
Abstract
Since the initial identification of TMEM106B as a risk factor for frontotemporal lobar degeneration (FTLD), multiple genetic studies have found TMEM106B variants to modulate disease risk in a variety of brain disorders and healthy aging. Neurodegenerative disorders are typically characterized by inclusions of misfolded proteins and since lysosomes are an important site for cellular debris clearance, lysosomal dysfunction has been closely linked to neurodegeneration. Consequently, many causal mutations or genetic risk variants implicated in neurodegenerative diseases encode proteins involved in endosomal–lysosomal function. As an integral lysosomal transmembrane protein, TMEM106B regulates several aspects of lysosomal function and multiple studies have shown that proper TMEM106B protein levels are crucial for maintaining lysosomal health. Yet, the precise function of TMEM106B at the lysosomal membrane is undetermined and it remains unclear how TMEM106B modulates disease risk. Unexpectedly, several independent groups recently showed that the C-terminal domain (AA120-254) of TMEM106B forms amyloid fibrils in the brain of patients with a diverse set of neurodegenerative conditions. The recognition that TMEM106B can form amyloid fibrils and is present across neurodegenerative diseases sheds new light on TMEM106B as a central player in neurodegeneration and brain health, but also raises important new questions. In this review, we summarize current knowledge and place a decade’s worth of TMEM106B research into an exciting new perspective.
Similar content being viewed by others
Introduction
Neurodegenerative disorders are characterized by misfolding and aggregation of proteins such as tau, amyloid-β (Aβ), α-synuclein, and TDP-43. These disorders are commonly referred to as proteinopathies and are named more specifically after the aggregating protein, e.g., tauopathies, synucleinopathies, and TDP-43opathies [12, 65]. Filamentous aggregates and inclusions in neuronal and glial cell types will ultimately lead to toxicity and cell death resulting in brain atrophy. Depending on the implicated brain region(s), the degeneration may disrupt core human characteristics such as memory, speech, behavior, personality, and movement. Improving our understanding of disease pathomechanisms that underlie the formation of aggregates as well as detailed knowledge of the structure of filamentous aggregates may offer disease insight and are crucial to aid in both biomarker and therapy development. Recent advances in cryogenic electron microscopy (cryo-EM) have enabled researchers to identify the structure of fibrils extracted from postmortem brain tissue, and over the past years the structure of pathological forms of filaments formed by tau (reviewed in [60]), amyloid-β [31], α-synuclein [58], and TDP-43 [2] have been determined. Several independent cryo-EM groups now report amyloid fibrils in brain tissue of a diverse set of neurodegenerative disorders as well as older neurologically normal individuals to comprise the C-terminal domain (AA120-254/274) of transmembrane protein 106B (TMEM106B), a protein previously shown to modulate disease risk in neurodegeneration and implicated in healthy aging [10, 17, 27, 57].
The identification of TMEM106B amyloid fibrils in postmortem brain tissue offers a new perspective on the involvement of TMEM106B in neurodegeneration and brain health. Here, we review these findings as well as summarize the current understanding of TMEM106B biology and function in both health and disease. Finally, we discuss and speculate on the implications of TMEM106B amyloid fibrils on disease and highlight the potential for biomarker development and therapeutic approaches related to TMEM106B.
TMEM106B in health and disease
Frontotemporal lobar degeneration (FTLD) is a group of heterogeneous, progressive neurodegenerative disorders representing 10–20% of all dementias with an early disease onset, making FTLD the second most common dementia in people under the age of 65 years [48, 72]. Pathologically, TAR DNA-binding protein 43 (TDP-43) is the most commonly aggregated protein (~ 50% of all cases) found in the brain of FTLD patients (FTLD-TDP), where TDP-43 forms hyperphosphorylated, ubiquitinated inclusions [24, 39]. According to the morphology and distribution of the TDP-43 inclusions, FTLD-TDP is further classified into types A through E [42]. In 2010, TMEM106B was identified as a risk-associated gene for FTLD-TDP, where the disease-modulating effect was especially prominent in FTLD-TDP patients harboring pathogenic mutations in the progranulin (GRN) gene [14]. Heterozygous loss-of-function mutations in GRN (resulting in a 50% loss of progranulin (PGRN) protein) represent ~ 20–25% of cases of FTLD-TDP, yet individuals with GRN mutations who also carry a TMEM106B ‘protective’ haplotype have approximately 50% lower odds of developing FTLD symptoms [51]. In later studies, the risk-modulating effect was also extended to patients carrying the C9orf72 hexanucleotide GGGGCC repeat expansion [4, 23].
Since then, multiple studies have established genetic variants in TMEM106B as important modifiers of disease risk in a variety of neurodegenerative disorders including other TDP-43 proteinopathies as well as tauopathies, reviewed in [47] and [19]. Moreover, TMEM106B’s risk-modifying capabilities go beyond disease protection alone, as it has been linked to brain aging even in the absence of known brain disease [54]. In addition, TMEM106B has been associated with neuronal proportion, conferring neuronal protection against general aging [38]. The importance of TMEM106B in brain health is further highlighted by its association with cognition [36, 52, 54, 66, 67, 70, 71] and the recent associations with mood disorders such as depression [13, 16, 45].
TMEM106B genetic haplotypes
In and around TMEM106B on chromosome 7p21, several single nucleotide polymorphisms (SNPs) were identified in high linkage disequilibrium, resulting in two common TMEM106B haplotypes in the human population [14]. Since it is not currently known which variant on the haplotype is functionally responsible for modulating disease risk, they are collectively referred to as either the risk or protective haplotype. It is the most frequent of these two haplotypes which has consistently been associated with an increased risk for neurodegenerative diseases and poor brain health. However, the functional effect of the risk haplotype, as well as the responsible disease-modifying variant, has been a topic of active discussion.
