, 7], leads to the activation of glycogen synthase (GS) [8, 9], and ultimately triggers glycogen synthesis [10]. Hepatic glycogen synthesis is upregulated in response to the elevated blood glucose and insulin upon feeding [5, 9], whereas gluconeogenesis becomes the predominant source of fuel under prolonged fasting [11]. Under the fasting state in vivo, glucagon is the dominant hormone stimulating hepatic gluconeogenesis. Glucagon accumulates cAMP and induces PKA, which in turn phosphorylates cAMP response element binding (CREB) protein to stimulate transcription of gluconeogenic genes glucose-6-phosphatase (G6pc) and phosphoenolpyruvate carboxykinase (Pck1) [12]. In addition, kinds of transcriptional regulators and kinases, such as hepatic nuclear factor 4α, forkhead box O1, AMP-activated protein kinase (AMPK) and salt inducible kinase 1 (SIK1) were also reported to control hepatic gluconeogenesis via regulating the Pck1 and G6pc expression, and thus modulated the hyperglycemia of diabetic mice [13]. Recently, more transcriptional regulators and coactivators such as heat shock transcription factor-1 (HSF1) [14] and the Wnt/β-catenin/transcription factor 7-like 2 (TCF7L2) signaling pathway were proved to be vital in controlling Pck1 and G6pc expression [15, 16].
As T2DM develops, insulin resistance shifts the major form of energy storage from glycogen to triglycerides (TGs) in the liver [17, 18], which further impede the hepatic insulin resistance [9, 19]. Meanwhile, excessive gluconeogenic substrates such as glycerol, lactates and pyruvates are released into the circulation, and accelerated the rate of gluconeogenesis. Moreover, insulin resistance caused an inability of insulin to appropriately stimulate glycogen synthesis and suppress gluconeogenesis [20]. Insulin receptor (IR) responses to insulin and processes metabolic and mitogenic effects through IR substrate (IRS) proteins and key mediators, such as PI3K and Akt, MEK and ERK. The pathogenesis of insulin resistance is complex and has not been fully understood. mTORC1 is hyperactivated under insulin resistant states and obesity, which could further impair insulin signaling and aggravate insulin resistance via phosphorylating IRS1 at serine residues [21]. Thus, antagonizing mTORC1 might ameliorate insulin resistance and normalize the impaired hepatic glycogen synthesis and the excessive gluconeogenesis in T2DM.
Considering that glycogen synthesis and gluconeogenesis are two essential physiological processes for the liver to maintain glucose homeostasis, we screened our natural products library to identify active molecules which could simultaneously promote glycogen synthesis and inhibit gluconeogenesis in primary cultured rat hepatocytes, and discovered the active compound coronarin A. As a natural product isolated from rhizomes of Hedychium gardnerianum, coronarin A has been reported to possess anti-inflammatory activity in bone marrow-derived dendritic cells [22] and cytotoxic effects in human small cell lung cancer (NCI-H187) cells [23, 24]. However, its beneficial effect on hepatic glucose modulation has never been reported. In the present study, we discovered that coronarin A stimulated the glycogen synthesis by activating PI3K/Akt/GSK3β and suppressed the gluconeogenesis by enhancing ERK/Wnt/β-catenin pathway. Moreover, we elucidated the novel molecular mechanisms that coronarin A concurrently modulates glycogen synthesis and gluconeogenesis by activating tyrosine phosphorylation of IRS1 through inhibiting mTORC1/S6K1 signaling, and identified the unrecognized anti-hyperglycemic efficacy of coronarin A in ob/ob mice.
Materials and methods
Chemicals and materials
Coronarin A was purchased from BioBioPha Co., Ltd (Kunming, China). Forskolin, metformin, insulin, dexamethasone, amyloglucosidase, pyruvate sodium and lactate sodium were purchased from Sigma-Aldrich (St Louis, MO, USA). MK-2206, LY294002, IWR-1-endo and SCH772984 were purchased from Selleckchem (Houston, TX, USA). Trichloroacetaldehyde hydrate was purchased from Sinopharma Chemical Reagent (Shanghai, China). Collagen and horseradish peroxidase-conjugated secondary antibody were purchased from Dingguo (Beijing, China). Minimum Essential Medium (MEM), glucose-free Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum, TRIzol reagent and collagenase IV were purchased from Life Technologies (Carlsbad, CA, USA). PrimerScriptTM RT reagent kit and SYBR Premix Ex Taq were purchased from TaKaRa Biotechnology (Dalian, China). The glucose assay kit was purchased from Rongsheng Biotech (Shanghai, China). The non-fat dry milk was purchased from Meilunbio (Dalian, China). Percoll and ECL plus Western Blotting Detection Reagent were purchased from GE Healthcare (Buckinghamshire, UK). ACCU-CHEK Advantage II Glucose Monitor was purchased from Roche (Indianapolis, IN, USA). The TG kit was purchased from Dongou Diagnostics (Wenzhou, China). The glycogen kit was purchased from Comin Biotechnology (Suzhou, China). The ultra-sensitive mouse insulin kit was purchased from Crystal Chem (Downers Grove, IL, USA). All primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), including p-mTOR Ser2448 (#2976), mTOR (#2983), p-S6K1 Thr389 (#9234), S6K1 (#2708), p-S6 Ser235/236 (#2215), S6 (#2217), p-IRS1 Ser1101 (#2385), p-IRS1 Tyr1222 (#3066), IRS1 (#2390), p-IR Tyr1135/1136 (#3024), IR (#3025), IRS1 (#2390), p-ERK Thr202/Tyr204 (#4370), ERK (#4695), p-β-catenin Ser675 (#4176), p-Akt Ser473 (#9271), p-Akt Thr308 (#9275), Akt (#9272), p-GSK3β Ser9 (#9323), GSK3β (#9315), GAPDH (#2118). All other chemicals and reagents were purchased of the highest grade available commercially.
