
Abstract
Regulated cell death (RCD), also well-known as programmed cell death (PCD), refers to the form of cell death that can be regulated by a variety of biomacromolecules, which is distinctive from accidental cell death (ACD). Accumulating evidence has revealed that RCD subroutines are the key features of tumorigenesis, which may ultimately lead to the establishment of different potential therapeutic strategies. Hitherto, targeting the subroutines of RCD with pharmacological small-molecule compounds has been emerging as a promising therapeutic avenue, which has rapidly progressed in many types of human cancers. Thus, in this review, we focus on summarizing not only the key apoptotic and autophagy-dependent cell death signaling pathways, but the crucial pathways of other RCD subroutines, including necroptosis, pyroptosis, ferroptosis, parthanatos, entosis, NETosis and lysosome-dependent cell death (LCD) in cancer. Moreover, we further discuss the current situation of several small-molecule compounds targeting the different RCD subroutines to improve cancer treatment, such as single-target, dual or multiple-target small-molecule compounds, drug combinations, and some new emerging therapeutic strategies that would together shed new light on future directions to attack cancer cell vulnerabilities with small-molecule drugs targeting RCD for therapeutic purposes.
Similar content being viewed by others

Introduction
The biennial report 2020–2021 issued by the international agency for research on cancer (IARC) of the World Health Organization points out that the reality of high incidence of cancer and the rising trend also make cancer one of the main reasons that threaten human life. Cancer has become one of the most common diseases in China,1 which also has unique epidemiological characteristics and patient types. For example, the epidermal growth factor receptor (EGFR) mutation rate of lung adenocarcinoma patients in China is 61%, while that in the United States is only 11%.2 Cancer is a heterogeneous disease characterized by cell death disorder. In the face of the high incidence of cancer and the rising trend, it is urgent to clarify its deep pathogenesis and carry out the targeted treatment.
Cell death can be classified according to the morphological criteria, cellular context and triggering stimulus. In 2018, hundreds of scientists in the field of cell death jointly published an article in the journal Cell Death & differentiation, entitled “molecular mechanisms of cell death: recommendations of the Nomenclature Committee on cell death 2018”.3 Scientists divided the types of cell death into regulated cell death (RCD) and accidental cell death (ACD).3 ACD is an uncontrolled process of cell death, which is triggered by accidental injury stimuli. These injury stimuli exceed the adjustable ability of cells, resulting in cell death. RCD refers to the autonomous and orderly death of cells controlled by genes in order to maintain the stability of the internal environment. Its induction and execution are mainly regulated by the formation of signal amplification complexes that play an evolutionarily important role in development and immune response.4 RCD, which occurs under physiological conditions, is also known as programmed cell death (PCD).3 Currently known RCD types mainly include: autophagy-dependent cell death, apoptosis, necroptosis, pyroptosis, ferroptosis, parthanatos, entosis, NETosis, lysosome-dependent cell death (LCD), alkaliptosis, and oxeiptosis. Mammalian cells exposed to unrecoverable disturbances in the intracellular or extracellular microenvironment can activate one of many signal transduction cascades and eventually lead to their death. Each of these RCD patterns is initiated and transmitted by molecular mechanisms that show a considerable degree of interconnection. In addition, each type of RCD can show the full spectrum of morphological characteristics from complete necrosis to complete apoptosis, as well as the immunomodulatory characteristics from anti-inflammatory and tolerance to promoting inflammation and immunogenicity.3,5
Different lethal subroutines during RCD can affect cancer progression and response to treatment. In the early stage of onset, cancer cells may have the characteristics of anti-cancer treatment because of the mutation that destroys the RCD pathway, and avoiding RCD is one of the important signs of cancer. The application of RCD signal to a specific cancer type or multiple target drugs can be avoided by single or combined application of RCD signal. Based on the current research results, paying attention to the crosstalk between different RCD pathways may be a new direction of cancer treatment in the future. This manuscript will briefly describe the characteristics of the regulatory cell death mechanism and its application in tumor treatment, in order to provide new targets and new ideas for tumor treatment.
Crucial signaling pathways of RCD subroutines in cancer
Generally, apoptosis and autophagy-dependent cell death are considered as crucial subroutines of RCD, which could induce degradation of organelles or cell death under the cellular stress and play a vital role in targeted therapy and regulation of cancer cell death.6 Apoptosis has been recognized as a critical intracellular process that maintains organism homeostasis and controls cell population. Several morphological characteristics of apoptosis include cell shrinkage, chromatin condensation, membrane blebbing, deoxyribonucleic acid (DNA) fragmentation, and apoptotic body formation.7,8 Apoptosis mainly occurs in two canonical pathways: the extrinsic pathway, stimulated by the activation of death receptors, and the intrinsic pathway, mediated by mitochondria. Binding of death ligands, namely, tumor necrosis factor α (TNFα), Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL), to the homologous death domain of its target cell surface receptors, i.e., TNF receptor 1(TNFR1), Fas, and death receptor (DR) 4/5, respectively, triggers the activation of the extrinsic apoptosis pathway, activating caspase-8 and then initiating the terminal phase or execution phase of apoptosis9 The intrinsic pathway is initiated when irreparable damage to cellular components occurs and is commonly modulated by B-cell lymphoma 2 (Bcl-2) family proteins. These proteins regulate the release of cytochrome c (Cyt-C) and second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI (SMAC/DIABLO). Cyt-C interacts with apoptotic protease activating factor 1 (Apaf-1) proteins, which then activates caspase 9 to induce apoptosis in cancer cells.10,11
Autophagy, a phagocytic biological process, can disintegrate damaging proteins or organelles through lysosomal fusion and is essential for maintaining cell function and homeostasis.12 Autophagy has been proved to exert the dual functions in tumor progression, and it could promote or inhibit cancer development according to tumor subtype and mutation status.13 In the precancerous stage, the inhibition of autophagy will lead to the accumulation of reactive oxygen species (ROS), and genomic dysfunction, which collectively results in the endoplasmic reticulum (ER) pressure increased and DNA damaged, thus promoting the formation of tumors. However, when stimulated by starvation or oxidative stress, autophagy can provide energy and nutrients to tumors, which can elicit the survival of cancer cells.14,15 The autophagic process is controlled by autophagy-related genes. Unc-51-like kinase 1 (ULK1), Beclin-1, light chain 3 (LC3), p62, forkhead box O (FoxO), and other autophagy-related genes are involved in the regulation of autophagy, among which ULK1 acts as a promoter of autophagy and regulates the initiating function of autophagy.16 Autophagy-associated signaling pathways, including phosphatidylinositol 3 kinase complex 1 (PI3KC1)- protein kinase B (Akt)-mammalian target of rapamycin complex 1 (mTORC1), Ras-Raf-mitogen activated protein kinases (MAPKs) and nuclear factor kappa-B (NF-κB) pathways, also play a vital role in combating tumor progression and metastasis.
Apoptosis and autophagy are the central mechanisms that maintain cellular homeostasis and regulate cell fate. Meanwhile, there is a certain interaction between apoptosis and autophagy, which can promote cell death through an independent or complementary relationship.17 The targeted regulation of apoptosis and autophagy by small-molecule compounds has fully demonstrated its therapeutic potential in cancer agent development.18 For example, Ampelopsin (Amp) has been shown to trigger apoptosis and autophagy-dependent cell death by promoting ROS generation and the activation of c-Jun N-terminal kinase (JNK) in glioma cells.19 Galectin-1 is a member of the galactose lectin family with multiple biological activities. It is highly expressed in numerous tumors and regulates the proliferation, migration, and growth of tumor cells.20 Shikonin could be a promising anti-colorectal agent to attenuate tumor growth. It was shown that shikonin could target galectin-1 and activate the JNK signaling pathway to induce apoptosis and autophagy in colorectal carcinoma (CRC) cells.21 Interestingly, dihydroartemisinin (DHA), as an active metabolite, regulated apoptosis, and autophagic cell death by blocking the Wnt/β-catenin signaling pathway and stimulating the p38/MAPK pathway in multiple myeloma (MM).22 F1012-2, an active component isolated from Eupatorium lindleyanum DC., effectively inhibited cell growth by triggering apoptosis via intrinsic and extrinsic pathways in triple negative breast cancer (TNBC) cells. Besides, the induced apoptosis could be increased by inhibiting autophagy.23
Necroptosis is a regulatory cell death mode driven by receptor-interacting serine/threonine kinase protein (RIPK) 1 through its kinase function to form complex IIB, which leads to cell necroptosis. It has the morphological characteristics of necroptosis cells and a signal mechanism similar to apoptotic cells. Morphologically, it is characterized by cell membrane perforation, increased intracellular osmotic pressure, resulting in cell rounding and swelling, organelle swelling, mitochondrial dysfunction, loss of mitochondrial membrane potential, loss of nuclear chromatin, and explosive rupture of the plasma membrane. The cancer cell contents released after cell rupture exacerbate the peripheral inflammatory response. The difference from necrosis is that necroptosis strictly follows the cancer intracellular signal regulation and has the characteristics of active energy consumption. After TNF-α binds to TNFR1 on the plasma membrane, downstream protein molecules are recruited to form complex I. The protein of RIPK1 is transformed into the cytoplasmic receptor of RIPK1.24 Depending on the stimuli or the cancer cellular microenvironment, complex I activates different signaling pathways downstream through the regulation of RIPK1, resulting in two death modes, apoptosis, and necroptosis. Polyubiquitination of the Lys63 domain of RIPK1 promotes the recruitment of Ikappa B kinase (IKK) and transforming growth factor kinase (TAK) into a complex, and both TAK and IKKα/IKKβ complexes activate NF-κB and promote cancer cell survival (NF-κB dependent).25,26 In addition, IKKα/IKKβ, TANK-binding kinase 1 (TBK1), and IKKε also inactivate the phosphorylation of RIPK1 and prevent its translocation into complex II, thereby preventing RIPK1-dependent cell death (Non-NF-κB dependent).27,28 Intracellular death promoting protein RIPK1 promotes the recruitment of pro-caspase-8 and produces activated caspase-8, which leads to apoptosis.29,30 If caspase-8 is inhibited or not expressed in cancer cells, RIP3 is recruited to form RIPK1-rip3 complex, which causes ripk3 phosphorylation to recruit executive protein mixed lineage kinase domain-like pseudokinase (MLKL), form necrotic body (also known as complex IIB), and trigger necroptosis in cancer.31,32 Activated RIPK1 acts as an intermediate bridging complex I and complex II during TNF-α-induced apoptosis.33 Linking complex I and complex II through the formation of RIPK1, which is also observed in partial necroptosis, a marker that can be used to determine the outcome of complex I disruption.33 Complex I will ultimately determine whether cancer cells survive, apoptosis or necroptosis by regulating the functional conversion of RIPK1. Apoptosis and necrosis are the two earliest and most classical ways of cell death. As an autonomous and orderly death mode of cells controlled by genetic genes, the former is not only a phenomenon that occurs in the specific growth and development stage of most cells in organisms, but also an essential process for cells in organisms to maintain normal activity and function. Different from apoptosis, necrosis is generally considered to be uncontrollable, which is a way of death defined by morphological characteristics. Excessive external inhibitory factors can directly cause necrosis, which is a passive cell death affected by the environment. However, recent studies have shown that necrosis can also be regulated by the intracellular signal transduction pathway, which cannot be mediated by caspase. Therefore, it can still play a role when the apoptotic pathway is inhibited, and its cell morphology is consistent with conventional necrosis. In 2005, regulated necrosis was first found. This includes many ways, such as secondary necrosis, self-death, iron apoptosis, pyroptosis, parp-1-dependent cell death, necrotic apoptosis, cyclophilin necrosis, and so on. It has the morphological characteristics of necrosis, such as nuclear fragmentation, swelling of cells and organelles, rupture of the plasma membrane, and so on. Many studies have proved that necrotic apoptosis or programmed necrosis plays an important role in the occurrence, development, invasion, metastasis, and drug resistance of malignant tumors. Cell resistance to necroptosis is often mediated by oncogenes, suggesting that escape from necroptosis may be a potential tumor marker similar to escape from apoptosis. Tumor therapy based on necroptosis is a new strategy of cancer therapy, but its feasibility is still controversial. Supporters believe that because necroptosis and apoptosis play a role through different signal pathways, inducing necroptosis of tumor cells has the potential to be used as an alternative therapy for anti-apoptotic malignant tumors. According to the current research, this hypothesis has been preliminarily verified. However, skeptics believe that congenital or acquired defects in the necrosis mechanisms have been observed in many cancer cells. Whether the used necrosis inducers can selectively kill cancer cells without interfering with normal cell activities and whether they will lead to de inflammation in organisms need further research. Apoptosis is a form of programmed cell death; its autonomous cell lysis will not cause inflammation and actively participate in the process of life and death balance of tumor cells.