TMEM106B structure and function
Structure and interactions
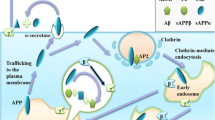
TMEM106B is a highly glycosylated, single-pass, type II transmembrane protein comprising a total of 274 amino acids. It is localized in the membrane of late endolysosomal compartments, with the N-terminus in the cytosol and the C-terminus within the lumen [34]. The cytoplasmic N-terminus (residues 1–96) is intrinsically disordered without a well-defined secondary or tertiary structure which may offer the ability to dynamically interact with diverse binding partners [29, 37]. A limited number of interaction partners have been reported for TMEM106B [21, 28, 30, 59, 63] (https://opencell.czbiohub.org/target/CID002001), which are schematically presented in Fig. 1. Additionally, TMEM106B can form homo- and heterodimers with its homolog TMEM106C, through a CxxCxGxG motif that is capable of forming a zinc-binding site. The interaction with TMEM106C has been confirmed and the presence of dimers was also observed on western blot; however, the functional importance of the dimers is unknown [37, 63]. The lysosomal sorting of TMEM106B is mediated by an extended dileucine signal located in the N-terminal region (ENQLVALI), and abrogation of this signal leads to a diffuse cytosolic distribution of TMEM106B [9].

Schematic presentation of reported interaction partners of TMEM106B. All interaction partners are listed in a black box with a short functional description near the respective implicated cellular pathways. A detailed schematic of the lysosomal membrane is depicted in the red box. PM, plasma membrane; ER, endoplasmic reticulum; EE, early endosome; LE, late endosome; MVB, multivesicular body
Following the transmembrane domain (residues 97–117), the C-terminal domain (residues 118–274) resides within the lumen and contains five N–X–T/S glycosylation motifs at N145, N151, N164, N183, and N256. The first three glycosylation sites are reported to result in simple N-glycosylation, while the last two sites are reported to result in complex N-glycosylation. The transport of TMEM106B to the lysosomes is also modulated by its post-translational modifications and depends on its fourth and fifth N-glycosylation sites. The loss of the N183 or N256 complex glycans was shown to result in impaired transport to the endosomes and lysosomes. In these instances, TMEM106B was retained in the endoplasmic reticulum (ER) or mislocalized to the plasma membrane instead [9, 34, 64].
Proteolytic processing
Importantly, TMEM106B is proteolytically processed by an unknown protease, most likely a lysosomal protease, to release its C-terminal domain in the lysosomal lumen. This process generates a residual N-terminal fragment (NTF) anchored to the lysosomal membrane. The remaining NTF is further cleaved by signal peptide peptidase-like 2A (SPPL2A), a GxGD aspartyl protease, by intramembrane proteolysis releasing an intracellular domain (ICD) in the cytosol as well as a small C-domain into the lumen [6]. TMEM106B processing likely occurs to modulate its levels on the lysosomal membrane to regulate its function, but a separate function for the ICD or luminal domain cannot be excluded. Many questions remain on (i) which protease(s) is responsible for cleaving the luminal domain, (ii) the precise cleavage site, and (iii) the relevance of the generated peptides beyond the degradation of full-length TMEM106B. The study of this process has been hampered by the lack of antibodies recognizing the luminal domain and the assumption that, at least under normal conditions, the luminal domain would be degraded by the lysosome along with other lysosomal content.
Lysosomal function
TMEM106B plays an important role in lysosome function which is demonstrated by the observation that both knock-down and overexpression of TMEM106B affect lysosomal morphology, pH, maturation, trafficking, and exocytosis. Aberrant changes in TMEM106B levels lead to an accumulation of enlarged lysosomes in the perinuclear region which ultimately induces cytotoxicity. In the lysosome, TMEM106B interacts with several partners (Fig. 1) that are critical for proper lysosome formation including CHMP2B (another FTLD-related protein), which is part of the ESCRT-III complex, with v-ATPase, which is crucial for lysosome acidification and with cathepsin D, a lysosomal enzyme [5, 19, 21, 33, 41, 59, 63]. TMEM106B also functionally interacts with MAP6 to control lysosomal trafficking [59]. Overexpression of TMEM106B causes translocation of transcription factor EB (TFEB) to the nucleus and thus induces the upregulation of the coordinated lysosomal expression and regulation (CLEAR) network. The CLEAR network regulates genes involved in lysosomal function and autophagy, identifying TMEM106B as a critical regulator of lysosomal function [33, 63].
Functional effect of the TMEM106B haplotypes
Variants on the TMEM106B haplotype alter TMEM106B levels
Available experimental evidence suggests that variants on the TMEM106B haplotypes exert their effect by altering TMEM106B expression, where an increased expression correlates with the risk haplotype. First, the levels of TMEM106B mRNA and protein were significantly increased in GRN mutation carriers [8, 11]. Second, the A-allele of a non-coding variant (rs1990620) located on the TMEM106B risk haplotype was shown to preferentially recruit the chromatin-organizing protein CCTC-binding factor (CTCF), modulating TMEM106B expression through transcriptional activation due to altered long-range chromatin-looping interactions [22]. Third, also within the haplotype block, there is one coding variant (rs3173615) encoding a threonine to serine change at amino acid position 185 (p.T185S) located in the fourth N–X–T/S glycosylation motif, which has been suggested to contribute to the disease-modifying effect. In vitro, it was shown that TMEM106B carrying the risk allele (T185) had higher expression levels than the protective allele (S185), potentially due to differences in glycosylation at N183 affecting the protein stability and degradation rate [46]. However, another study observed no such effect [5].
The hypothesis that increased levels of TMEM106B may drive the disease-modifying effect was further supported by the observation that TMEM106B contains miRNA-132 and miRNA-212 binding sites in its 3′ UTR, which inhibit TMEM106B expression upon binding [11]. In neurodegeneration (including Alzheimer’s disease (AD), FTLD-TDP, etc.) the expression of the microRNA132/212 cluster is decreased [18, 25, 50, 56], suggesting an upregulation of TMEM106B expression in disease.
Variants on the TMEM106B haplotype alter TMEM106B biology and function
Alternatively, it was suggested that the coding p.T185S variant might influence disease risk irrespective of TMEM106B levels by altering either TMEM106B biology (cleavage, dimerization, etc.) or by affecting binding (or binding affinity) to interaction partners which could both lead to lysosomal dysfunction. Jun and colleagues showed an enhanced binding of the T185 variant as compared to the S185 variant to CHMP2B, especially with its mutant form, which led to a decrease in autophagic flux [28].