Animals
Male Sprague-Dawley rats were purchased from SLAC Laboratory Animals (Shanghai, China). B6.V-Lepob/Lepob mice (Jackson Laboratory, Bar Harbor, ME, USA) were bred and maintained at the Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences (CAS) (Shanghai, China). All animals were housed in a temperature- and humidity-controlled environment with a 12 h light/dark cycle, free access to water and the ob/ob mice were fed with a high-fat diet (Cat. P1400F, Puluteng, Shanghai, China). The animal experiments were approved and conducted in accordance with the guidelines of the Institutional Animal Care and Utilization Committee, SIMM, CAS.
Primary hepatocytes culture
Hepatocytes were isolated from male SD rats using the two-step collagenase manner as previously reported with minor modifications [25]. Briefly, SD rat was anesthetized and then the liver was perfused with collagenase IV digestion medium through the portal vein. The isolated primary hepatocytes were collected after 100 μm mesh filtration and Percoll centrifugation. Hepatocytes were plated in 6- or 48-well plates (1 or 0.125 × 106 cells per well) pre-coated with collagen in MEM containing 10% FBS, 100 nM insulin, 10 nM dexamethasone, and were incubated in a humidified incubator at 37 °C and 5% CO2.
Cell viability measurement
Cell viability was measured by MTT assay. Primary rat hepatocytes were seeded in 96-well plates for overnight and then incubated with different doses of coronarin A for 5.5 h or 12 h. After that, the medium was removed and 100 mL DMSO was added to dissolve the formazan. The light absorption was measured at 570 nm and the cell viability was calculated and normalized to control group.
Glycogen synthesis measurement
Cultured primary hepatocytes were treated with coronarin A or insulin for the indicated time and then harvested with distilled water using the cell scraper. After sonification and centrifugation at 12,000 × g, 4 °C for 10 min, the supernatant was collected and incubated with 1 mg/mL amyloglucosidase in 0.2 M sodium acetate buffer (pH 4.8) for 2.5 h with shaking in 40 °C. The reaction was stopped by 0.05% trichloroacetic acid and the glucose content in the supernatant was detected by the glucose assay kit. The result was normalized to cellular protein concentration.
Gluconeogenesis measurement
After seeded for 4 h, rat primary hepatocytes were treated with compounds in serum-free, glucose-free DMEM medium for 1.5 h, with or without 20 μM forskolin stimulation. After this, gluconeogenic substrates (2 mM pyruvate sodium plus 20 mM lactate sodium) were added into the medium for another incubation of 4 h. For inhibitor studies, hepatocytes were pre-treated with pharmacological inhibitors for 0.5 h before coronarin A treatment. Medium was collected to determine glucose production and normalized to cellular protein concentration.
Real-time PCR
Total RNA was extracted from rat primary hepatocytes or mouse liver by TRIzol reagent. cDNA synthesis was conducted using PrimerScriptTM RT reagent kit. The indicated gene mRNA levels were qualified using SYBR Premix Ex Taq. 18 S (hepatocytes) or β-actin (liver samples) were applied as normalizing controls using the 2−ΔΔCT method. All oligonucleotide primers were synthesized by Invitrogen and the sequences were listed in Table 1.
Western blot
Protein preparation and Western blot analysis were performed as described previously [26]. All the primary antibodies used in this study were diluted by 1:1000 except the antibody anti-GAPDH was diluted by 1:4000. The blots were blocked in 7.5% milk for 2 h and were then incubated with the primary antibody overnight at 4 °C; this was followed by an incubation with the goat anti-rabbit antibody (dilution 1:10,000). The blot bands were detected by ECL plus chemiluminescence and were quantified via densitometry using Quantity One (Bio-Rad, Hercules, California, USA).
RNA interference
Primary rat hepatocytes were transfected with 100 nM selective siRNA of IRS1 or non-target siRNA using LipofectamineTM 2000 (Thermo Fisher Scientific, Waltham, MA, USA) for 48 h for follow-up glycogen synthesis and gluconeogenesis test. The siRNAs of IRS1 and non-target siRNA were obtained from Genomeditech (Shanghai, China).
Pharmacokinetic study of coronarin A in ob/ob mice
Female ob/ob mice (10–11 weeks) were fasted overnight and fed 2 h post dose with free access to water. Coronarin A was dissolved in 0.5% Tween 80 or 0.25% CMC-Na and intraperitoneally or orally administered to ob/ob mice with a volume of 10 mL/kg of body weight at a dose of 30 mg/kg. The blood was obtained at 1, 2, 4, 8, and 12 h after dosing. Serum was harvested by centrifugation and concentrations of coronarin A in serum were determined by LC-MS/MS.
Chronic treatment of coronarin A in ob/ob mice
Male ob/ob mice (7–8 weeks) were assigned into four groups based on body weight and blood glucose (n = 8), and subjected to intraperitoneally treatment with 30 mg/kg coronarin A or the vehicle (0.1% Tween 80) or gavage with 100 mg/kg coronarin A or the vehicle (0.25% CMC-Na) once daily for 22 days. The non-fasting and fasting blood glucose of all mice were monitored on day 4, 8, 12, 16, and 22. The food intake and body weight of mice were measured once daily. Oral glucose tolerance tests (OGTT, 1.5 g/kg) were performed at 6 h fasting ob/ob mice on day 19 and the blood samples were collected to determine the serum glucose and insulin concentrations. On the last day, all mice were anaesthetized (trichloroacetaldehyde hydrate, 500 mg/kg) after 6 h fasting. The HOMA index of insulin resistance (HOMA-IR) was calculated as fast serum insulin (pM) × fast plasma glucose (mM)/156, which reflects the whole-body insulin sensitivity. The livers of gavage mice were collected and stored at −80 °C for Western blot analysis, gene expression measurement and the hepatic glycogen detection. Hepatic glycogen content was determined using the anthrone colorimetry method by the corresponding commercial kit following the manufacturer’s protocol.