Pyroptosis is a form of programmed cell death associated with an inflammatory response. Gasdermins family is the primary executor of pyroptosis, including gasdermin-a (GSDMA), gasdermin-b (GSDMB), gasdermin-c (GSDMC), gasdermin-d (GSDMD), gasdermin-e (GSDME, also known as DNFA5), DFNB59 and other proteins. The characteristics of pyroptosis in cancer are mainly the cleavage and polymerization of gasdermins family proteins, the cleavage of N-terminal and C-terminal junction domains of gasdermins, and the release of activated N-terminal regions. The N-terminal binds to membrane lipids, phosphatidylinositol, and cardiolipin, and forms pore in the cell membrane, resulting in cell osmotic swelling, plasma membrane rupture, and death.34,35 Gasdermins family proteins form 10 to 20 nm holes in the cell membrane, and the cell contents are slowly released through the membrane holes (which can trigger amplified inflammatory reactions). The cells gradually flatten and produce 1–5 μm apoptotic body-like protrusions (focal dead bodies). The cells gradually swell to the rupture of plasma membrane, with nuclear concentration and chromatin DNA breakage.36 The pyroptosis pathway can be divided into classical and non-classical pyroptosis pathways in cancer. The activation of the classical pyroptosis pathway is initiated by pathogen-associated molecular patterns (PAMPs) or sterile molecular patterns (DAMPs).37,38 They are recognized by cytoplasmic pattern recognition receptors (PRRS). After recognizing specific stimuli, nod-like receptors (NLRs) or melanoma deficiency factor 2-like receptors (ALRs) initiate assembly to form inflammatory bodies and process to form activated caspase-1. Caspase-1 cleaves GSDMD, and the N-terminal of GSDMD is localized and aggregated into pores on the cell membrane. In addition, caspase-1 cleaves pro-IL-1β and pro-IL-18 to form mature IL-1β and IL-18, and the intracellular contents are secreted outside the membrane through the membrane pore. The nonclassical pyrolytic pathway depends on the activation of caspase-4/caspase-5/caspase-11. After the cytoplasm is stimulated by lipopolysaccharide (LPS), caspase-4/caspase-5/caspase-11 (the human counterpart of mouse caspase-11 caspase-4/caspase-5) can directly bind to the conserved structure of LPS, lipoprotein A, causing oligomerization, leading to activation, further cutting GSDMD, causing the N-terminal of GSDMD to be cleaved and localized to the cell membrane to form membrane pores.39 Compared with apoptosis, pyroptosis occurs faster and more violently, accompanied by the release of many pro-inflammatory factors. Inflammatory corpuscles and GSDM family proteins are the key substrates causing cell scorch. At present, in the research on digestive system tumors, hematological system tumors, respiratory system tumors, and reproductive system tumors, it has been found that the above two are involved in inhibiting the growth of tumor cells and promoting tumor cell death. In addition, some research results also suggest that cell scorch can also promote the growth of tumors in different kinds of tumor cells. This shows that cell pyroptosis plays a dual role in promoting and inhibiting tumors. It is also necessary to further study the relationship between cell pyroptosis and tumor occurrence and progression.
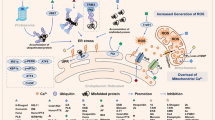
Ferroptosis is a new form of oxidative and non-apoptotic programmed cell death found in recent years. It is different from other cell death modes such as apoptosis, autophagy, and necrosis in morphology, genetics, and molecular biology.40 The main characteristics of ferroptosis are cell death induced by iron-dependent lipid peroxide injury in mitochondria, accompanying with the deficiency of activity of the lipid repair enzyme glutathione peroxidase 4 (GPX4). Its biochemical characteristics are mainly manifested in the inhibition of cystine/glutamate transporter system, the increase of nicotinamide adenine dinucleotide phosphate (NADPH) oxidation, and the accumulation of ROS caused by iron overload, and the increase of lipid peroxidation products. Ferroptosis has unique morphological and bioenergetic characteristics. Its morphological characteristics are that the cell membrane is not broken, the plasma membrane blisters, mitochondria shrink, the density of the mitochondrial membrane increases, the mitochondrial cristae decreases or disappears, and the nuclear size is normal, but the chromatin does not condense. The level of intracellular lipid peroxidation is finely regulated: on the one hand, the highly expressed polyunsaturated fatty acids on the cell membrane are very vulnerable to the attack of lipid ROS induced by divalent iron or oxidized by lipoxygenase, leading to the cascade reaction of lipid peroxidation and the accumulation of a large number of lipid peroxides; On the other hand, GPX4 of the antioxidant system will reduce the lipid peroxide to the corresponding lipid alcohol, so as to reduce the burden of lipid peroxidation and protect the cell membrane from damage. Only when this regulation system is out of balance, and the accumulation of lipid peroxide reaches a lethal amount will ferroptosis occur. The occurrence of ferroptosis is closely related to the accumulation of iron in cells, the production of free radicals, the supply of fatty acids, and lipid peroxidation. Extracellular Fe3+ binds to transferrin (TF) and is transported into cells through transferrin receptor 1 (TfR1) and reduced to Fe2+.41 After that, it was stored in the intracellular labile iron pool (LIP) with the help of intracellular divalent metal transporter 1 (DMT1) and zinc transporter 8/14 (ZIP8/14).42 Fe2+ can transfer electrons through Fenton reaction with peroxide to produce free radicals with oxidation ability. When intracellular iron is overloaded, a large number of free radicals can react with polyunsaturated fatty acid (PUFA) of cell membrane phospholipids under the catalysis of ester oxygenase and iron to produce a large number of lipid peroxides, resulting in cell death.8 At the same time, the intracellular antioxidant stress system mainly relies on GPX4 to remove excess lipid peroxides. Cystine/glutamate antiport, also known as system xc− is responsible for transporting glutamate out of cells and the same amount of cystine into cells. When it is blocked by system xc− inhibitors such as erastin, it prevents cystine from entering cells, resulting in the reduction of the content of cysteine necessary for the synthesis of glutathione (GSH) and the obstruction of the synthesis of GSH. GSH is involved in the process of GPX4 hydrolyzing lipid peroxide. The inhibition of GSH synthesis or the inactivation of GPX4 can make the excess lipid peroxide in cells unable to be removed, resulting in cell oxidative damage and inducing ferroptosis.43 Therefore, inhibiting system xc−, consuming GSH, and inactivating GPX4 are the key nodes to induce ferroptosis. Therefore, many ferroptosis inducers have been developed. Ferroptosis, non-apoptotic regulatory cell death found in recent years, is a hot issue in biological and medical research. However, at present, the compounds that induce ferroptosis are only effective for some tumor cells, and different kinds of cancer seem to have different sensitivity to ferroptosis. Loading ferroptosis inducer, reactant of ferroptosis process or traditional Chinese medicine preparation through nanotechnology to target the tumor site and make the drug concentration gather at the tumor site, which may bring new options for cancer treatment based on ferroptosis.