Identification of TMEM106B Fibrils
Diseases
Over the past months, several research groups have reported the cryo-EM structures of TMEM106B filaments derived from the brains of a variety of neurodegenerative diseases as well as older neurologically normal individuals [10, 17, 27, 57]. The cryo-EM reports included fibrils obtained from sarkosyl-insoluble fractions of postmortem tissue of individuals with Alzheimer’s disease (AD), argyrophilic grain disease (AGD), amyotrophic lateral sclerosis (ALS), aging-related tau astrogliopathy (ARTAG), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), dementia with Lewy bodies (DLB), early-onset Alzheimer’s disease (EOAD), sporadic and inherited Parkinson’s disease (PD), PD dementia (PDD), inherited and sporadic frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) type A, B, C, D, familial frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17T), limbic-predominant neuronal inclusion body 4R tauopathy (LNT), multiple system atrophy (MSA), pathological aging (PA), as well as neurologically normal controls (Fig. 2).

Overview of brain material used in the cryo-EM reports which identified TMEM106B fibrils, including brain region and disease status
Cryo-EM
Although all groups used sarkosyl to extract the fibrils, there was some variability in the fractionation protocol with variations in the stage of sarkosyl addition, the use of ultracentrifugation or low-speed centrifugation, and heating or pronase treatment of the extract. Nevertheless, all reports observed amyloid fibrils with an ordered core comprising residues S120-G254 of TMEM106B. Interestingly, the identification of TMEM106B was distinct between groups. Whereas Chang et al. [10] complemented the cryo-EM with mass spectrometry to identify TMEM106B peptides present in the sarkosyl-insoluble fraction, Jiang et al. [27] modeled two query sequences based on the cryo-EM densities, and Schweighauser and colleagues [57] found their way to TMEM106B based on the fibril’s distinctive glycosylation pattern.
Despite that TDP-43 filaments have been identified in ALS/FTD [2], none of the current studies identified fibrils constituted of TDP-43. Only Jiang and colleagues report abundant non-filamentous aggregates of TDP-43 in FTLD-TDP extracts [27]. Considering Arseni and colleagues [2] used a different extraction method than the current studies, it is plausible that the TDP-43 filaments were lost during the extraction process and reside within another fraction that was not analyzed in the present studies. This emphasizes the importance of the sample preparation protocol in cryo-EM studies and indicates that extraction methods need to be carefully compared, as relatively small changes may influence the purified content. Moreover, the isolation of TDP-43 filaments in future cryo-EM studies aiming to resolve TDP-43 aggregates might require a different approach.
TMEM106B fibril structure, polymorphisms, and ultrastructural polymorphs
The TMEM106B fibrils comprise either a single protofilament forming rod-like structures or a doublet formed by two protofilaments forming a twisted ribbon. The structure of several polymorphisms of the protofilaments, four singlet polymorphisms and two doublet polymorphisms, have been reported (Fig. 3). Unlike other amyloid fibrils, no clear relationship between the different polymorphisms and disease status was observed. All polymorphisms share a similar five-layered ordered core consisting of 17–19 β-strands with a highly conserved N-terminal region. All cryo-EM studies report TMEM106B to be fully glycosylated in all folds at the glycosylation sites present within the fibrillar structure (N145, N151, N164, and N183), and the presence of a disulfide bond between C214 and C253. The structural variation between polymorphisms is mainly located in the middle region and C-terminal region [10, 27, 57]. The high number of β-strands results in a highly stable fibril core, which could potentially be irreversible once formed, as suggested by Jiang et al. [27]. The S120 is buried deep within the fibril core, leaving no space for additional amino acids. Therefore, fibrillization may only occur when TMEM106B is cleaved at residue 119.

Schematic representation of the distinct forms of TMEM106B fibrils identified in the cryo-EM reports. a Singlet polymorphisms and b doublet polymorphisms. Boxed singlets and doublet polymorphisms represent the predominant polymorphism(s) identified in multiple reports and/or several disorders. Fibril structures were compared in PDB and extracted from PDB entries 7U16, 7U17, 7QWL, 7QWG, 7SAS, and 7SAR
Based on TMEM106B genotypes, singlet I can be formed by either the T185 or S185 isoform (encoded by the risk and protective TMEM106B haplotypes, respectively) considering it is present in TT, TS, and SS individuals, while singlet II is only observed in TS and SS individuals. This leaves the possibility that the packing of the singlet II fibril leaves insufficient space for a threonine residue and can only be formed by the S185 isoform, as suggested by Schweighauser et al. [57].
Though Schweighauser et al. [57] report all polymorphisms to be capable of forming doublets, only the complete cryo-EM structure of two polymorphs of doublets comprising two protofilaments of singlet I have been fully resolved. In doublet I, the two singlets are arranged with a twofold symmetry centered around the positively charged residues K178 and R180 with an unidentified non-proteinaceous anionic cofactor in the middle, facilitating doublet formation [10, 27, 57]. Alternatively, doublet II is centered around Glu206, Met207, Tyr209, and Tyr211 [27].
Of note, the normal C-terminal portion of TMEM106B comprises AA118-274, meaning that the last 20 amino acids are not present within the fibril core. It is not clear whether the last amino acids are cleaved off, or alternatively reside outside of the fibril. While it was proposed to be potentially present as a fuzzy coat by Jiang et al. [27], similar to what has been observed for Tau [60], α-synuclein [58], TDP-43 [2], and Aβ[31], Schweighauser et al. described TMEM106B fibrils as a fibril that seemed to lack a fuzzy coat based on the electron micrographs [57].