Statistical analysis
The results were expressed as the mean ± SEM and analyzed with GraphPad Prism software. All statistical analyses were evaluated using a two-tailed unpaired Student’s t test. P < 0.05 was considered statistically significant.
Results
Coronarin A stimulated glycogen synthesis and inhibited gluconeogenesis in rat primary hepatocytes
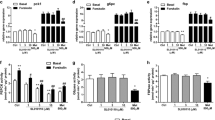
The structure of coronarin A is shown in Fig. 1a. Since the hepatocytes were treated with coronarin A for a total 5.5 h in the gluconeogenesis assay and for 4 or 12 h in the glycogen synthesis experiment, we detected the effect of coronarin A on cell viability of rat primary hepatocytes after 5.5 h or 12 h-incubation, respectively. As shown, coronarin A (1–30 μM) showed no toxicity on cell viability of rat primary hepatocytes after 5.5 h (Fig. 1b) or 12 h (Fig. 1c) incubation, whereas 100 μM coronarin A decreased cell viability after 12 h incubation (Fig. 1c). Thus, the maximum concentration used to study the effects of coronarin A on glucose metabolism in primary hepatocytes was 30 μM. Significant enhancement of glycogen synthesis was observed within 4 h of coronarin A treatment (10 μM) and was strengthened for up to 12 h, with 1.38- and 1.90-fold increase respectively (Fig. 1d). The glycogen synthesis was stimulated in a dose-dependent manner after 4 h (Fig. 1e) or 12 h (Fig. 1f) treatment with 1, 3 or 10 μM coronarin A. Coronarin A also exerted a dose-dependent inhibition on gluconeogenesis in rat primary hepatocytes under basal state, with 10 μM coronarin A leading to comparative inhibitory effect with 500 μM metformin (Fig. 1g). Forskolin, a potent activator of adenylate cyclase, is widely used to mimic the effect of glucagon for stimulating gluconeogenesis in the in vitro experiments [27]. Here, the forskolin-stimulated gluconeogenesis was significantly suppressed, with 3 and 10 μM coronarin A causing a reduction of 28.2% and 43.3%, respectively (Fig. 1h). The mRNA expressions of gluconeogenic genes Pck1 and G6pc were decreased by coronarin A under both basal and forskolin-stimulated state (Fig. 1i, j).

a Chemical structure of coronarin A. b and c Cell viability was measured in rat primary hepatocytes after treatment with different doses of coronarin A for 5.5 h (b) or 12 h (c). d Effect of 10 μM coronarin A on glycogen synthesis for the indicated time points. e and f Glycogen synthesis was measured in primary hepatocytes after treatment with different doses of coronarin A for 4 h (e) or 12 h (f). 100 nM insulin was set as a positive control. g Dose-dependent inhibition on gluconeogenesis by coronarin A in rat primary hepatocytes. 500 μM metformin was used as a positive control. Forskolin (20 μM) was applied to stimulate gluconeogenesis in primary hepatocytes, and the effects of coronarin A on gluconeogenesis (h), the expression levels of gluconeogenic genes Pck1 (i) and G6pc (j) were determined. All results were presented as mean ± SEM (n = 4). *P < 0.05, **P < 0.01 versus control under basal condition; #P < 0.05, ##P < 0.01 versus control under forskolin-induced condition.
Coronarin A stimulated glycogen synthesis through activating PI3K/Akt/GSK3β signaling in primary hepatocytes
As glycogen synthesis is mainly regulated by insulin signaling, we first investigated the effects of coronarin A on key mediators of insulin signaling, Akt and GSK3β, and insulin was set as a positive control. As shown here, coronarin A dose-dependently increased the Akt (Ser473 and Thr308) and GSK3β (Ser9) phosphorylation in rat primary hepatocytes (Fig. 2a–c). Treatment with 10 μΜ coronarin A for 4 h significantly increased the Akt phosphorylation at Ser473 and Thr308 to 1.9- and 1.5-fold of control, respectively. The phosphorylation of GSK3β at Ser9 was significantly increased to 2.4-fold at the same concentration. The effects of the Akt inhibitor MK-2206 and the PI3K inhibitor LY294002 on coronarin A stimulated Akt signaling and glycogen synthesis were investigated in rat primary hepatocytes. As shown in Fig. 2d, both MK-2206 and LY294002 pretreatment abolished the 10 μΜ coronarin A-stimulated Akt and GSK3β phosphorylation. Correspondingly, the glycogen synthesis stimulation caused by 10 μΜ coronarin A was completely blocked by MK-2206 and LY294002 pretreatment (Fig. 2e). These data indicated that coronarin A promoted glycogen synthesis in cultured primary hepatocytes dependent on PI3K/Akt signaling.

a–c Coronarin A increased the Akt and GSK3β phosphorylation dose-dependently. d and e Cultured hepatocytes were pre-treated with the Akt inhibitor MK-2206 (2 μM) or PI3K inhibitor LY294002 (10 μM) and then co-treated with coronarin A. The phosphorylation levels of Akt and GSK3β (d) and glycogen synthesis (e) were detected. 100 nM insulin was set as a positive control. All results were presented as mean ± SEM (n = 3). *P < 0.05, **P < 0.01 versus the corresponding control.