Poly (ADP-ribose) polymerase-1 (PARP-1)-dependent cell death (parthanatos) is a new type of regulatory cell death. It was named parthanatos in 2008.44 “Par” stands for par (poly ADP ribose), and the suffix “Thanatos” comes from ancient Greek mythology, which means “death”. The process of parthanatos is different from apoptosis and other regulatory necrosis. It is mainly manifested in: when parthanatos occurs, PARP-1 is abnormally activated and produces a large amount of par; When the mitochondrial membrane is depolarized, the levels of ATP and NADPH decrease, and the apoptosis-inducing factor (AIF) enters the nucleus from mitochondria; Although caspase is activated in the late stage of parthanatos, caspase inhibitor can not inhibit the regulatory cell necrosis, but PARP-1 inhibitor or PARP-1 gene knockout can prevent its occurrence;45 AIF is transferred to the nucleus, where chromatin condenses and produces a large number of DNA fragments ranging from 15 KB to 50 KB. Parthanatos widely occurs in many pathological processes such as inflammatory injury, ROS-induced injury and tumor. The occurrence process will lead to abnormal activation of PARP-1 and produce a large number of ADP ribose polymers (PAR) connected by glycosidic bonds. Par itself has toxic effects on cells. Therefore, the signal transduction of par polymer to mitochondria and the transfer of AIF from mitochondria to the nucleus is the crucial way to causing parthanatos. Parthanatos is a new type of programmed death different from apoptosis and necrosis. Its main feature is that caspase is not involved in this process. PARP-1 inhibitor or PARP-1 gene deletion can completely block the occurrence of parthanatos, while caspase inhibitors can not inhibit the occurrence of parthanatos. In addition to brain injury diseases, many factors such as ROS, ultraviolet irradiation and alkylating agents can also cause DNA breakage, which can lead to the over activation of PARP-1 and the occurrence of parthanatos. The overactivation of PARP-1, par accumulation, and AIF nuclear displacement are the main signs of parthanatos. Parthanatos has some of the same characteristics as necroptosis, apoptosis, and autophagy, but the molecular mechanism is different. Compared with apoptosis, parthanatos could not form small fragments of DNA fragments and apoptotic bodies; Compared with cell necrosis, parthanatos could not cause organelle swelling; Compared with autophagy, parthanatos did not form autophagic vesicles and lysosomal degradation; Compared with necroptosis, parthanatos did not cause swelling of cell membrane and organelles, cell lysis, and RIPK1 activation. Different from apoptosis and necrosis, they are the process of chromatin degradation and particle release into extracellular space.
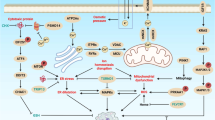
In this review, we not only focus on apoptosis and autophagy, two pivotal pathways that regulate cell death, but also involve other RCD subroutines such as necroptosis, pyroptosis, ferroptosis, parthanatos, entosis, NETosis and Lysosome-dependent cell death (LCD). Therefore, we summarize some representative small-molecule compounds which can target these subroutines, control cancer cells’ survival, and thus improve the efficacy of cancer therapy (Fig. 1).

Crucial signaling pathways of RCD subroutines in cancer
Apoptotic signaling pathways in cancer
Apoptosis refers to the spontaneous and orderly death of cells controlled by multiple genes in order to maintain the stability of the internal environment. Inhibition or resistance of cell death often leads to the occurrence of tumors.46,47 Therefore, the regulation of the apoptosis signaling pathway is one of the crucial methods to improve cancer treatment.48 Then, we focus on summarizing some representative small-molecule compounds that ultimately induce cancer cell death through the regulation of some crucial apoptotic signaling pathways and targets, such as TNF-related ligands and their receptors, Bcl-2 family, Apaf-1 and Cyt-C, NF-κB pathway, p53, etc. (Fig. 2).

Small-molecule compounds targeting apoptosis-related pathways in cancer. There are two core apoptosis pathways, intrinsic and extrinsic. The extrinsic pathway is initiated by multiple death receptors, such as TNFR1, Fas, and DR4/5. The intrinsic pathway is mediated by Bcl-2 family proteins. Activation of either pathway ultimately triggers a cascade of caspases, thus inducing caspase-dependent nucleosome fragmentation leading to cell death. In addition, NF-κB, JAK-STAT3, and MAPKs signaling pathways play an essential role in regulating cell apoptosis
Targeting TNF-related ligands and their receptors
Tumor necrosis factor (TNF) is a regulatory cytokine, as well as an essential signal transduction protein. The death receptor-mediated apoptosis pathway could be triggered by binding of TNF-related ligands such as FasL, TNFα, and TRAIL to their corresponding receptors Fas, TNFR1, and DR4/5, respectively.11,49 This interaction subsequently activates the recruitment of death domain-containing adaptor proteins, like Fas-associated protein with death domain (FADD) and TNFR1-associated death domain (TRADD), which could bridge the death effector domain (DED) to pro-caspase 8, forming the death-inducing signaling complex (DISC). DISC promotes the activation of pro-caspase-8 by cleavage and then activates other caspase proteins, leading to apoptosis execution.50,51 In this review, we have summarized some small-molecule compounds that promoted cell apoptosis and suppressed the growth of cancer cells by targeting TNF-related receptors and their ligands.
TRAIL, also called TNF superfamily 10, is a pleiotropic cytokine from the TNF superfamily. TRAIL has been shown to selectively induce apoptosis in various tumor cells without affecting normal cells.52 In this report, Shishodia et al. found that tetrandrine (TET) could sensitize resistant and mildly sensitive prostate cancer cells to TRAIL-induced apoptosis, and these effects were regulated by upregulating the messenger ribonucleic acid (mRNA) expression of DR4/DR5.53 ONC201, a TRAIL-inducing compound expressed the potential anti-cancer activity in numerous cancer cell lines. ONC201 bound to dopamine receptors DRD2 and DRD3, as well as mitochondrial caseinolytic protease P (ClpP), resulting in activation of activating transcription factor 4 (ATF4), which leads to DR5 upregulation and cell death dependent on C/EBP homologous proteins (CHOP).54,55 ABBV-621 is a TRAIL agonist that could enhance caspase-8 aggregation and the formation of death signal complexes independent of FcγR-mediated cross-linking. The research showed that ABBV-621 could induce cell death in ~36% of solid cancer cell lines in vitro at subnanomolar concentrations. ABBV-621 could overcome the resistance in ABBV-621 therapy when combined with chemotherapeutic agents or selective inhibitors of B-cell lymphoma-extra large (Bcl-xL). In a word, ABBV-621 shows therapeutic antitumor efficacy in phase 1 clinical trial (NCT03082209).56 Tannic acid (TA), as a natural polyphenol compound, has a more effective anticancer activity. TA was shown to arrest sub-G1 cell cycle arrest and induce apoptosis through enhancing the generation of mitochondrial (mROS) and further stimulating the TRAIL-induced extrinsic apoptosis pathway in NCCIT cells.57
Traditional Chinese Medicine (TCM) has been considered a new source of candidate small-molecule agents due to natural products possessing diverse bioactivities. Scutellarein (SCU), a flavone compound isolated from the perennial herb Scutellaria baicalensis, was reported to present antitumorigenic effects by promoting Hep3B cell apoptosis. SCU increased the expression level of Fas and FasL to activate caspase 8 and caspase 3, eventually causing Fas-mediated extrinsic apoptosis.58 CPT211, a novel camptothecin derivative, had been reported to suppress the proliferation and induce apoptosis of MDA-MB-231 cells effectively by activating Fas/FADD/caspase-8 signaling.59 Moreover, Apigetrin, a flavonoid glycoside compound, was shown to exert an antiproliferation effect on AGS cells. It could upregulate extrinsic apoptosis proteins expression like Fas, FasL, and DR4, as well as induce autophagy by increasing the beclin-1 and p62 proteins.60 Imipramine was a tricyclic antidepressant that triggered extrinsic apoptosis by upregulating FasL and y activating caspase 8/3 in glioblastoma cells. In an in vivo experiment, imipramine could also effectively attenuate tumor growth by suppressing the extracellular signal-regulated kinase (ERK)/NF-κB pathway activation.61 Gentian violet (GV) has an inhibitory effect on the survival and growth of cutaneous T-cell lymphoma (CTCL) tumors. GV could kill the CTCL cells by upregulating DR4/5, TRAIL, and FasL expression, triggering the extrinsic apoptosis pathway.62
Epigenetic inheritance is critical for gene expression and stability, and its disruption is thought to play an important role in the development of many tumor types.63 Histone deacetylation is an essential epigenetic event involved in the development and progression of cancer by regulating DNA expression.64 Compound 3 was a potent histone deacetylase (HDAC) inhibitor that could inhibit lung cancer cell growth and is an effective compound for the epigenetic remodeling activity of A549 cells. Compound 3 induced cancer cell apoptosis through both extrinsic and intrinsic pathways. It facilitated the expression of procaspase 8, FasL/Fas, and TNF-α to activate the extrinsic pathway and upregulated Bax, downregulated Bcl-2, thereby releasing Cyt-C to activate the intrinsic pathway.65 In non-small cell lung cancer (NSCLC) cell lines, after pemetrexed treatment, the expression of TNFRSF10B and a vesicular trafficking regulator protein, yip domain family 2 (YIPF2), was increased. YIPF2 facilitated chemotherapeutic drug-mediated apoptosis by promoting TNFRSF10B cell membrane circulation in NSCLC.66 ASTX660 as a cIAP1/2 and X-linked inhibitor of apoptosis protein (XIAP) antagonist could sensitize murine oral cancer (MOC1) cells to TNF-α and induce apoptosis of TNFR superfamily downstream cells. Besides, ASTX660 combined with cisplatin and PD-1 blockade could delay tumor growth.67
Recently, with the development of metal complexes as anti-cancer agents, their mechanism of action has gained more attention. The research has shown that ruthenium (Ru) complex 2c could target death receptors like DR5 and Fas to trigger the extrinsic apoptosis pathway. In addition, complex 2c entered the nucleus and interacted with DNA to activate p53 protein, ultimately promoting apoptosis. Compared with the chemotherapeutic drug cisplatin, complex 2 possessed lower toxicity in vivo and had considerable antitumor activity.68 Furthermore, the MnIII complex was reported to enhance the activity of caspase 8 and caspase 9, upregulate the Bax/Bcl-2 ratio expression, and promote the binding of TNF-α to its receptor, indicating a simultaneous activation of both intrinsic and extrinsic apoptotic pathways in MDA-MB-231 cells.69 Apart from the compounds mentioned above, other small-molecule compounds that induce apoptosis by targeting TNFR-related proteins are also summarized in Table 1.70,71,72,73,74,75,76,77
Targeting Bcl-2 family
The B-cell lymphoma-2 (Bcl-2) family is an essential regulatory factor in the mitochondria-mediated apoptosis pathway, controlling apoptosis and survival through the interaction of pro-apoptotic and anti-apoptotic molecules, and is the most extensive class of proteins in apoptosis research.78 Bcl-2 family proteins are divided into three groups, which are comprised of anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-w, and Mcl-1), pro-apoptotic proteins (Bax, Bak, and Box), and Bcl-2 homology domain 3 (BH3)-only proteins (Bad, Bim, and Bid).79,80 Overexpression of anti-apoptotic Bcl-2 family proteins or loss of pro-apoptotic proteins are frequently observed in various human tumors.81 Therefore, targeting these proteins with small molecules was proving to be an attractive strategy for anticancer therapy. Meanwhile, targeting anti-apoptotic proteins could also restore the sensitivity of cancer cells to apoptotic stimulation.