Updated view on TMEM106B biology
Proteolytic processing
Considering that the proteolytic cleavage of TMEM106B is a critical event to form fibrils, further studies on factors modulating proteolytic processing such as the involved protease(s) and physiological environment will be crucial. While Chang et al. queried the neighboring sequence to identify potential proteases (granzyme A, kallikrein-related peptidase 4, cathepsin P) [10], Schweighauser et al. proposed that, in the native structure, the globular domain is located too close to the lysosomal membrane. They argue that the lack of a flexible linker and the hydrophobic surface patch at the end of the domain make it highly unlikely that S120 is accessible to a lysosomal protease. Rather, the authors propose the existence of a non-canonical shedding pathway [57]. The C-terminal domain shedding of TMEM106B may therefore occur through canonical shedding of the C-terminal domain of TMEM106B by an unidentified lysosomal protease at position G127, as described before [6], and/or at position S120. Alternatively, the S120 C-terminal domain is released through non-canonical shedding of TMEM106B. Interestingly, the existence of non-canonical shedding by SPPL2A has recently been reported for TNF-α [62]. After shedding the C-terminal domain, SPPL2A may further cleave TMEM106B within the membrane (Fig. 4).

An updated view on TMEM106B biology and processing at the endolysosomal membrane. Prior to intramembrane proteolysis by SPPL2a, the C-terminal domain of TMEM106B may be released by canonical shedding through a lysosomal protease. Chang et al. proposed three novel candidate enzymes, which are indicated in the pink box. Conversely, Schweighauser et al. suggest that this region may not be accessible to lysosomal proteases. Instead, TMEM106B’s C-terminal domain may be released through non-canonical shedding. This may potentially also occur by SPPL2a, similar to non-canonical shedding of TNF-α [62]
It is interesting to speculate that canonical shedding by a lysosomal protease indeed occurs at position G127, as reported in [6], rendering C-terminal fragments generated by this process incapable of forming fibrils. Canonical shedding at position G127 and non-canonical shedding at position S120 may both occur in physiological circumstances with different efficiencies. In disease, the shedding process might be skewed more toward the non-canonical way, for example, due to lysosomal dysfunction and/or reduced activity of the lysosomal protease responsible for canonical shedding. This would increase the amount of protein that is processed into the fibrillogenic C-terminal peptide (S120) to maintain the amount of functional full-length protein present on the lysosomal membrane.
Fibril formation
Due to the lack of detailed intracellular stainings such as co-stainings with organelle markers, it is not clear whether the fibrils form within the lysosomal lumen or whether the C-terminal domain first needs to escape the lumen. This leaves two possibilities (Fig. 5): (I) the fibrils form within the lumen and potentially later escape the lysosome, or (II) the fibrils can only form within the cytosol once the C-terminal domain is released from the lumen. In this regard, Jiang et al. remarked that the presence of negatively charged amino acids directed at each other suggests that the fibrils can only form in an acidic environment, where the negative charges are attenuated, as would be the case in the acidic lumen of the lysosome [27].

Fibril formation of TMEM106B. After shedding the C-terminal domain, fibrils may form within the acidic lysosomal lumen. Lysosomal leakage or rupture, as a consequence of lysosomal dysfunction, may allow them to escape to the cytosol. Alternatively, fibrils may form within the cytosol once the C-terminal domain escaped the lysosomal lumen. In normal circumstances, TMEM106B’s C-terminal domain or fibrils will be degraded along with other lysosomal content. Factors that may modulate fibrillization are related to lysosomal function (pH, protease activity, lysosomal integrity, etc.) or as a result of the TMEM106B isoforms (polymorphism skewing, glycosylation, levels)
Lysosomal health will be an important factor mediating the fibrillar burden as TMEM106B is primarily localized and processed within the lysosome. In addition, the lysosome will likely also be responsible for the degradation of the C-terminal fragment and/or TMEM106B fibrils. Lysosomal dysfunction may alter fibrillization by affecting pH, and protease activity, or may lead to an increment in fibrils within the lumen due to impaired degradative function. Impaired lysosomes may also release the fibrils within the cytosol in case of lysosomal rupture, or within the extracellular space due to lysosomal exocytosis. Interestingly, lysosomal exocytosis is increased when TMEM106B levels are elevated [21, 33], which might induce spreading of TMEM106B fibrils to neighboring cells.
TMEM106B haplotypes
The discovery of TMEM106B fibrils revives the question of the functional effect of the TMEM106B risk/protective haplotype. Considering all three combinations of haplotypes (TT, TS, SS) were represented in the study population, it is apparent that both the T185 isoform and the S185 isoform can form fibrils. Because it is not known whether the T185S variant located on the haplotype is responsible for the effect, we describe both the potential contribution of the T185 and S185 isoform as well as the described increase in expression levels. While in theory we can distinguish these hypotheses as separate possibilities, in all likelihood these mechanisms converge and are contributing to the process together.
-
(I)
Polymorphism skewing (p.T185S): as the current cryo-EM resolution was not able to distinguish a serine from a threonine, it is not known whether the isoforms are equally represented in the fibrils.
-
(II)
Glycosylation status (p.T185S): all fibril polymorphisms are stated to be fully glycosylated and Schweighauser et al. reported residues G177-N183 to form a conserved conformation in all folds. It was shown that N–X–T is glycosylated more efficiently than N–X–S [7], which raises the possibility that the p.T185S variant may alter TMEM106B fibrillization by altering the glycosylation status at position N183 and therefore fibril formation. After all, post-translational modifications are known to affect fibrillization of other aggregating proteins [1, 68].
-
(III)
Fibril burden (Levels/p.T185S): alternatively, it may be the described increase in TMEM106B protein levels that may directly modulate fibrillar burden which might underlie a potential modifying effect of the risk/protective haplotype, either due to expression changes (chromatin looping) [22] or decreased stability of the S185 isoform [46].
-
(IV)
A final possibility is that the proteolytic processing of the S185 isoform is different from that of the T185 isoform due to altered binding affinity or substrate recognition.