Coronarin A inhibited gluconeogenesis by activating ERK-dependent Wnt/β-catenin/TCF7L2 pathway in primary hepatocytes
Though activating the Akt signaling might also affect the hepatic gluconeogenesis, the coronarin A-suppressed gluconeogenesis was not influenced by the PI3K and Akt inhibitors (Fig. 3a, b), suggesting the PI3K/Akt signaling was dispensable here. The RNA-seq analysis of coronarin A showed that the gene expression of TCF7L2 was stimulated. Coronarin A (10 μM) increased the mRNA level of TCF7L2 to a 1.66-fold of control in rat primary hepatocytes (Fig. 3c), and the mRNA levels of TCF target genes including Axin2, c-Myc and Ccnd1 were also significantly upregulated (Fig. 3d). The Wnt signaling has been reported to regulate gluconeogenesis, and TCF7L2 and β-catenin are key components of Wnt pathway. Here, coronarin A dose-dependently increased the phosphorylation of β-catenin at Ser675, with significant increase by 84.0% after 10 μM treatment (Fig. 3e). In previous reports, ERK was proved to be involved in the activation of β-catenin by insulin in Hepa1-6 cells [28]. The phosphorylation of both ERK1 and ERK2 were dose-dependently stimulated by coronarin A, with increase to 1.8-fold of control after 10 μM of coronarin A treatment (Fig. 3f). The phosphorylation of mitogen-activated protein kinase kinase (MEK) was also dose-dependently stimulated by coronarin A, with increase to 1.6-fold of control after 10 μM of coronarin A treatment (Fig. 3g). Pretreatment with ERK inhibitor SCH772984 abolished the effects of coronarin A on ERK1/2 and β-catenin phosphorylation (Fig. 3h), and the inhibition of coronarin A on gluconeogenesis was also blocked (Fig. 3i). The Wnt inhibitor IWR-1-endo blocked the effect of coronarin A on β-catenin phosphorylation and the inhibition on gluconeogenesis, but did not affect the coronarin A-stimulated phosphorylation ERK1/2 (Fig. 3h, i). These data suggested coronarin A inhibited gluconeogenesis through activating the ERK-dependent Wnt/β-catenin/TCF7L2 pathway.

a and b The inhibition on gluconeogenesis by coronarin A could not be attenuated by PI3K inhibitor LY294002 (a) and the Akt inhibitor MK2206 (b) in rat primary hepatocytes. c and d The mRNA expression level of TCF7L2 and β-catenin/TCF targeted genes Axin2, c-Myc and Ccnd1 were measured in primary hepatocytes after coronarin A treatment. e–g Effects of coronarin A on β-catenin, ERK and MEK phosphorylation were detected using Western blot analysis and quantitated. h and i Cultured hepatocytes were pre-treated with β-catenin inhibitor IWR-1-endo (10 μM) or ERK inhibitor SCH772984 (1 μM) and then co-treated with coronarin A. The ERK/β-catenin pathway (h) and gluconeogenesis (i) were detected. All results were presented as mean ± SEM (n = 3). *P < 0.05, **P < 0.01 versus control.
Coronarin A increased tyrosine phosphorylation of IRS1 through inhibiting mTOR/S6K1 signaling
As coronarin A showed stimulation on both the PI3K/Akt and MEK/ERK signaling, we wonder whether coronarin A affected the same upstream regulation of these two signaling. IRS proteins regulate metabolic and mitogenic functions mainly through the PI3K/Akt and Ras/MEK/ERK signaling cascades [29]. As expected, the tyrosine phosphorylation of IRS1 at Tyr1222 was dose-dependently enhanced by coronarin A, with a significant increase to 1.8-fold of control after 10 μM of coronarin A (Fig. 4a), whereas the serine phosphorylation of IRS1 was dose-dependently inhibited (Fig. 4b), indicating the activity of IRS1 was strengthened. The tyrosine phosphorylation of insulin receptor (IR) at Tyr1135/1136 was unchanged after coronarin A treatment (Fig. 4c). Since the mTOR/S6K1 signaling was reported to negatively regulate IRS activity [30], we evaluated the activity of this signaling in rat primary hepatocytes here. As shown in Fig. 4d and e, coronarin A dose-dependently reduced the phosphorylation of mTOR, S6K1 and S6, suggesting that the mTOR/S6K1 signaling was inhibited. These data indicated that coronarin A increased the tyrosine phosphorylation of IRS1 by downregulating mTOR/S6K1 pathway and inhibiting the IRS1 serine phosphorylation. To further verify the role of IRS1 in the regulation of glycogen synthesis and gluconeogenesis by coronarin A, we knocked down the expression of IRS1 in primary rat hepatocytes by using IRS1 siRNA. Figure 4g showed that the IRS1 expression was effectively knocked down. The stimulation of glycogen synthesis in response to coronarin A and insulin was all abolished by knockdown of IRS1 (Fig. 4h), and the inhibitory effect of coronarin A on forskolin-induced gluconeogenesis was also blocked (Fig. 4i). These results suggested that IRS1 is indispensable in the coronarin A regulated glycogen synthesis and gluconeogenesis.

a and b Coronarin A dose-dependently enhanced the phosphorylation of IRS1 at Tyr1222 in rat primary hepatocytes, and decreased the serine phosphorylation of IRS1 at Ser1101. c Coronarin A showed no influence on the tyrosine phosphorylation of IR in rat primary hepatocytes. d–f Coronarin A dose-dependently decreased the phosphorylation of mTOR, S6K1 and S6 in rat primary hepatocytes. g–i Primary rat hepatocytes were pre-treated with IRS1 siRNA or non-target siRNA (NC) for 48 h and the IRS1 protein (g) was detected; or the hepatocytes were followed with or without coronarin A treatment for further 4 h for glycogen synthesis (h) or 5.5 h for gluconeogenesis (i) measurement. Results were presented as mean ± SEM (n = 3–4). *P < 0.05, **P < 0.01 versus control.