Some small-molecule inhibitors targeting Bcl-2 family proteins have been developed as classic therapy for cancers. For example, ABT-263 (navitoclax) is a Bcl-2/Bcl-xL inhibitor, which could block Bcl-xL to sequester activator BH3-only molecules (BH3s) without the obvious effect on Bax. The response to ABT-263 is closed to the expression of Mcl-1 protein in small cell lung cancer (SCLC) cells.82 However, low expression of Mcl-1 in NSCLC cells could not imply that ABT-263 is of therapeutic significance. It was found that increased the expression of intracellular ROS could upregulate the sensitivity of NSCLC cells to a certain extent, which could be used as a new marker for diagnosis and treatment.83 In addition, ABT-263 was found to induce apoptosis in human oral cancer-derived cell lines via increasing the expression of C/EBP-homologous protein (CHOP) and its mRNA.84 A selective Bcl-2 inhibitor, ABT-199 (Venetoclax), has received FDA approval for the treatment of chronic lymphocytic leukemia (CLL) and acute myelocytic leukemia (AML). Meanwhile, numerous trials have been conducted on other malignancies.80 Lochmann et al. reported that ABT-199 could trigger Bim-dependent apoptosis in SCLC cell lines via the disruption of Bim and Bcl-2 complexes. Besides, ABT-199 could also inhibit tumor growth and promote tumor regression in vivo. And ABT-199 combined with doxorubicin (DOX) or dinaciclib could effectively improve the therapeutic outcome of SCLC.85 Another Bcl-2 inhibitor, AT-101, was used to explore its antitumor activity and the mechanism of targeting cancer stem cells (CSCs) and anti-apoptotic proteins in gastro-esophageal cancers (GEC).86 AT-101 could induce apoptosis of cells with Bcl-2/Mcl-1 high expression in gastric cancer tissues and then inhibit cell proliferation and growth. In vivo studies had shown that AT-101 combined with docetaxel increased antitumor activity and significantly decreased CSCs biomarkers (YAP1/SOX9). In a preliminary clinical trial, 13 patients received AT-101 in combination with chemoradiotherapy for locally advanced esophageal or gastroesophageal junction cancer. (NCT00561197) The results showed relief of clinical symptoms and improved overall survival.86
Yes-associated protein 1 (YAP) is an essential downstream factor in the Hippo signal cascade that regulates cell proliferation, apoptosis, and angiogenesis. YAP is overexpressed in several types of malignant tumors and is involved in the occurrence and development of tumors, which may be a potential therapeutic target for cancer therapy.87 Verteporfin as a photosensitizer could inhibit the proliferation of pancreatic ductal adenocarcinoma (PDAC) PANC-1 and SW1990 cells, blocking cells at the G1 phase and further inducing apoptosis. It could suppress the interaction in YAP and TEAD by downregulating the expression of cyclinD1, cyclinE1, and Bcl-2 protein.88 Furthermore, (E)-2-benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one (BCI) is a phosphatase 1/6 and MAPK inhibitor that could inhibit the viability of lung cancer cells. In NCI-H1299 cells, BCl could downregulate the level of Bcl-2 protein and upregulate the Bax protein expression, thereby promoting the release of Cyt-C and activating caspase 8 to trigger apoptosis.89
McL-1 is an anti-apoptotic protein in the Bcl-2 family proteins, which is essential for the survival of normal cells. It is overexpressed in various cancers, such as lung cancer, colon cancer, multiple myeloma, etc., and is closely associated with poor prognosis.90,91 Therefore, targeting McL-1 is a promising therapeutic strategy for cancer. Zhu et al. had developed 3,5-dimethyl-4-sulfonyl-1H-pyrrole-based compound 40 as an Mcl-1 inhibitor and found that it could trigger the apoptosis pathway in Mcl-1 dependent way. Additionally, oral compound 40 could attenuate tumor growth in MV4-11 xenograft models.92 Similarly, AZD5991 had shown robust antitumor activity in multiple myeloma and acute myeloid leukemia (AML) models. It could induce apoptosis through activating the Bak-dependent intrinsic apoptosis pathway to activate caspase 3 by binding to Mcl-1. Based on these experimental data, a phase 1 clinical trial (NCT03218683) was initiated to evaluate the validity of AZD5991 in hematological malignant tumors.93
It was worth noting that natural compounds or their synthetic derivatives have gradually become a new source for discovering antitumor drug candidates. Garciniaxanthone I (GXI) was a novel active compound isolated from the bark of G. xanthochymus. It could trigger HepG2 cell apoptosis via upregulating Bax protein levels and decreasing the Bcl-2, Bcl-xL, and Mcl-1 levels. Besides, GXI could inhibit cell migration by downregulating the expression of MMP-7 and MMP-9.94 Ampelopsin, a plant-derived natural compound, possesses various pharmacology properties, including anticancer, anti-inflammatory, antibacterial, and so far. Ampelopsin was shown to activate the apoptosis pathway by regulating Bcl-2 family proteins in K563 and HL60 leukemia cells. Furthermore, it could inhibit cell growth by suppressing the Akt and NF-κB pathways.95 It could be a potential agent for the treatment of leukemia. A natural flavone compound, 5,3′-dihydroxy-3,6,7,8,4′-pentamethoxyflavone (PMF), was reported to induce an intrinsic apoptotic pathway in MCF-7 cells via enhancing the expression of Bax, Cyt-C, and PARP-1, decreasing the Bcl-2 level.96 Some other natural products, like Tracheloside (TCS) and Deoxypodophyllotoxin (DPT), could significantly inhibit the growth of CRC cells.97,98
Fu et al. synthesized a series of trimethoxyphenyl-1,2,3-triazole compounds, among which triazole containing coumarin structure 19c possessed the best antitumor activity, superior to colchicine. Compound 19c was shown to trigger apoptosis via upregulating the expression of Bax and DR5, as well as downregulating the Bcl-xL and XIAP protein levels. Further, the research showed that compound 19c could bind to the colchicine site to inhibit tubulin polymerization.99 A recent study showed compound 8 as a novel steroidal-chalcone compound was synthesized, which used two hydrophilic amide linkages to synthesize a steroidal hybrid molecule and exerted anti-TNBC activity.100 It triggered apoptosis by downregulating the Bcl-2/Bax protein ratio and activating caspase-3. In addition, upregulating ROS levels could accelerate the apoptosis of MDA-MB-231 cells.100
Among the new anticancer drugs currently studied, metal-based drugs have become an important one. More and more metal-based complexes with high efficiency, low toxicity, and high anticancer activity have been synthesized.101 The Pt(IV) complexes 14 and 17 designed and synthesized by Huang et al., showed better anticancer activity in human cancer cells than the mother Pt(II) counterparts, but their antitumor activity was different due to the difference in carbon chain lengths. They could induce apoptosis of HepG-2 cells through the release of Cyt-C, downregulation of Bcl-2, upregulation of Bax, and activation of caspase 9/3.102 In addition to platinum-based complexes, other metal cores have been explored, such as Ir, Co, Zn, and Cu, which were considered to be new therapeutic drug candidates.103 For example, iridium (III) complexes possess strongly anticancer activity. [Ir(ppy)2(THPDP)]PF6 (Ir-1) was synthesized to induce cell apoptosis by activating ROS to cause mitochondrial dysfunction, which was indicated by the expression level of the Bcl-2 family and the release of Cyt-C. Besides, the experiment result showed that Ir-1 could also induce apoptosis by suppressing the PI3K/Akt/mTOR pathway.104 Moreover, copper complexes containing benzimidazole exhibit favorable anticancer and antimicrobial activities but with toxic side effects. Therefore, Qi et al. introduced dipeptides into Cu(II) complex to alleviate its toxicity. [Cu(Gly-L-leu)(HPBM)(H2O)]ClO4 had been synthesized and shown excellent stability in the buffer solutions. The group had further explored its anticancer mechanism, and the results indicated that it could regulate Bcl-2 family proteins level and activate ROS to trigger apoptosis of HeLa cells.105 Likewise, numerous small-molecule compounds could trigger apoptosis of cancer cells by regulating Bcl-2 family proteins106,107,108,109,110,111,112,113 (Table 2).