Disease implications
TMEM106B in brain aging and neuronal health
The observation of TMEM106B fibrils across multiple neurodegenerative disorders is not surprising given the extensive repertoire of disease associations previously reported for TMEM106B. It is nevertheless intriguing that all cryo-EM reports draw markedly different conclusions on the role of TMEM106B fibrils in the process of neurodegeneration. While Jiang et al. define the fibrils of FTLD-TDP patients as being solely constituted of TMEM106B with a possible central role in disease [27], Schweighauser et al. claim that TMEM106B fibrils form in an age-dependent manner in the brain without a mechanistic connection to disease [57]. It is important to note that neurodegenerative disease hallmarks such as amyloid plaques or TDP-43 aggregates are also observed in apparently healthy individuals, though to a more limited extent [53, 54]. The mere presence of TMEM106B fibrils in neurologically normal individuals is therefore not sufficient to exclude a pathogenic effect of the fibrils and it may thus be premature to classify the fibrils as generic byproducts similar to lipofuscin. Rather, the age-dependent accumulation of TMEM106B fibrils also in healthy controls may potentially underlie the age-dependent association of TMEM106B risk haplotype with differential aging and neuronal proportion [38, 54]. The post-mitotic nature of neurons makes them highly vulnerable to the stressors accompanied by the aging process such as oxidative stress and buildup of damaged proteins [32, 40, 44]. These stressors put a strain on the endolysosomal system, which could result in the accumulation of TMEM106B fibrils even in the absence of disease which might be neurotoxic in a high enough concentration. Consequently, fibrillization of TMEM106B by itself may contribute to neuronal loss. Additionally, the presence of TMEM106B fibrils might diminish the brain’s resilience against neurodegeneration by influencing neuronal health.
Given the large number of genetic associations of the TMEM106B haplotypes to neurodegenerative disorders and general brain aging, it seems unlikely that the fibrils are benign. Yet, TMEM106B has only been identified as a risk factor for neurodegenerative disorders and a potentially toxic accumulation of a protein only associated with disease risk has not been described before. This leaves the possibility that disease-causing mutations in TMEM106B still need to be identified or that the risk- and disease-modifying effect of the TMEM106B is unrelated to TMEM106B fibrils and potentially occurs prior to their formation. TMEM106B regulates several aspects of lysosomal functioning and alterations in TMEM106B’s biology and function, be it TMEM106B expression levels or a functional consequence of p.T185S, may therefore underlie the disease-modifying effect regardless of fibril formation. Considering TMEM106B C-terminal fragments are likely degraded within the lysosome under normal circumstances, it should be noted that the formation and presence of TMEM106B fibrils may be representative of lysosomal dysfunction in general and not necessarily actively contribute to neurotoxicity or pathology. Regardless, the findings of TMEM106B amyloid fibrils in disease and normal brains bring a new dimension to the involvement of TMEM106B in brain health.
TMEM106B in proteinopathies: lysosomal dysfunction as the central hub
Given that neurodegeneration and brain diseases are complex disorders with several contributing factors, there might not be one answer to explain the role of TMEM106B fibrils in all implicated disorders. The contribution of TMEM106B fibrils to disease may depend on the affected brain region as well as the inflicted cell types or underlying pathomechanisms. It is also possible that in some neurodegenerative disorders, TMEM106B fibrils will be active drivers of the pathogenic process, while in other disorders TMEM106B fibrils might act as secondary bystanders that may promote or accelerate disease pathology and aggravate disease phenotype. A variable contribution of TMEM106B fibrils to disease pathogenesis is also supported by the nature of the observed genetic associations of TMEM106B with disease. For example, in FTLD-TDP caused by GRN mutations, the TMEM106B risk haplotype modifies disease risk to such an extent that people who are homozygous for the protective TMEM106B haplotype may remain lifelong symptom free, whereas in Parkinson’s disease and ALS the TMEM106B haplotype association is restricted to an effect on the degree of cognitive decline [36, 51, 67]. However, it remains to be determined whether TMEM106B fibrils are more frequently associated with certain proteinopathies, which will require detailed unbiased studies of large cohorts.
While it is not clear where exactly the fibrils reside, within lysosomes or in the cytosol, TMEM106B is localized in lysosomes and is proteolytically processed at the lysosomal membrane defining the lysosome as a central hub. Lysosomes have gradually been recognized as important players in neurodegenerative disorders [3, 26, 44, 55, 75], thus it is plausible that the contribution of TMEM106B fibrils to disease might depend on the importance of the lysosome within the specific pathomechanism (Fig. 6).

Potential pathomechanism and disease contribution of TMEM106B fibrils. Lysosomal dysfunction is represented as central hub within the disease mechanism. The contribution of TMEM106B fibrils to disease might depend on the importance of the lysosome within the specific pathomechanism. The p.T185S variant might modulate disease by affecting lysosomal health through modulation of TMEM106B levels or by affecting TMEM106B fibrillization. TMEM106B fibrils may actively drive the disease mechanism in FTLD-GRN, as PGRN and TMEM106B functionally converge within the lysosome and are thus affected early in the disease mechanism. In other disorders or during aging, the lysosome is only affected in a later stage and TMEM106B fibrils may play a more secondary role, potentially promoting or accelerating disease pathology
FTLD-TDP caused by GRN mutations: TMEM106B fibrils as primary disease protein
The exceptionally strong disease-modifying effect in GRN mutation carriers likely occurs within the endolysosomal system as PGRN and TMEM106B are both important players in maintaining lysosomal health. In fact, homozygous mutations in GRN have been shown to cause a lysosomal storage disorder, neuroid lipofuscinosis (NCL) [61]. PGRN is cleaved within the lysosome into functional granulins and affects several aspects of lysosomal function, including the activity of lysosomal enzymes such as cathepsin D [49, 75]. Mutations in GRN result in dysfunctional lysosomes and it is tempting to speculate that loss of PGRN/granulins will directly affect TMEM106B processing or fibril formation by modulating lysosomal health.
Interestingly, knock-out of Tmem106b worsens disease pathology in Grn-/- mouse models and induces the accumulation of phosphorylated TDP-43 in an age-dependent manner [20, 69, 74]. A recent report also showed TMEM106B knock-out to result in an increase in TDP-43 cytoplasmic aggregates in a cellular model for TDP-43 proteinopathy [43]. These findings together with the identification of TMEM106B fibrils in FTLD-TDP highlight a direct, seemingly complex relationship between TMEM106B and TDP-43. At a minimum, it confirms the importance of the lysosome in the disease mechanism. Two of the main TDP-43 degradation pathways are mediated by lysosomes, e.g., chaperone-mediated autophagy (CMA) occurring in the lysosomes and the autophagosome–lysosome pathway itself [55], and TMEM106B knock-out disrupts normal lysosomal functioning [6, 63]. Second, it suggests that the loss of function of TMEM106B, potentially caused by TMEM106B fibrillization, may contribute to TDP-43 mislocalization and aggregation.