Intraperitoneal injection of coronarin A ameliorated hyperglycemia and insulin resistance in ob/ob mice
The ob/ob mice were intraperitoneally administered coronarin A (30 mg/kg, qd) for 22 days to investigate its effects on metabolic abnormalities. Coronarin A significantly decreased the non-fasting and fasting blood glucose levels with an average reduction rate of 37.1% and 37.0%, respectively (Fig. 5a, b). After 19 days of treatment, coronarin A remarkably improved the oral glucose tolerance of ob/ob mice (Fig. 5c). This was accompanied by a reduction in serum insulin concentrations at 0 and 60 min after glucose loading in the coronarin A-treated mice, with a 36.4% and 41.0% reduction, respectively (Fig. 5d). The whole body insulin sensitivity indicator, HOMA of insulin resistance (HOMA-IR) was significantly reduced by coronarin A, with a 68.3% reduction (Fig. 5e). During the whole treatment, the food intakes and the body weight were unaffected (Fig. 5f, g). After administration of coronarin A, the liver glycogen content was increased by 1.6-fold compared to the vehicle mice (Fig. 5h), and the expression of gluconeogenic gene Pck1 and G6pc was significantly decreased (Fig. 5i).

Intraperitoneal injection of coronarin A (30 mg/kg) improved both non-fasting blood glucose (a) and fasting blood glucose (b) during the 22 days’ treatment. Oral glucose tolerance test (OGTT, 1.5 g/kg) was determined on day 19. The blood glucose (c) and serum insulin (d) were detected at the indicated time points. e HOMA-IR was calculated. Food intake accumulation (f) and body weight (g) were monitored during treatment. h Liver glycogen content was detected. i The expression levels of hepatic gluconeogenic genes Pck1 and G6pc were detected by real-time PCR. All results were presented as mean ± SEM (n = 8). *P < 0.05, **P < 0.01 versus vehicle mice.
Oral administration of coronarin A ameliorated hyperglycemia in ob/ob mice
We wonder whether oral administration of coronarin A can also improve glucose homeostasis of ob/ob mice. A pharmacokinetic study of coronarin A by intraperitoneal injection and oral gavage respectively was conducted in ob/ob mice before chronic oral treatment. As shown in Table 2, intraperitoneal injection of coronarin A exhibited higher plasma exposure than oral gavage at the same dose of 30 mg/kg, with Cmax value of 1073 and 388 ng/mL, respectively. According to the Cmax data, we decided to gavage the ob/ob mice with a dose of 100 mg/kg for chronic treatment.
As expected, the results showed that oral treatment with 100 mg/kg coronarin A exerted similar effects to intraperitoneal injection with 30 mg/kg coronarin A in ob/ob mice. Gavage with 100 mg/kg coronarin A once daily significantly decreased the non-fasting and fasting blood glucose with an average reduction rate of 30.3% and 34.0% (Fig. 6a and b), respectively. After 19 days of treatment, oral administration of coronarin A significantly improved the oral glucose tolerance of ob/ob mice (Fig. 6c) and significantly reduced the serum insulin concentration at 15 min after glucose loading (Fig. 6d). The HOMA-IR showed a 51.7% reduction (Fig. 6e). During the whole treatment, oral administration of coronarin A resulted in a 23.1% reduction of the average daily food intake (Fig. 6f), while the body weight was unaffected (Fig. 6g). After 22 days’ oral administration of coronarin A, the hepatic glycogen content was increased (Fig. 6h) and the expression levels of gluconeogenic gene Pck1 and G6pc were significantly decreased (Fig. 6i), consistently to the effects by intraperitoneal injection of coronarin A.

Oral administration of coronarin A (100 mg/kg) also improved both non-fasting blood glucose (a) and fasting blood glucose (b) during the 22 days’ treatment. Oral glucose tolerance test was determined on day 19. The blood glucose (c) and serum insulin (d) were detected at the indicated time points. e HOMA-IR was calculated. Food intake (f) and body weight (g) were monitored during treatment. h Liver glycogen content was detected. i The expression of hepatic gluconeogenic genes Pck1 and G6pc were detected by real-time PCR. All results were presented as mean ± SEM (n = 8). *P < 0.05, **P < 0.01 versus vehicle mice.
Coronarin A inhibited the mTOR/S6K1 pathway to activate PI3K/Akt and ERK/β-catenin signaling in livers of ob/ob mice
After chronic gavage treatment with coronarin A, the livers of ob/ob mice were used for further investigation. As shown in Fig. 7a, the chronic coronarin A treatment caused a 65.3% decrease in the phosphorylation of mTOR in the liver, and the phosphorylation of S6K1 and S6 was also significantly inhibited. More importantly, coronarin A decreased the serine phosphorylation of IRS1 at Ser1101 by 49.0% (Fig. 7b), whereas the tyrosine phosphorylation of IRS1 in livers was increased to 1.57-fold of the vehicle group in the liver of ob/ob mice (Fig. 7c). The phosphorylation of Akt at Ser473 and Thr308 was significantly increased by 79.2% and 80.6%, respectively (Fig. 7d). Correspondingly, the phosphorylation of GSK3β at Ser9 was also elevated by 116.4% (Fig. 7d). Meanwhile, coronarin A caused a 69.5% and 98.9% increase in the phosphorylation of ERK1 and ERK2, respectively, and phosphorylation of β-catenin was increased by 40.6% (Fig. 7e). The mRNA expression of TCF7L2 (Fig. 7f) and the β-catenin/TCF target genes Axin2, c-Myc and Ccnd1 (Fig. 7g) were also significantly increased.