Targeting Apaf-1 and Cyt-C
Cyt-C, a member of the mitochondrial electron transport chain, is a vital factor promoting cell apoptosis.114 Apaf-1 is a cytoplasmic protein with a cysteine protease recruitment domain at its amino-terminal, which is the core of apoptosome and plays a pivotal role in activating the caspase cascade. Cyt-C/Apaf-1 are the essential classical pathway in the initiation of apoptosis in mitochondria.115,116 In the process of apoptosis, the permeability of mitochondrial intima was increased, and the mitochondrial membrane potential was reduced, which promoted the release of pro-apoptotic factors such as Cyt-C, Apaf-1, and AIF from the mitochondria membrane space into the cytoplasm and then bound with Apaf-1 to activate downstream caspase 9 and caspase 3, and finally, lead to the occurrence of apoptosis.117
Macrophage migration inhibitory factor (MIF) is a pro-inflammatory factor overexpressed in several solid tumors to accelerate tumor development and metastasis. CPSI-1306 as a MIF inhibitor was found to decrease tumor growth and metastasis both in vitro and in vivo. CPSI-1306 enhanced ROS levels in TNBC cells and promoted the release of Cyt-C and AIF from mitochondria, leading to the induction of cell apoptosis.118 Ginseng is a kind of traditional Chinese medicine with various pharmacological activities. Piperazine groups introduced into ginsenoside can improve ROS generation and stimulate the apoptosis of cancer cells.119 In this work, xiao et al. synthesized a series of novel ginsenoside piperazine derivatives and tested the antiproliferative activity against PC-3 cells. Ginsenoside piperazine derivative 6g with an IC50 value of 1.98 ± 0.34 μM was identified as the most potent compound. It could induce apoptosis in PC-3 cells, and this induction was mediated by the enhanced ROS production and Cyt-C release. It could also upregulate the expression of Cl-PARP, Cl-Caspase-3, and Cl-Caspase-9, leading to apoptosis.119 Moreover, ((E)-2-(3-benzyl-4,4-dimethyl-2-oxooxazolidin-5-ylidene)-N,N-diethylacetamide (OI), the derivative of oxazolidinones, exhibited good antitumor activity. OI treatment could activate ROS generation, causing the reduction of mitochondrial membrane potential, further increasing the expression of caspase 9 and the release of Cyt-C, leading to apoptosis of MCF-7 and HeLa cells.120 Compound 19b as a boehmeriasin A derivative was found to trigger apoptosis of liver cancer cell lines by promoting the release of Cyt-C, the cleavage of PARP, and arrest of the cell cycle at the SubG1 phase.121
Additionally, ethyl 5-(2-cyano-3-(furan-2-yl)acrylamido)-1,3-diphenylpyrazole-4-carboxylate 5 as a synthesized compound had shown significant cytotoxic effect against colon cancer and initiated the intrinsic apoptosis through increasing the protein level of Cyt-C, Apaf-1, and SMAC/DIABLO. Besides, other apoptosis-related genes had also been stimulated, such as Bax, Bcl-2, p53, MMP-1, etc.122
Traditional Chinese medicine or natural medicine possesses tremendous development potential in anticancer drug research. For example, baicalein (BA), a natural flavonoid compound, was shown to inhibit the viability of A549 and H1299 lung cancer cells and trigger apoptosis. BA made the mitochondrial impairment, which would cause changes in mitochondrial membrane potential, as well Cyt-C and AIF in mitochondria would be released into the cytoplasm. In addition, BA could also activate the adenosine 5′-monophosphate-activated protein kinase (AMPK)/mitochondrial fission pathway to elicit apoptosis and autophagy.123 Hexokinase 2 (HK2) is overexpressed in human cancers, accelerating glucose uptake and forming the HK2-VDAC complex to generate apoptosis resistance.124 Liu et al. found that xanthohumol exerted a significant antitumor effect on CRC cells by downregulating HK2 expression and suppressing glycolysis. Besides, it could promote the release of Cyt-C to activate apoptosis.125 Lycorine, as a natural compound isolated from the Amaryllidaceae plant, was shown to induce mitochondrial apoptosis in HepG2 cells via the promotion of Cyt-C release into the cytosol.126 Natural products, curcumin and emodin, also exhibited significant antitumor effects against melanoma and liver cancer cells, respectively.127,128 Except as mentioned above, oroxyloside, the metabolite of oroxylin A, was reported to be a promising agent for human glioma treatment. It was found that apoptosis was suppressed by oroxyloside by improving the cleavage of caspase 9, caspase 3, and PARP, as well as promoting the release of Cyt-C into the cytoplasm and increase of Apaf-1129 (Table 3).
Targeting NF-κB
NF-κB, a crucial nuclear transcription factor in cells, participates in various complex biological processes such as cell proliferation, invasion, and angiogenesis.130,131 NF-κB is also a crucial regulatory member of the anti-apoptosis signaling pathway. Typically, NF-κB exists in the cytoplasm in the form of dimer and binds to the specific inhibitor IκBα. Upon certain stimuli, IκBα is phosphorylated and degraded in a proteasome-dependent manner, which releases NF-κB and ultimately transfers to the nucleus, regulating the transcription of target genes, thus affecting the occurrence and development of tumors.132,133 Blocking the activity of NF-κB could change the survival/death balance of tumor cells. Therefore, targeting NF-κB to induce apoptosis of cancer cells has been considered to be an effective way to treat human cancers. We have collected several potential small-molecule compounds to treat human cancers by targeting the NF-κB pathway.
Selinexor has been shown to inhibit XPO1-mediated nuclear export from exerting anticancer activity. Survivin was an inhibitor of apoptosis, which could be regulated by the transcription of NF-κB. Selinexor attenuates the growth of cancer cells and induces selinexor-resistant cells to become sensitive to selinexor by stabilizing IκB to inhibit the NF-κB pathway and then downregulating the expression of survivin protein. Nair et al. reported that selinexor A has an inhibitory effect on various sarcoma cells with IC50 values ranging from 50 nM to 2.5 μM, among them the liposarcoma (LS141) showing the most substantial sensitivity. Besides, when combined with proteasome inhibitor carfilzomib, it caused resistant cell lines to be more sensitive to Selinexor.134 A phase I clinical trial (NCT01607905) showed that selinexor possessed antitumor activity against advanced solid tumors. Among the 157 patients treated, 7 had reduced target lesions, and 27 had stable disease control for four months or longer.135 MicroRNAs (miRNAs), as a class of non-coding ribonucleic acid (RNA) molecules, could directly participate in mRNA expression or inhibit the translation processes from regulating the related-protein level, which was also associated with cell apoptosis, proliferation, and metastasis. Puerarin, a flavonoid compound, had been observed in bladder cancer cell lines to significantly inhibit cell viability and proliferation, inactivating the NF-κB pathway by upregulating the mRNA level of mir-16 and thereby enhancing cell apoptosis.136
The emergence of multidrug resistance (MDR) was a major obstacle to treating cancer diseases.137 Studies have shown that NF-κB could increase the expression of the MDR1 gene, and then inhibition of NF-κB activity could enhance the sensitivity of drug-resistant cancer cells to chemotherapy drugs.138 Abdin et al. proposed that the combination of NF-κB inhibitors (Pentoxifylline/Bortezomib) and DOX could reduce DOX resistance in breast cancer cells. DOX/NF-κB inhibitor combination therapy could inhibit the migration of cancer cells, activate apoptosis-related proteins, and induce cell apoptosis. It could be an effective therapeutic strategy for overcoming multidrug resistance in cancer cells.139 Radiotherapy was the main treatment for non-small cell carcinoma, while radioresistance would be induced, leading to a low reactivity.140 The research showed that isorhamnetin (ISO), a flavonoid extracted from Hippophae L., could be a potent natural radiosensitizer to increase the incidence of apoptosis, the change of MMP, as well as suppress the upregulation of NF-κB p65 triggered by irradiation in A549 cells. Additionally, the expression of interleukin-13 (IL-13) was related to the strength of ISO-mediated radiosensitization. Therefore, ISO treatment could enhance the expression of IL-13.141
Aurantoside C (C828), a natural product obtained from marine sponge Manihinea lynbeazleyae, had shown potent cytotoxic activity against TNBC cells.142 C828 was found to trigger apoptosis by inhibiting the phosphorylation of Akt/mTOR and NF-κB pathways and upregulating the expression of p38/MAPK and SAPK/JNK pathways without cytotoxic effects on normal cells. Furthermore, C828 exhibited a 20-fold and 35-fold higher cytotoxic effect than chemotherapeutic drugs DOX and cisplatin. Therefore, C828 could be a promising lead compound for developing anti-TNBC agents.142 Another natural flavonoid glycoside product, hyperoside (quercetin 3-o-β-d-galactopyranoside), exhibited antitumor, antidepressant, and anti-inflammatory effects. It was reported to suppress the viability and migration ability of MCF-7 and 4T1 cells, as well as promote apoptosis through the inhibition of ROS generation, further suppressing the activation of the NF-κB signaling pathway. Besides, the finding from the in vivo study suggested that it could decrease the size of tumors in mouse model.143 Arnicolide D (Ar-D), a sesquiterpene lactone compound, was shown to exert an anti-melanoma effect. This effect was mediated by inhibiting the expression of IKKα/β, the degradation of IκBα, and the phosphorylation of NF-κB p65, ultimately eliciting the apoptosis of melanoma cells.144 Furthermore, Glaucocalyxin A (GLA) could also induce mitochondrial apoptosis through the inhibition of the NF-κB/p65 pathway.145 Other natural small-molecule compounds such as cardamonin, ginsenoside Rk1, and betulinic acid exhibited potent anticancer effects and triggered cancer cell apoptosis through the inhibition of the NF-κB pathway146,147,148 (Table 4).
Targeting p53
The p53 protein, a notable tumor suppressor, plays a principal role in the regulation of cell cycle arrest, cell differentiation, cell metastasis, and apoptosis.149 In response to cellular stress, p53 is activated and then exerts its transcriptional regulatory function to influence the level of Bcl-2 family protein, resulting in increased cellular level of pro-apoptotic members of the Bcl-2 family and concomitantly decreased level of anti-apoptotic proteins, thereby preventing malignant transformation and leading to apoptotic cell death.150 A large number of studies had reported that the dysfunction of p53 owing to the mutation of p53 protein was observed in ~50% of all human tumors, which was always accompanied by angiogenesis, tumor progression, and drug resistance.151 Mutations in tumor suppressor proteins could lead to the loss of tumor suppressor function or promote the acquisition of new cancer phenotypic functions, such as excessive proliferation, invasive enhancement, and chemo-resistance.152,153 Therefore, targeting mutant p53 with small-molecule compound to induce apoptosis could be an attractive strategy for the development of anticancer therapy.