Of note, TDP-43 knock-out in its turn also affects lysosomal biology and induces the abnormal accumulation of lysosomes in the perinuclear region, a decrease in cathepsin L and PGRN to the lysosome, abnormal cathepsin B processing, and secretion of undigested proteins from the lysosome [35, 39, 55]. This means that the mislocalization and/or accumulation of TDP-43 may further induce the formation of TMEM106B fibrils by disrupting lysosomal function, which will further compromise the degradation of the cleaved C-terminal domain of TMEM106B as well as clearance of TDP-43 aggregates which could set in motion a feedback loop.
Other proteinopathies: TMEM106B fibrils as secondary disease protein
Alternatively, protein aggregation and subsequent lysosomal dysfunction, caused by other environmental or genetic factors, may be upstream of the formation of TMEM106B fibrils which could potentially contribute to or aggravate disease pathology. A contribution of TMEM106B fibrils to disease may also present itself more as a disease modifier where the formation of TMEM106B fibrils contributes to disease manifestation, for example modifying cognitive decline in Parkinson’s disease and ALS [36, 67]. A potential effect of TMEM106B in contributing to the formation of other aggregating proteins may be supported in TDP-43 proteinopathies by the observation that the TMEM106B risk variants have been associated with increased TDP-43 aggregates in neuropathology-based association studies of apparently healthy older individuals [15, 73] and the observation that having the protective allele reduced TDP-43 burden in C9orf72 expansion carriers [4].
Conclusion and future directions
The identification of TMEM106B amyloid fibrils will undoubtedly transform research on TMEM106B and its involvement in neurodegeneration, but simultaneously raises a lot of new questions. First, the pathogenicity and disease contribution of the fibrils need to be determined, especially since fibrils were also observed in aged individuals without neurodegenerative diseases. Second, detailed studies on the contribution of the haplotype are required. Regardless of the precise mechanism, it is possible that the TMEM106B risk haplotype either indirectly affects the fibrillar burden by modulating TMEM106B levels or processing, or directly by affecting glycosylation at position N183. Third, it will be important to elucidate the precise mechanism, the involved proteases, and cellular conditions required for TMEM106B fibrillization as well as to determine the precise location of the fibrils and the identity of the cofactor within the doublets. Finally, since TMEM106B fibrils were found in neurodegenerative disorders that are typically characterized by aggregation of other proteins, such as TDP-43, it will be important to determine whether there could be a synergistic effect on the burden of other aggregated proteins.
However, the most urgent challenge the field currently encounters is the need to generate suitable antibodies and other tools to be able to study TMEM106B and its fibrillization mechanism. Antibodies will be necessary to perform correlation studies in large patient cohorts to investigate the contribution of TMEM106B pathology to disease as well as to be able to study TMEM106B biology such as its proteolytic processing. Depending on the outcome of these vital studies, TMEM106B fibrils may be pursued as a biomarker or even as a therapeutic target. Yet, this might not be straightforward considering the tight regulation of TMEM106B levels within the cell, with both knock-down and overexpression of TMEM106B resulting in lysosomal dysfunction. This means that therapeutic interventions targeting TMEM106B will need to be precisely monitored and requires detailed knowledge of TMEM106B fibril formation, as well as its normal function.
References
Alquezar C, Arya S, Kao AW (2021) Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol 11:1826. https://doi.org/10.3389/fneur.2020.595532
Arseni D, Hasegawa M, AnG M, Kametani F, Arai M, Yoshida M et al (2022) Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 601:139–143. https://doi.org/10.1038/s41586-021-04199-3
Bain HDC, Davidson YS, Robinson AC, Ryan S, Rollinson S, Richardson A et al (2019) The role of lysosomes and autophagosomes in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol 45:244–261. https://doi.org/10.1111/nan.12500
van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC et al (2014) TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol 127:397–406. https://doi.org/10.1007/s00401-013-1240-4
Brady OA, Zheng Y, Murphy K, Huang M, Hu F (2013) The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet 22:685–695. https://doi.org/10.1093/hmg/dds475
Brady OA, Zhou X, Hu F (2014) Regulated intramembrane proteolysis of the frontotemporal lobar degeneration risk factor, TMEM106B, by signal peptide peptidase-like 2a (SPPL2a). J Biol Chem 289:19670–19680. https://doi.org/10.1074/jbc.M113.515700
Breitling J, Aebi M (2013) N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a013359
Busch JI, Martinez-Lage M, Ashbridge E, Grossman M, van Deerlin VM, Hu F et al (2014) Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol Commun. https://doi.org/10.1186/2051-5960-1-36
Busch JI, Unger TL, Jain N, Skrinak RT, Charan RA, Chen-Plotkin AS (2016) Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet 25:2681–2697. https://doi.org/10.1093/hmg/ddw127
Chang A, Xiang X, Wang J, Lee C, Arakhamia T, Simjanoska M et al (2022) Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 185:1346-1355.e15. https://doi.org/10.1016/j.cell.2022.02.026
Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L et al (2012) TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci 32:11213–11227. https://doi.org/10.1523/JNEUROSCI.0521-12.2012
Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem 86:27–68. https://doi.org/10.1146/annurev-biochem-061516-045115
Dall’Aglio L, Lewis CM, Pain O (2021) Delineating the genetic component of gene expression in major depression. Biol Psychiatry 89:627–636. https://doi.org/10.1016/j.biopsych.2020.09.010
van Deerlin VM, Sleiman PMA, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239. https://doi.org/10.1038/ng.536