a After coronarin A (100 mg/kg, once daily, p.o.) treated for 22 days, livers of ob/ob mice were collected and phosphorylations of mTOR at Ser2448, S6K1 at Thr389 and S6 at Ser235/236 were detected by Western blot analysis. Phosphorylation of IRS1 at Ser1101 (b) and Tyr895 (c) were determined. d Phosphorylation of Akt at Ser473 and Thr308, and GSK3β at Ser9 were increased by coronarin A treatment. e Phosphorylations of ERK1/2 at Thr202/Tyr204 and β-catenin at Ser675 were also significantly increased. The gene expression of TCF7L2 (f) and Wnt target genes Axin2, c-Myc and Ccnd1 (g) were stimulated by coronarin A treatment. All results were presented as mean ± SEM (n = 8). *P < 0.05, **P < 0.01 versus vehicle mice.
Discussion
Hepatic glucose homeostasis is essential in keeping up the whole body blood glucose level [31]. The disruption of glucose metabolism in diabetic patients is manifested as the impaired postprandial glucose tolerance and high fasting blood glucose [32]. Therapeutic approaches modulating hepatic glycogen synthesis and gluconeogenesis would bring beneficial effects in normalizing hyperglycemia [12, 33]. In this study, we discovered a natural product, coronarin A could increase glycogen synthesis by activating the PI3K /Akt/GSK3β signaling and suppress gluconeogenesis by activating the ERK/Wnt/β-catenin signaling, and thus significantly decreased the non-fasting and fasting blood glucose and ameliorated glucose tolerance in diabetic mice. Moreover, we identified that IRS1 might be the upstream regulator of these two signaling activated by coronarin A, and the inhibition on mTORC1/S6K1 signaling was involved.
Hepatic glycogen synthesis is stimulated by feeding and contributes to postprandial blood glucose maintenance [34]. Glycogen synthesis is mainly controlled by insulin signaling, which is impaired in diabetic patients due to insulin resistance [6]. In our research, coronarin A effectively promoted glycogen synthesis in the primary hepatocytes, and increased the Akt and GSK3β phosphorylation, implying that activation of Akt pathway might contribute to coronarin A-induced glycogen synthesis. Though Akt phosphorylation was dose-dependently enhanced by coronarin A, the same doses of coronarin A did not affect the cell viability of primary rat hepatocytes, this might be due to multiple signaling cascade besides Akt are involved in cell proliferation and survival [35]. Both the Akt inhibitor MK2206 and PI3K inhibitor LY294002 abrogated the activation of Akt pathway and coronarin A-stimulated glycogen synthesis, which evidenced that PI3K and Akt was indispensable.
In addition to promoting the hepatic glycogen synthesis, coronarin A could also inhibit gluconeogenesis, which might further ameliorate the persistent hyperglycemia in T2DM [36,37,38]. Although coronarin A activated Akt and thus might inhibit gluconeogenesis via phosphorylating Foxo1, the Akt and PI3K inhibitors can not block the coronarin A-suppressed gluconeogenesis. With the help of RNA-seq analysis, we found the gene expression of TCF7L2 was stimulated by coronarin A. During the last decade, several researches suggested that the Wnt signaling pathway modulated hepatic gluconeogenesis through its key effector β-catenin and TCF7L2 [16]. Here, we verified that coronarin A increased the phosphorylation of β-catenin, and did also elevate the mRNA levels of other Wnt target genes including Axin2, c-Myc and Ccnd1. Moreover, the coronarin A-inhibited gluconeogenesis could be blocked by β-catenin inhibitor IWR-1-endo, suggesting that the β-catenin/TCF7L2 pathway was involved. A few reports showed that ERK could regulate Wnt activity by interaction with β-catenin [28, 39]. Hence, we wondered whether ERK pathway was involved here. As expected, the phosphorylation of ERK1/2 and its upstream kinase MEK were dose-dependently stimulated by coronarin A in primary rat hepatocytes. Meanwhile, the coronarin A-induced β-catenin phosphorylation and inhibition on gluconeogenesis could be blocked by the ERK inhibitor SCH772984. These results suggested that coronarin A suppressed hepatic gluconeogenesis via ERK/β-catenin/TCF7L2 pathway.
Since coronarin A stimulated glycogen synthesis by activating PI3K/Akt/GSK3β signaling and suppressed gluconeogenesis via ERK/Wnt/β-catenin signaling, we suspected that coronarin A might regulate common upstream modulators of these two signaling pathways. In the classic insulin signaling, IR and its substrates IRS proteins regulate metabolic and mitogenic functions mainly through the PI3K/Akt and Ras/MEK/ERK signaling cascades [29], and the IR and IRS proteins are generally activated by tyrosine phosphorylation and IRS proteins are inactivated by serine/threonine phosphorylation [23, 31]. Hepatocytes-specific knockout of IRS proteins showed IRS1 is more valuable in regulating hepatic glucose metabolism [40, 41]. Hence, IRS1 was firstly taken into consideration. Here, our data showed that coronarin A had no effect on tyrosine phosphorylation of IR, but increased tyrosine phosphorylation and reduced serine phosphorylation of IRS1 in primary hepatocytes, leading to the reasonable proposal that IRS1 might be the key upstream modulator in the regulation of coronarin A on hepatic glucose metabolism. It is well-studied that mTORC1 and S6K1 could phosphorylate multiple serine/threonine residues to attenuate IRS1 signaling transduction [23, 30, 42, 43], while inhibition of mTORC1 and S6K1 would activate IRS1 and rescue the PI3K/Akt signaling [30]. Here, coronarin A decreased phosphorylation of mTOR, S6K1, and the phosphorylation of S6, a substrate of S6K1 was also reduced. Accordingly, the phosphorylation of IRS1 at Ser1101 [7], a site which could be phosphorylated by S6K1 was suppressed by coronarin A, further convincing that the enhanced tyrosine phosphorylation of IRS1 was caused by inhibition on mTORC1/S6K1. Moreover, the coronarin A-induced stimulation on glycogen synthesis and inhibition on gluconeogenesis was fully blocked by downregulation of IRS1, indicating the dependence on IRS1. In addition, although AMPK, the central regulator of energy homeostasis, is known to enhance insulin signaling and suppress mTORC1 activity [44], we found that coronarin A did not affect the phosphorylation level of AMPK and ACC (data not shown), which suggested the AMPK signaling was not involved in the suppressed mTORC1 by coronarin A. Taken together, coronarin A activated IRS1 through inhibition of mTORC1/S6K1, and then enhanced the downstream PI3K/Akt/GSK3β and ERK/Wnt/β-catenin signaling pathway, led to stimulation of glycogen synthesis and inhibition on gluconeogenesis.