Murine double minute 2 (MDM2) is a negative regulator directly bound to p53 protein and inhibits the activation of p53, causing aberrant cell proliferation and growth.154 The report showed that p53 protein could be degraded by the complex of human papillomaviruses (HPV) oncoprotein E6 and E6-associated protein (E6AP) ubiquitin ligase.155 MDMX could inhibit the trans-activation of p53-mediated target genes and increase the activity of MDM2 through stabilization.156,157 XI-011 (NSC146109) was an MDMX inhibitor that could trigger apoptosis and suppress the growth of cervical cancer cells by restoring the stability of p53 and enhancing its transcription activity.158 And a new tryptophanol-derived oxazoloisoindolinone, DIMP53-1, bound to p53 suppressing its interaction with MDM2 and MDMX, which inhibit the migration and invasion of colon adenocarcinoma HCT116 cell.159 p73 was a tumor suppressor whose structure and function were similar to p53 protein, but it rarely mutated in cancers and was easily inactivated when combined with MDM2, MDM4, etc.160 Protoporphyrin IX (PpIX), as a metabolite of aminolevulinic acid, could inhibit the p53/MDM2 and p53/MDM4 interactions, and further elicit the accumulation of p53 and Tap73 to promote apoptosis in CLL cells.161 Therefore, targeting MDMX-mediated inhibition of p53 functions as a novel anticancer drug development strategy. Additionally, there are also numerous small molecule compounds that target to inhibit MDM2-activated p53. For example, APG-115 was a novel MDM2/p53 inhibitor, and the report showed that it could improve the radiosensitivity effects in gastric cancer cells by regulating the expression of MDM2-p53 pathway-related proteins.162 It also observed that cell apoptosis increased and cell cycle arrested after treatment of APG-115. Besides, APG-115 combined with radiosensitivity could enhance the antitumor activity both in vitro and in vivo.162 HL001 was a Cyclophilin A (CypA) inhibitor, which restored the expression of p53 by suppressing the MDM2-mediated ubiquitination of p53, thus inducing the cell cycle arrest and apoptosis of NSCLC cells.163 Similarly, anthraquinone (AQ) analog, AQ-101, promoted the apoptosis of acute lymphoblastic leukemia (ALL) cells via downregulating the MDM2 level to activate p53 protein.164
Mortalin/GRP75 belongs to the heat-shock protein (Hsp70) family, which is found to overexpress in several cancers like ovarian, colorectal, and hepatocellular carcinoma. p53 protein binding to mortalin could inhibit its translocation, further eliminating its tumor suppression function.165 Therefore, Sari et al. synthesized a new small compound, Mortaparib (Plus), which was shown to block the interaction between mortalin and p53, as well as reactivate the p53 expression, causing the induction of apoptosis in CRC cells.166 Moreover, Mortaparib (Plus) could also activate the p53 pathway to induce apoptosis in MCF-7 breast cancer cells.167 In some cancers, c-Myc could induce stem cells, suppress cell senescence and differentiation, and promote the survival of eliminating leukemic stem cells (LSCs) in leukemia. A small-molecule compound, DJ34, was reported to inhibit the transcription of c-Myc and activate p53 protein to selectively and synergistically eliminate LSCs.168 In human ovarian cancer, placenta-specific protein 1 (PLAC1) was overexpressed and could cause cell proliferation and metastasis. p53 inhibited PLAC1 transcription, while mutation of p53 attenuated this effect. Studies have shown that the p53 agonist, HO-3867, could restore the transcription inhibition of PLAC1 by mutant p53 in ovarian cancer cells, inhibit cell growth, and ultimately induce apoptosis. Treatment of ovarian cancer with HO-3867 may be used as adjunctive therapy to improve outcomes in these patients.169 Another compound, andrographlide (ANDRO), was reported to suppress the expression of mutant p53 and promote the combination of the Hsp70 and mutant p53, further inducing proteasomal degradation of p53 and ultimately inhibiting cell growth.170
Some natural and semi-synthetic compounds were reported to target p53 protein to induce apoptosis. For example, renieramycin T (RT) was a tetrahydroisoquinoline alkaloid obtained from the Thai blue sponge Xestospongia sp. RT treatment could significantly activate the expression of p53 and induce the degradation of McL-1 in lung cancer cells, thereby triggering apoptosis.171 Protopine was a natural isoquinoline alkaloid that could stabilize p53 protein to enhance the p53-mediated transcriptional level, leading to the induction of apoptosis and inhibition of HCT116 colon cancer cells proliferation.172 Furthermore, Actinomycin V and TCCP could also activate p53 expression to trigger cancer cell apoptosis.173,174 Reddy et al. had synthesized a series of methyl β-orsellinate based 3, 5-disubstituted isoxazole hybrids and tested the proliferative activity against four human cancer cell lines. Among them, compound 12 was the most active hybrid with an IC50 value of 7.9 ± 0.07 μM against MCF-7 cells. It could block the cell cycle at the G2/M phase and induce apoptosis through the activation of p53 and PTEN, further promoting the expression of Bax and Cyt-C.175 It was also found that indolizine derivatives, compound 3, and resveratrol derivative, trans-3, 5, 4′-trimethoxystilbene (TMS), promoted the activation of p53, causing the apoptosis of cancer cells.176,177 TMS cotreatment with TRAIL could reverse the resistance to apoptosis in cells.177
Several studies proved that the p53 pathway could involve gold complexes mediated apoptosis. [di-(1,3-diethylbenzylimidazol-2-ylidene)] gold(I) iodide (MC3) as a gold(I) NHC complex had shown potent cytotoxic effects against CRC cell lines with different p53 profiles. MC3 treatment could upregulate ROS level and p21 expression, whatever the status of p53, but with WT p53 cells exhibiting the highest pro-apoptotic activity.178 Ma et al. introduced 6-bromocoumarin-3-carboxylic acid into Pt(IV) complex to synthesize bromocoumarinplatin 1. It was found that bromocoumarinplatin 1 could also activate p53 protein to enhance the anticancer activity and overcome the resistance of cisplatin through the p53 pathway.179 Diplatin, a novel platinum complex, had shown that the antitumor activity of diplatin against lung cancer cell lines was superior to that of carboplatin. In the mouse xenotransplantation model, diplatin could significantly improve some therapeutic indicators and inhibit the growth of lung cancer cells that were resistant to cisplatin. Diplatin induced tumor cell apoptosis by activating ROS/JNK/p53 signaling pathway. Accordingly, compared with cisplatin and carboplatin, diplatin had better therapeutic efficiency and safety180 (Table 5).
Other targets
The mitogen-activated protein kinase (MAPK) signaling pathway has a critical role in regulating cell growth, proliferation, differentiation, and apoptosis.181 Abnormal expression of MAPK in tumor cells may cause the uncontrolled proliferation and resistance to apoptosis of tumor cells. MAPK could be classified into three distinct cascades: ERK1/2, JNK1/2, and p38 MAPK.182,183 Therefore, some small-molecule compounds that target MAPK related signaling pathway provide a promising strategy for cancer treatment. Cudraflavone C has been shown to activate the MAPKs pathway (the phosphorylation of p38, ERK, and JNK) to enhance the expression of apoptotic proteins, thus leading to the apoptosis of melanoma cells.184 Celastrol, a natural triterpene compound, could induce apoptosis of nasopharyngeal carcinoma cells and oral cancer cells by increasing the expression level of p38/MAPK, ERK1/2, and activating the JNK1/2 signaling pathway, respectively.185,186 Diallyl trisulfide (DATS)-mediated antitumor activity involved multiple pathways that induced apoptosis by activating the JNK/p38 MAPK pathway and decreasing Nrf2 and Akt protein expression. When DAT was combined with cisplatin (DDP), the antitumor activity was increased, with side effects decreased.187 Another one, Avicequinone-B, a furanonaphthoquinone derivative with poor water solubility, was prepared into liposomes to improve the anticancer activity against cutaneous squamous cell carcinoma (SCC) cells. Liposomal Avicequinone-B promoted the induction of apoptosis by activating the ERK, p38, and JNK pathways.188
AMPK is a serine/threonine protein kinase that regulates downstream signal molecules by phosphorylation to exert its biological activity. AMPK could induce tumor cell apoptosis through different signaling pathways.189 In CRC cell lines, gomisin A was shown to trigger caspase-dependent apoptosis through the regulation of the AMPK/p38 pathway.190 Metformin is a hypoglycemic agent in treating type 2 diabetes, and previous studies have shown that it could exert antitumor effects by regulating the AMPK pathway.191 Lu et al. investigated the anti-proliferation and pro-apoptotic activities of metformin. It showed that metformin could trigger apoptosis in AGS cells by increasing phosphorylation of AMPK, inhibiting phosphorylation of ERK, p38, and JNK.192
The phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin PI3K/Akt/mTOR pathway is one of the most important signaling pathways regulating cell progression,193 which is overactivated in the occurrence and development of cancers194 PKI-402 was a dual PI3K/mTOR inhibitor that inhibited the growth of cisplatin-resistant ovarian cancer epithelial cell SKOV3. It could enhance the degradation of anti-apoptotic protein Mcl-1 via the inhibition of the PI3K/Akt/mTOR signaling pathway, thus promoting the apoptotic pathways in SKOV3 cells.195 1,7-Bis(4-hydroxyphenyl)-1,4-heptadien-3-one (EB30) was a curcumin analog that could block the cell cycle and activate ROS production, which induced lung cancer cells apoptosis by inhibiting the PI3K/Akt pathway and activating the ERK1/1 pathway.196 Apigenin 7-O-glucoside (AGL) was shown to trigger apoptosis of HeLa cells via the inhibition of the PTEN/PI3K/Akt pathway in a concentration-dependent manner.197
Small molecules inhibit the overexpression of the Wnt/β-catenin pathway in cancer cells to regulate cell proliferation and apoptosis.198 C644-0303, a small-molecule inhibitor, was shown to suppress the Wnt/β-catenin pathway to inhibit cell migration and induce apoptosis in CRC cell lines.199 Obatoclax as a pan-Bcl-2 inhibitor had been shown to obstruct BH3-mediated binding of BH3-only proteins or Bax/Bak to induce apoptosis. In this study, obatoclax could suppress the expression of anti-apoptotic protein surviving and inhibit the activation of the Wnt/β-catenin signaling pathway, further triggering apoptosis in HCT116 cells.200
Generally, the expression, phosphorylation, or activation of signal transducer and activator of transcription 3 (STAT3) would induce apoptosis and prevent tumor progression.201 CYT997 could enhance the ROS accumulation and trigger apoptosis by inhibiting the JAK2-STAT3 pathway.202 Licochalconce H (LCH), a synthesized compound, exhibited a potent antiproliferative effect and induced apoptosis in oral squamous cell carcinoma (OSCC) cells via the suppression of JAK2-STAT3 signaling pathway.203 AH057, a JAK inhibitor, was reported to induce apoptosis by blocking the JAK-STAT pathways204 (Table 6).
Autophagy-dependent cell death signaling pathways in cancer
Autophagy is a process of degrading aging and damaged organelles, recycling degradation products, and renewing organelles.205 Autophagy plays a dual role in the occurrence and development of tumors. In the early stage of tumor occurrence, autophagy exerts a preventive effect in controlling or killing cancer cells, while in the formed tumor cells, autophagy could maintain the survival of cancer cells and promote development.205,206 In the following section, we will focus on the autophagy-related signaling cascade, namely, ULK1 complex, PI3KC1-Akt-mTORC1, Ras-Raf-MAPKs, p53, p62, FoxO, NF-κB, Beclin-1, etc. Meanwhile, we have discussed the research progress of the interaction between small molecule compounds and related signaling pathways in cancer treatment (Fig. 3).