Dickson DW, Rademakers R, Nicholson AM, Schneider JA, Yu L, Bennett DA (2015) The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 85:1354–1355. https://doi.org/10.1212/01.wnl.0000472918.79256.a9
Fabbri C, Pain O, Hagenaars SP, Lewis CM, Serretti A (2021) Transcriptome-wide association study of treatment-resistant depression and depression subtypes for drug repurposing. Neuropsychopharmacology 46:1821–1829. https://doi.org/10.1038/s41386-021-01059-6
Fan Y, Zhao Q, Xia W, Tao Y, Yu W, Chen M et al (2022) Generic amyloid fibrillation of TMEM106B in patient with Parkinson’s disease dementia and normal elders. Cell Res 32:585–588. https://doi.org/10.1038/s41422-022-00665-3
el Fatimy R, Boulaassafre S, Bouchmaa N, el Khayari A, Vergely C, Malka G et al (2021) The emerging role of miRNA-132/212 cluster in neurologic and cardiovascular diseases: neuroprotective role in cells with prolonged longevity. Mech Ageing Dev. https://doi.org/10.1016/j.mad.2021.111566
Feng T, Lacrampe A, Hu F (2021) Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol 141:327–339. https://doi.org/10.1007/S00401-020-02246-3
Feng T, Mai S, Roscoe JM, Sheng RR, Ullah M, Zhang J et al (2020) Loss of TMEM 106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep. https://doi.org/10.15252/embr.202050219
Feng T, Sheng RR, Solé-Domènech S, Ullah M, Zhou X, Mendoza CS et al (2020) A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 143:2255–2271. https://doi.org/10.1093/brain/awaa154
Gallagher MD, Posavi M, Huang P, Unger TL, Berlyand Y, Gruenewald AL et al (2017) A dementia-associated risk variant near TMEM106B alters chromatin architecture and gene expression. Am J Hum Genet 101:643–663. https://doi.org/10.1016/j.ajhg.2017.09.004
Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, van Swieten JC et al (2014) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 127:407–418. https://doi.org/10.1007/s00401-013-1239-x
Goldman JS, Farmer JM, van Deerlin VM, Wilhelmsen KC, Miller BL, Grossman M (2004) Frontotemporal dementia: genetics and genetic counseling dilemmas. Neurologist 10:227–234. https://doi.org/10.1097/01.nrl.0000138735.48533.26
Gong X, Huang M, Chen L (2022) Mechanism of miR-132–3p promoting neuroinflammation and dopaminergic neurodegeneration in Parkinson’s disease. eNeuro. https://doi.org/10.1523/ENEURO.0393-21.2021
Götzl JK, Lang CM, Haass C, Capell A (2016) Impaired protein degradation in FTLD and related disorders. Ageing Res Rev 32:122–139. https://doi.org/10.1016/j.arr.2016.04.008
Jiang YX, Cao Q, Sawaya MR, Abskharon R, Ge P, DeTure M et al (2022) Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 605:304–309. https://doi.org/10.1038/s41586-022-04670-9
Jun MH, Han JH, Lee YK, Jang DJ, Kaang BK, Lee JA (2015) TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol Brain. https://doi.org/10.1186/s13041-015-0177-z
Kang J, Lim L, Song J (2018) TMEM106B, a risk factor for FTLD and aging, has an intrinsically disordered cytoplasmic domain. PLoS ONE. https://doi.org/10.1371/journal.pone.0205856
Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TKT et al (2017) Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 95:281-296.e6. https://doi.org/10.1016/j.neuron.2017.06.026
Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A et al (2019) Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat Commun. https://doi.org/10.1038/s41467-019-12683-8
Krisko A, Radman M (2019) Protein damage, ageing and age-related diseases. Open Biol. https://doi.org/10.1098/rsob.180249
Kundu ST, Grzeskowiak CL, Fradette JJ, Gibson LA, Rodriguez LB, Creighton CJ et al (2018) TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat Commun. https://doi.org/10.1038/s41467-018-05013-x
Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D et al (2012) Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 287:19355–19365. https://doi.org/10.1074/jbc.M112.365098
Leibiger C, Deisel J, Aufschnaiter A, Ambros S, Tereshchenko M, Verheijen BM et al (2018) TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum Mol Genet 27:1593–1607. https://doi.org/10.1093/hmg/ddy066
Lemprière S (2019) Frontotemporal dementia risk variant accelerates cognitive decline in Parkinson disease. Nat Rev Neurol 15:307. https://doi.org/10.1038/s41582-019-0196-y
Levine TP (2022) TMEM106B in humans and Vac7 and Tag1 in yeast are predicted to be lipid transfer proteins. Proteins Struct Funct Bioinform 90:164–175. https://doi.org/10.1002/prot.26201
Li Z, Farias FHG, Dube U, Del-Aguila JL, Mihindukulasuriya KA, Fernandez MV et al (2020) The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol 139:45–61. https://doi.org/10.1007/s00401-019-02066-0
Liao YZ, Ma J, Dou JZ (2022) The Role of TDP-43 in neurodegenerative disease. Mol Neurobiol. https://doi.org/10.1007/s12035-022-02847-x
Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D et al (2018) Oxidative stress, aging, and diseases. Clin Interv Aging 13:757–772. https://doi.org/10.2147/CIA.S158513
Lüningschrör P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D et al (2020) The FTLD risk factor TMEM106B regulates the transport of lysosomes at the axon initial segment of motoneurons. Cell Rep 30:3506-3519.e6. https://doi.org/10.1016/j.celrep.2020.02.060
Mackenzie IRA, Neumann M, Baborie A, Sampathu DM, du Plessis D, Jaros E et al (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113. https://doi.org/10.1007/s00401-011-0845-8
Mao F, Robinson JL, Unger T, Posavi M, Amado DA, Elman L et al (2021) TMEM106B modifies TDP-43 pathology in human ALS brain and cell-based models of TDP-43 proteinopathy. Acta Neuropathol 142:629–642. https://doi.org/10.1007/s00401-021-02330-2
Monaco A, Fraldi A (2020) Protein aggregation and dysfunction of autophagy-lysosomal pathway: a vicious cycle in lysosomal storage diseases. Front Mol Neurosci 13:37. https://doi.org/10.3389/fnmol.2020.00037
Monereo-Sánchez J, Schram MT, Frei O, O’Connell K, Shadrin AA, Smeland OB et al (2021) Genetic overlap between Alzheimer’s disease and depression mapped onto the brain. Front Neurosci 15:653130. https://doi.org/10.3389/fnins.2021.653130
Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB, Castanedes-Casey M et al (2013) TMEM106B p. T185S regulates TMEM106B protein levels: Implications for frontotemporal dementia. J Neurochem 126:781–791. https://doi.org/10.1111/jnc.12329