Although coronarin A modulates hepatic glucose metabolism via inhibiting mTORC1/S6K1 signaling, the mTORC1 inhibitor rapamycin did not exert similar effects [23]. Rapamycin failed to suppress the expression of PEPCK in hepatocytes in previous report [45] and did not affect gluconeogenesis in rat primary hepatocytes in our lab (data not shown). The possible reason might be that rapamycin suppressed not only mTORC1 but also mTORC2 [46], an important enzyme which could activate Akt by phosphorylating the site of Ser473. Rapamycin treatment would suppress mTORC2 and inhibit phosphorylation of Akt at the Ser473 residue, leading to insulin resistance and severe glucose intolerance [47]. Our data showed that coronarin A increased the phosphorylation of Akt at both Thr308 and Ser473 residue, indicating that coronarin A might exert different effects on mTORC2 compared with rapamycin.
Further in vivo studies were performed to evaluate the effect of coronarin A on the whole-body glucose homeostasis in ob/ob mice. As expected, both intraperitoneal injection and oral administration of coronarin A remarkably reduced the non-fasting and fasting blood glucose. The oral glucose tolerance was significantly improved and the corresponding insulin levels were decreased by coronarin A treatment, indicating the improved whole-body insulin sensitivity, which was further convinced by the reduced HOMA-IR, a fundamental indicator of overall insulin sensitivity [48]. Consistent with the results in primary rat hepatocytes, chronic coronarin A treatment increased liver glycogen content, decreased gluconeogenic gene expression, suppressed the mTOR/S6K1 pathway and the serine phosphorylation of IRS1, whereas enhanced tyrosine phosphorylation of IRS1 and activated the PI3K/Akt/GSK3β and ERK/Wnt/β-catenin signaling pathway. These findings supported that coronarin A improved the overall glucose homeostasis and insulin sensitivity by modulating hepatic glycogen synthesis and gluconeogenesis through inhibiting mTOR/S6K1 pathway in ob/ob mice.
Conclusion
In conclusion, we identified that coronarin A could increase the tyrosine phosphorylation of IRS1 by inhibiting mTORC1/S6K1, thereby activate PI3K/Akt/GSK3β signaling to stimulate hepatic glycogen synthesis and enhance ERK-dependent Wnt/β-catenin/TCF7L2 pathway to suppress gluconeogenesis both in vitro and in vivo (Fig. 8). Chronic administration of coronarin A significantly reduced blood glucose, improved glucose tolerance and insulin sensitivity in ob/ob mice. These findings discovered an unrecognized anti-hyperglycemic effect of coronarin A with a novel mechanism by stimulating glycogen synthesis and suppressing gluconeogenesis simultaneously, and highlighted coronarin A as a valuable lead compound for the development of new T2DM therapeutics.

Coronarin A reduced the serine phosphorylation of IRS1 by inhibiting mTOR/S6K1, which led to enhancement of IRS1 tyrosine phosphorylation, and subsequently activated the PI3K/Akt/GSK3β and ERK/Wnt/β-catenin pathway to stimulate hepatic glycogen synthesis and suppress gluconeogenesis, respectively.
References
Ekberg K, Landau BR, Wajngot A, Chandramouli V, Efendic S, Brunengraber H, et al. Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes. 1999;48:292–8.
Sharabi K, Tavares CD, Rines AK, Puigserver P. Molecular pathophysiology of hepatic glucose production. Mol Asp Med. 2015;46:21–33.
Hwang JH, Perseghin G, Rothman DL, Cline GW, Magnusson I, Petersen KF, et al. Impaired net hepatic glycogen synthesis in insulin-dependent diabetic subjects during mixed meal ingestion. A 13C nuclear magnetic resonance spectroscopy study. J Clin Invest. 1995;95:783–7.
Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–7.
Rines AK, Sharabi K, Tavares CD, Puigserver P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat Rev Drug Discov. 2016;15:786–804.
Krssak M, Brehm A, Bernroider E, Anderwald C, Nowotny P, Dalla Man C, et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes. 2004;53:3048–56.
Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. 2018;19:31–44.
Dangana EO, Michael OS, Omolekulo TE, Areola ED, Olatunji LA. Enhanced hepatic glycogen synthesis and suppressed adenosine deaminase activity by lithium attenuates hepatic triglyceride accumulation in nicotine-exposed rats. Biomed Pharmacother. 2019;109:1417–27.
Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126:12–22.
Florez JC. Pharmacogenetics in type 2 diabetes: precision medicine or discovery tool? Diabetologia. 2017;60:800–7.
An H, He L. Current understanding of metformin effect on the control of hyperglycemia in diabetes. J Endocrinol. 2016;228:R97–106.
Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13:572–87.