Small-molecule compounds targeting autophagy-dependent cell death pathways in cancer. Autophagy is a complex process regulated by multiple signaling pathways. ULK1 complex is essential during the early-stage initiation of autophagy. ULK1 could be phosphorylated by mTOR or AMPK, which promotes the binding of Beclin-1 to vacuolar protein sorting 34 (VPS34) and ultimately participates in the regulation of autophagy. Autophagy-related signaling pathways, including the Ras/Raf/MEK/ERK pathway, PI3KC1/Akt/mTORC1 pathway, and NF-κB pathway, are significant to autophagy signal transduction and mediate the occurrence of autophagic cell death. p53 is a tumor suppressor protein. Nuclear p53 stimulates autophagy through translocation activation, and cytoplasmic p53 represses autophagy. FoxO regulates autophagy by transcriptional dependent mechanism. p62 is also a key regulator of autophagy that can directly bind to LC3 to promote the formation of autophagosomes
Targeting ULK1 complex
ULK1 is the only serine/threonine (S/T) kinases in mammals, as well as a core component of the autophagy pathway.207 ULK1 could form a protein complex with focal adhesion kinase interacting protein of 200 KD (FIP200), autophagy-related protein 13 (ATG13), and ATG101, which plays a crucial role in autophagy induction. ULK1 is associated with early autophagosome formation and activates downstream Beclin-1 protein to initiate the autophagy cascade.208,209 As a potential multifunctional target, ULK1 plays different roles according to the types and stages of tumors. Both inhibition and activation of ULK1 show significant effects on tumor treatment. For example, blocking early autophagy by inhibiting ULK1 contributed to attenuating cell growth and overcoming the development of drug resistance in tumor therapy, while the activation of ULK1 could mediate the poor prognosis of tumors.210 At present, several small-molecule compounds that regulate the ULK1 have been extensively employed in cancer therapy (Table 7).
SBI-0206965 is a highly selective ULK1 inhibitor (ULK1: IC50 = 108 nM; ULK2: IC50 = 711 nM), which could downregulate the ULK1-mediated phosphorylation to inhibit autophagy and survival of NSCLC cells.211 Moreover, SBI-0206965 also inhibits the growth of neuroblastoma cell lines and promotes apoptosis by targeting the autophagic kinase ULK1.212 Subsequently, the group reported another ULK1/2 dual inhibitor, SBP-7455, with higher activity than SBI-0206965, which could inhibit survival and proliferation of MDA-MB-468 cells via the inhibition of starvation-induced autophagic flux.216 MRT68921 is a dual NUAK family SNF1-like kinase 1 (NUAK1)/ULK1 inhibitor with a strong anticancer effect, which could block ULK1-dependent protective autophagy.213
Recently, Sun et al. designed and synthesized a series of new ULK1 inhibitors based on the structure of previously reported ULK1 inhibitors. Among them, compound 3s, 5-bromo-4-(2-fluoro-4-nitrophenoxy)-N-(3,4,5-trimethoxyphenyl) pyrimidin-2-amine showed potent anticancer activity and blocked A549 cell autophagy by inhibiting ULK1 kinase activity, which was accompanied by increasing the expression of p62 and decreasing the level of beclin-1.214 Moreover, some natural compounds were shown potent anticancer activity via targeting ULK1. For example, rocaglamide (RocA), a natural product, was reported to inhibit autophagy in NSCLC cells, thus enhancing the sensitivity of NSCLC cells to natural killer (NK) cell-mediated killing. This effect was induced by the suppression of ULK1.215 Phloretin (PH) effectively inhibited cytoprotective autophagy via the downregulation of the mTOR/ULK1 pathway, further restoring the sensitivity of breast cancer cells to chemotherapeutic drugs.216 Additionally, the research showed that bromodomain and extraterminal domain (BET) inhibitor, JQ1, was considered a hopeful epigenetic agent for the treatment of various tumor types. Studies reported that pro-survival autophagy could cause drug resistance of stem cells to BET inhibitors, and inhibition of autophagy by targeting AMPK-ULK1 could improve the sensitivity of drug-resistant cells to therapeutic drugs (BET inhibitors).217
Several types of research have shown that ULK1 was under-expressed in some tumor tissues like breast cancer. Therefore, it suggested that the activation of ULK1 to induce autophagy and thus inhibit tumor growth could be an effective treatment strategy for some cancers.209,218 Zhang et al. found a ULK1 activator, LYN-1604, through silico screening and chemical synthesis, which targeted ULK1 and interacted with activating transcription factor 3 (ATF3), RAD21, and caspase 3 to induce autophagy-dependent cell death. Besides, LYN-1604 activated ULK1 with a median effective concentration (EC50) value of 18.94 nm in an in vitro kinase assay.219 Liu et al. found that a dual PI3K/mTOR, NVP-BEZ235, promoted autophagy colon cancer cell death via the upregulation of the AMPK/ULK1 pathway.220 Disulfiram (DSF) was an anti-alcohol drug that could suppress malignant tumor growth. Hu et al. reported that DSF combined with Cu could significantly inhibit the viability of CRC cells and trigger autophagic cell death instead of apoptosis via enhancing the ULK1 level.221
Targeting PI3KC1/Akt/mTORC1
PI3KC1/Akt/mTORC1 signaling pathway has been considered as the pivotal regulator in various cell processes. PI3K, a class of lipid kinases, could be divided into three types, including PI3KC1, PI3KC2, and PI3KC3; among them, PI3KC1 is the most widely studied.222 mTOR is a highly conserved serine/threonine (Ser/Thr) kinases, which belong to the PI3K protein kinase family and is a downstream effector protein of the PI3K/Akt signaling pathway. mTOR exists as a form of two complexes, mTORC1 and mTORC2, of which mTORC1 is related to nutrients and growth factors and is an essential negative regulator of autophagy.223,224,225 This pathway regulates autophagy by phosphorylating various substrates downstream of mTORC1, such as ULK1 complex and beclin-1.222 Targeting PI3KC1/Akt/mTORC1-mediated autophagy was a promising therapeutic strategy for multiple tumors and exerted essential role in suppressing cell growth and improving the chemosensitivity of tumor cells.226 Numerous small-molecule compounds target PI3KC1/Akt/mTORC1-mediated autophagy, leading to tumor cell death.
ABTL0812, a novel first-in-class compound, was found to inhibit the Akt/mTORC1 pathway and induce autophagy-dependent cancer cell death by high-throughput silicon screening comparison. This mechanism was mediated by activating the expression of the Tribbles-3 pseudokinase (TRIB3) gene.227 ABTL0812 was in clinical evaluation in phase 1/1b trial in advanced cancer patients (NCT02201823). PP242 was an mTOR inhibitor with stronger pro-autophagy activity than rapamycin, which could activate lysosomal by the blockade of the mTORC1 level.228 In ovarian cancer SKOV3 cells, cardamonin was able to induce autophagy via the inhibition of mTORC1, and the downregulation of Raptor was involved.229 Additionally, the synthesized compound, N-(1-benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamide, could prevent the reactivation of mTORC1 and increase the accumulation of LC3-II to promote Induce autophagy-mediated cell death.230 Inhibition of the PI3K/Akt/mTOR pathway could promote tumor cell apoptosis by increasing the level of autophagy. The natural product, ginsenoside Rg5, was reported to trigger human osteosarcoma cell apoptosis via the LC3-mediated autophagy pathway by inhibiting the activation of PI3K/Akt/mTORC1.231
In addition to the small molecules mentioned above, more small molecules showed potent antitumor activity via the regulation of autophagy. For example, AZD2014, as a dual mTOR inhibitor, could suppress proliferation and induce autophagy to sensitize thyroid undifferentiated carcinoma (ATC) cells to paclitaxel (PTX).232 Another novel PI3K/Akt/mTOR inhibitor, W922, was shown to attenuate the growth of CRC cells via the induction of autophagy and when treated in combination with chloroquine, caused a large production of apoptotic cells.233 Chen et al. synthesized a quinazolinyl-arylurea derivative, compound 7j, which possessed a lower IC50 value and good selectivity. Compound 7j regulated PI3K/Akt/mTOR/ULK1 and Sxc(-)/GPX4/ROS pathways to induce autophagy and ferroptosis of cancer cells at high concentrations while inducing apoptosis at low concentrations.234 It had been reported that some active substances of traditional Chinese medicine had strong antitumor effects, such as Tanshinone IIA (Tan IIA), 20(S)-ginsenoside Rh2 (20 (S)-GRh2), could induce cancer cell cycle arrest and autophagy by inhibiting PI3K/Akt/mTOR pathway, accompanied by increased expression of LC3-II protein235,236 (Table 7).
Targeting Ras/Raf/MAPKs
MAPK pathway is the main pathway for transmitting signals from the cell membrane to the nucleus, which could be divided into three subtypes: ERK1/2, JNK, and p38 MAPK.237 Among them, ERK1/2 plays a critical role in autophagic regulation in various tumor cells, and ERK1/2 could be phosphorylated and activated to regulate autophagy through the Ras/Raf/MEK/ERK axis.12,238 The Ras/Raf/MEK/ERK pathway was closely associated with tumorigenesis. Therefore, small-molecule compounds induce autophagy-dependent cell death by the Ras/Raf/MAPKs pathway may be a potential therapeutic strategy for cancer.
For example, a small molecule 2′-dihydroxy-4,4′-dimethoxydihydrochalcone had been reported to induce autophagy-dependent cell death in MKN45 cells via activating ROS/MEK/ERK pathway, as well as upregulating Beclin-1, Atg5 and, Atg7 levels.239 CYT-Rx20, a synthetic β-nitrostyrene derivative, could induce autophagy which was regulated by the MEK/ERK pathway.240 In A549 and NCI–H292 cells, morusin triggered the inhibition of Akt and activation of JNK and ERK pathways to induce autophagy-mediated cell death and apoptosis.241 8-CEPQ was shown to attenuate the growth of colon cancer cells via triggering autophagy-dependent cell death by the activation of ERK.242 Furthermore, the administration of tetrandrine at the maximum noncytotoxic dose of 50 mg/kg could induce autophagy of nasopharyngeal carcinoma cells by inhibiting the MEK/ERK pathway and enhancing the sensitivity of nasopharyngeal carcinoma to radiotherapy without side effects occurs243 (Table 8).