Nicholson AM, Rademakers R (2016) What we know about TMEM106B in neurodegeneration. Acta Neuropathol 132:639–651. https://doi.org/10.1007/s00401-016-1610-9
Olney NT, Spina S, Miller BL (2017) Frontotemporal dementia. Neurol Clin 35:339–374. https://doi.org/10.1016/j.ncl.2017.01.008
Paushter DH, Du H, Feng T, Hu F (2018) The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. https://doi.org/10.1007/s00401-018-1861-8
Piscopo P, Albani D, Castellano AE, Forloni G, Confaloni A (2016) Frontotemporal lobar degeneration and microRNAs. Front Aging Neurosci 8:17. https://doi.org/10.3389/fnagi.2016.00017
Pottier C, Zhou X, Perkerson RB, Baker M, Jenkins GD, Serie DJ et al (2018) Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol 17:548–558. https://doi.org/10.1016/S1474-4422(18)30126-1
Premi E, Grassi M, van Swieten J, Galimberti D, Graff C, Masellis M et al (2017) Cognitive reserve and TMEM106B genotype modulate brain damage in presymptomatic frontotemporal dementia: a GENFI study. Brain 140:1784–1791. https://doi.org/10.1093/brain/awx103
Reas ET (2017) Amyloid and tau pathology in normal cognitive aging. J Neurosci 37:7561–7563. https://doi.org/10.1523/JNEUROSCI.1388-17.2017
Rhinn H, Abeliovich A (2017) Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst 4:404-415.e5. https://doi.org/10.1016/j.cels.2017.02.009
Root J, Merino P, Nuckols A, Johnson M, Kukar T (2021) Lysosome dysfunction as a cause of neurodegenerative diseases: lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2021.105360
Salta E, de Strooper B (2017) MicroRNA-132: a key noncoding RNA operating in the cellular phase of Alzheimer’s disease. FASEB J 31:424–433. https://doi.org/10.1096/fj.201601308
Schweighauser M, Arseni D, Bacioglu M, Huang M, Lövestam S, Shi Y et al (2022) Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 605:310–314. https://doi.org/10.1038/s41586-022-04650-z
Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B et al (2020) Structures of α-synuclein filaments from multiple system atrophy. Nature 585:464–469. https://doi.org/10.1038/S41586-020-2317-6
Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K et al (2014) The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J 33:450–467. https://doi.org/10.1002/embj.201385857
Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A et al (2021) Structure-based classification of tauopathies. Nature 598:359–363. https://doi.org/10.1038/s41586-021-03911-7
Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M et al (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90:1102–1107. https://doi.org/10.1016/j.ajhg.2012.04.021
Spitz C, Schlosser C, Guschtschin-Schmidt N, Stelzer W, Menig S, Götz A et al (2020) Non-canonical shedding of TNFα by SPPL2a is determined by the conformational flexibility of its transmembrane helix. iScience. https://doi.org/10.1016/j.isci.2020.101775
Stagi M, Klein ZA, Gould TJ, Bewersdorf J, Strittmatter SM (2014) Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci 61:226–240. https://doi.org/10.1016/j.mcn.2014.07.006
Staudt C, Puissant E, Boonen M (2017) Subcellular trafficking of mammalian lysosomal proteins: an extended view. Int J Mol Sci 18:47. https://doi.org/10.3390/ijms18010047
Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, Kopito R et al (2017) Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener. https://doi.org/10.1186/s40035-017-0077-5
Tropea TF, Mak J, Guo MH, Xie SX, Suh E, Rick J et al (2019) TMEM106B effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol 85:801–811. https://doi.org/10.1002/ana.25486
Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D et al (2011) Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 121:373–380. https://doi.org/10.1007/s00401-010-0782-y
Vugmeyster L, Au DF, Ostrovsky D, Kierl B, Fu R, Hu Z et al (2019) Effect of post-translational modifications and mutations on amyloid-β fibrils dynamics at N terminus. Biophys J 117:1524–1535. https://doi.org/10.1016/j.bpj.2019.09.004
Werner G, Damme M, Schludi M, Gnörich J, Wind K, Fellerer K et al (2020) Loss of TMEM 106B potentiates lysosomal and FTLD -like pathology in progranulin-deficient mice. EMBO Rep. https://doi.org/10.15252/embr.202050241
White CC, Yang HS, Yu L, Chibnik LB, Dawe RJ, Yang J et al (2017) Identification of genes associated with dissociation of cognitive performance and neuropathological burden: multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med. https://doi.org/10.1371/journal.pmed.1002287
Wise AH, Alcalay RN (2022) Genetics of cognitive dysfunction in Parkinson’s disease. Prog Brain Res 269:195–226. https://doi.org/10.1016/bs.pbr.2022.01.015
Young JJ, Lavakumar M, Tampi D, Balachandran S, Tampi RR (2018) Frontotemporal dementia: latest evidence and clinical implications. Ther Adv Psychopharmacol 8:33–48. https://doi.org/10.1177/2045125317739818
Yu L, de Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA (2015) The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 84:927–934. https://doi.org/10.1212/WNL.0000000000001313
Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC et al (2020) Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep. https://doi.org/10.15252/embr.202050197
Zhou X, Kukar T, Rademakers R (2021) Lysosomal dysfunction and other pathomechanisms in FTLD: evidence from progranulin genetics and biology. Adv Exp Med Biol. https://doi.org/10.1007/978-3-030-51140-1_14
Acknowledgements
J.P is supported by a fellowship from Research Foundation – Flanders (FWO, application 11M1622N). Dr. Rademakers is a member of the Scientific Advisory Board of Arkuda Therapeutics and receives invention royalties from a patent related to progranulin. Dr. Rademakers receives research support from the National Institutes of Health (NIH) and the Department of Defense (DOD). Figures were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Perneel, J., Rademakers, R. Identification of TMEM106B amyloid fibrils provides an updated view of TMEM106B biology in health and disease. Acta Neuropathol 144, 807–819 (2022). https://doi.org/10.1007/s00401-022-02486-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-022-02486-5