Liu S, Huang S, Wu X, Feng Y, Shen Y, Zhao QS, et al. Activation of SIK1 by phanginin A inhibits hepatic gluconeogenesis by increasing PDE4 activity and suppressing the cAMP signaling pathway. Mol Metab. 2020;41:101045.
Qiao A, Jin X, Pang J, Moskophidis D, Mivechi NF. The transcriptional regulator of the chaperone response HSF1 controls hepatic bioenergetics and protein homeostasis. J Cell Biol. 2017;216:723–41.
Oh KJ, Han HS, Kim MJ, Koo SH. CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013;46:567–74.
Liu H, Fergusson MM, Wu JJ, Rovira II, Liu J, Gavrilova O, et al. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011;4:ra6.
Palomo V, Martinez A. Glycogen synthase kinase 3 (GSK-3) inhibitors: a patent update (2014-2015). Expert Opin Ther Pat. 2017;27:657–66.
Couturier K, Qin BL, Batandier C, Awada M, Hininger-Favier I, Canini F, et al. Cinnamon increases liver glycogen in an animal model of insulin resistance. Metabolism. 2011;60:1590–7.
Kusunoki M, Tsutsumi K, Hara T, Ogawa H, Nakamura T, Miyata T, et al. Correlation between lipid and glycogen contents in liver and insulin resistance in high-fat-fed rats treated with the lipoprotein lipase activator NO-1886. Metabolism. 2002;51:792–5.
Rizza RA. Pathogenesis of Fasting and Postprandial Hyperglycemia in Type 2 Diabetes: implications for therapy. Diabetes. 2010;59:2697–707.
Yoon MS. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients. 2016;8:405.
Kiem PV, Anh Hle T, Nhiem NX, Minh CV, Thuy NT, Yen PH, et al. Labdane-type diterpenoids from the rhizomes of Hedychium coronarium inhibit lipopolysaccharide-stimulated production of pro-inflammatory cytokines in bone marrow-derived dendritic cells. Chem Pharm Bull (Tokyo). 2012;60:246–50.
Yoon MS. The role of mammalian target of rapamycin (mTOR) in insulin signaling. Nutrients. 2017;9:1176.
Shah OJ, Wang ZY, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–6.
Huang SL, Yu RT, Gong J, Feng Y, Dai YL, Hu F, et al. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia. 2012;55:1469–81.
Huang S, Ma S, Ning M, Yang W, Ye Y, Zhang L, et al. TGR5 agonist ameliorates insulin resistance in the skeletal muscles and improves glucose homeostasis in diabetic mice. Metabolism. 2019;99:45–56.
Liang JC, Liu CZ, Qiao AJ, Cui Y, Zhang HB, Cui AF, et al. MicroRNA-29a-c decrease fasting blood glucose levels by negatively regulating hepatic gluconeogenesis. J Hepatol. 2013;58:535–42.
Ip W, Shao W, Chiang YT, Jin T. The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. Am J Physiol Endocrinol Metab. 2012;303:E1166–76.
Das D, Arur S. Conserved insulin signaling in the regulation of oocyte growth, development, and maturation. Mol Reprod Dev. 2017;84:444–59.
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–23.
Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the regulation of hepatic metabolism by insulin. Trends Endocrinol Metab. 2017;28:497–505.
Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world–a growing challenge. N Engl J Med. 2007;356:213–5.
Bergman RN, Iyer MS. Indirect regulation of endogenous glucose production by insulin: the single gateway hypothesis revisited. Diabetes. 2017;66:1742–7.
Morales-Ruiz M, Santel A, Ribera J, Jimenez W. The role of Akt in chronic liver disease and liver regeneration. Semin Liver Dis. 2017;37:11–6.
Kenerson HL, Yeh MM, Kazami M, Jiang XY, Riehle KJ, Mcintyre RL, et al. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology. 2013;144:1055–65.
Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–56.
Zou ZP, Chen J, Yang J, Bai XC. Targeted inhibition of rictor/mTORC2 in cancer treatment: a new era after rapamycin. Curr Cancer Drug Tar. 2016;16:288–304.
Sharabi K, Lin H, Tavares CD, Dominy JE, Camporez JP, Perry RJ, et al. Selective chemical inhibition of PGC-1alpha gluconeogenic activity ameliorates type 2 diabetes. Cell. 2017;169:148–60.
Jin T. The WNT signalling pathway and diabetes mellitus. Diabetologia. 2008;51:1771–80.
Guo S, Copps KD, Dong XC, Park S, Cheng ZY, Pocai A, et al. The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol Cell Biol. 2009;29:5070–83.
Kubota N, Kubota T, Itoh S, Kumagai H, Kozono H, Takamoto I, et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 2008;8:49–64.
Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, Roden M, et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci USA. 2007;104:14056–61.
Zhang Z, Liu H, Liu J. Akt activation: a potential strategy to ameliorate insulin resistance. Diabetes Res Clin Pr. 2019;156:107092.
Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019;18:527–51.
Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA. 2010;107:3441–6.
Rosner M, Hengstschlaeger M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet. 2008;17:2934–48.
Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–43.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Acknowledgements
This work was supported by Foundation of Shanghai Science and Technology Committee (Grant No 21DZ2291100).
Author information
Authors and Affiliations
Contributions
YL, JHS, and SLH designed the research. SLH, WX, Yu Shen, YLY, TFX, JL, HQ, ZHZ, and Yu Shi performed the research. SLH, WX, and YL analyzed and interpreted the data. SLH, WX, and YL wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, Sl., Xie, W., Ye, Yl. et al. Coronarin A modulated hepatic glycogen synthesis and gluconeogenesis via inhibiting mTORC1/S6K1 signaling and ameliorated glucose homeostasis of diabetic mice. Acta Pharmacol Sin 44, 596–609 (2023). https://doi.org/10.1038/s41401-022-00985-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00985-5