Targeting p53
p53 protein is well-established as a tumor suppressor gene as well as an essential regulator of autophagy. As a nuclear transcription factor, p53 could activate the transcription of autophagy-related genes, thus inducing autophagy, while p53 in the cytoplasm has a negative regulatory role in inhibiting the occurrence of autophagy. p53 protein is involved in the initiation of autophagy. Unlike p53-induced apoptosis, many p53 target genes can stimulate autophagy, and this effect is usually achieved by inhibiting the mTOR signaling axis, a negative regulator of autophagy. Besides, p53 can also activate the expression of various autophagy-related proteins such as Atg7 and Atg10. Most cancer patients have mutations in the p53 gene, and the remaining possess defective wild-type p53 pathways.244 Accordingly, we primarily focus on inhibiting the expression of mutant p53 protein and restoring the function of wild-type p53 to retard further tumor development through small molecule compounds so as to achieve the goal of effective treatment of cancer.
Accordingly, blocking the expression of mutant p53 protein leads to inhibiting cell growth and proliferation, reducing the resistance to some anticancer agents as well as improving the poor prognosis.244 For example, small molecule gambogic acid (GA) was shown to trigger the degradation of mutant p53 and thus enhance the sensitivity of cancer cells to the chemotherapeutic agent.245 In cancer cells, the high expression of some proteins, such as heat shock protein 90 (HSP90), enhanced the stability of p53 mutant protein, while hsp90 inhibitors could reduce this stability and degrade mutant p53.246 Like Hsp90 inhibitor, 17-AAG, promoted mutant p53-R248Q degradation by stimulating autophagy.247 Similarly, suberoylanilide hydroxamic acid (SAHA), as an HDAC inhibitor, could induce autophagy and prevent the expression of mutant p53 protein.248
On the other hand, p53 protein presented a vital role in tumorigenic inhibition, and it was feasible to induce cell death by repairing the wild-type p53 pathway. Trans-chalcone (TC) was a stable isomer with various properties, such as antioxidant, anti-proliferation, and anti-inflammation. Treatment with TC could activate the expression of p53 gene and downregulate the β-catenin protein, thus inducing the autophagy death of hepatocellular carcinoma (HCC) cells.249 β-asarone activated the expression of p53 protein and p53 pathway-related proteins such as Beclin-1, AMPK, and LC3-II/I to induce autophagic cell death.250 Similarly, 6-Azauridine (6-AZA)-mediated autophagy death depended on the expression of p53 and AMPK.251 [4-NH2-2-Me(Q)H][VO(bcma)(H2O)]2H2O (T1) suppress proliferation and induced autophagy which had been shown to activate p53/p21 pathway.252 Furthermore, the combination of β-catenin inhibitor FH535 and Akt inhibitor AZD5363 could stimulate the expression and phosphorylation of p53 protein in the nucleus, further regulating the AMPK/mTOR/ULK1 pathway, ultimately inducing autophagy-dependent death of HCC cells253 (Table 8).
Targeting p62, FoxO, and NF-κB
p62, also known as sequestosome 1 (SQSTM1), is a ubiquitin-binding protein involved in cellular signal transduction as well as a selective substrate of autophagy.254 p62 directly interacts with LC3 through the LC3-interacting region (LIR) within p62 to promote the formation of autophagosomes, while the inhibition of autophagy is accompanied by insufficient degradation of the p62 protein.255 Therefore, p62 and LC3 are widely established as markers of autophagy flux in cancer research. p62 is regarded as a potential target for monitoring autophagy, and some small molecules could influence the expression level of p62, thus regulating autophagic cell death.256 Arzanol, as a novel compound, was observed to induce the accumulation of p62 and inhibit autophagy, thus improving the sensitivity of bladder cancer RT-112 cells to cisplatin.257 Autophinib, as a VPS34 inhibitor, could inhibit p62 degradation via suppression of starvation- or Rapamycin-triggered autophagy by targeting VPS34 in MCF7-LC3 cells.258 A dual inhibitor of both Hsp90 and late-autophagy, DCZ5248, promoted the LC3-II transformation and upregulated p62 level to inhibit colon cancer HCT116 cell late-autophagy.259 Furthermore, Shen et al. designed and synthesized a tetrahydroquinolin-2(1H)-one derivative 11k with an IC50 value of 4.9 μM in PANC-1 cell, which could enhance the expression of p62 to inhibit autophagy.260 7-aminobenzo[cd]indol- 2(1H)-one 33 as a novel Atg4B inhibitor was shown to inhibit autophagy via decreasing the degradation of p62, as well compound 33 could sensitize HT-29 cell to oxaliplatin.261
In mammals, the FoxO family includes FoxO1, FoxO3, FoxO3, and FoxO6, which act as transcription factors by binding to target DNA through the forkhead domain to activate or inhibit downstream genes, thus affecting the occurrence and development of cancer.262 FoxO proteins could participate in the cell autophagy process to regulate cancer growth and metastasis. FoxO-autophagy regulation plays a different role according to the tumor environment, which can promote or inhibit tumor progression.263 Accordingly, FoxO is also modulated by various signaling pathways, including acetylation, ubiquitination, and phosphorylation.
FoxO-mediated autophagy exhibited promising therapeutic potential in regulating cancer progression. The study found that histone deacetylase inhibitors (HDACIs) inhibited the mTOR expression by activating FoxO1 and SESN3, thereby activating autophagy and inducing drug resistance.264 When combined with autophagy inhibitors, autophagy could be inhibited, thus inhibiting drug resistance and enhancing the therapeutic effect.265 Xiao et al. also reported that HDACIs induced autophagy via the AMPK-FoxO1-ULK1-Snail axis in hepatoma cells and combination with FoxO1 inhibitor, AS1842856, was more effective in the treatment with HCC.266 When treating NSCLC with gefitinib, the FOXO1 protein expression would increase and induce protective autophagy, thus leading to drug resistance in NSCLC cells. The combination of gefitinib and a HIF-1α inhibitor, YC-1, could significantly inhibit autophagy, as well as acquired resistance could be overcome.267 WX20120108, as a novel IAP inhibitor, triggered autophagy in HeLa and MDA-MB-231 cells via upregulating the FOXO3 gene and promoting ROS production.268 Furthermore, in an acidic microenvironment, FOXO3 could trigger autophagy by enhancing the expression of LC3-II and Beclin-1 to inhibit tumor cell growth.269
NF-κB as a nuclear transcription factor was overexpressed in multiple malignancies and promoted tumor cell survival. Small-molecule compounds activated autophagy via the blocking of the NF-κB pathway to exert an antitumor biologic effect.270 Studies conducted by Zhu et al. demonstrated that olanzapine-induced autophagy via the suppression of the NF-κB pathway also had an inhibitory effect on the MGMT-positive gene, thus leading to the inhibition of proliferation.271 Camptothecin (CPT) was shown to promote esophageal cancer cells’ protective autophagy via the suppression of neddylation, the accumulation of IκBα, and the blockade of the NF-κB pathway. Besides, CPT could also inhibit the AMPK/mTOR/ULK1 axis from regulating autophagy272 (Table 8).
Targeting Beclin-1
Beclin-1, a crucial autophagy regulatory gene in mammals, is homologous to yeast autophagy-related gene Atg6/Vps30. Beclin-1 is an indispensable protein for the activation of autophagy and is controlled by different transcription factors.273 Beclin-1 has a BH3 binding domain, which interacted with Vps34 and promoted the formation of PI3KCIII core complexes, thereby regulating autophagy.274 Beclin-1 is a critical tumor suppressor that participates in the process of tumor development by regulating autophagy activity.275 It was found that Beclin-1 was under-expressed in some malignant tumors, such as lung cancer, breast cancer, cervical cancer, ovarian cancer, etc. Therefore, Beclin-1 has been considered a potential therapeutic target for tumor therapy.
Oseltamivir, an anti-influenza virus drug, was shown to increase Beclin-1 expression and decrease p62 expression to trigger autophagy-dependent death of Huh-7 cells.276 Moreover, regorafenib was reported to trigger autophagy-dependent cell death via blocking the interaction of Beclin-1-Bcl-2 and further promoting the dissociation of Beclin-1 from Bcl-2.277 4-Methoxydalbergione (4MOD) had been recently reported to upregulate Beclin-1 and LC3-II/LC3-I, inducing autophagy-dependent cell death and thus inhibiting the growth of J82 and UMUC cells, which accompanied with the suppression of Akt/ERK pathway.278 Interestingly, natural products, BA and isoliquiritigenin (ISL) were shown to stimulate autophagic cell death in ovarian cancer cells via the activation of Beclin-1279,280 (Table 9).
Other targets
AMPK, as a critical cell energy sensor, has been proved to regulate the development of tumors through autophagy. AMPK can directly activate ULK1 and thus stimulate autophagy via the phosphorylation of Ser317 and Ser777. In addition, AMPK inhibits the phosphorylation of downstream mTOR to promote the expression of ULK1, further initiating autophagy. Some small-molecule compounds were used to target AMPK for cancer treatment. In multiple myeloma, metformin was shown to induce autophagy and cell cycle arrest via the dual repression of mTORC1 and mTORC2 pathways mediated through AMPK activation.281 Fluoxetine was reported to cause autophagic cell death via activating the AMPK-mTOR-ULK1 axis in TNBC cell lines.282,283 Moreover, polyphyllin VI (PPVI) was a natural product with potent antitumor activity, which could regulate multiple pathways to induce autophagic cell death, such as PI3K/Akt/mTOR, MEK/ERK, and AMPK/ mTOR pathways.284 Apigenin (APG) elicited autophagic cell death by modulating the mTOR-AMPK-ULK1 signaling pathway in GC cell lines.285 In addition, Docosahexaenoic acid (DHA) was shown to increase the accumulation of oxidative stress-induced growth inhibitor 1 (OSGIN1) and ROS to activate AMPKα/Raptor and inhibit mTOR/ULK1 pathways in MCF-7 cells, leading to the formation of autophagosome.286
PTEN-induced putative kinase 1 (PINK1)/Parkin pathway is an important pathway to mediate mitochondrial autophagy (Mitophagy). Parkin is an E3-ubiquitin ligase that, when activated, drives mitochondrial proteins’ ubiquitination to induce autophagy. δ-Valerobetaine (δVB) as a dietary metabolite was reported to activate mitophagy in SW480 and SW620 colon cancer cells via PINK1/Parkin/LC3B axis.287 F0911-7667, a sirtuin-1 (SIRT1) activator, could also induce mitophagy via the SIRT1–PINK1–Parkin pathway in U87MG and T98G cells, and induce autophagic cell death through regulating the AMPK-mTOR-ULK pathway.288 Additionally, thioridazine (THD), an antipsychotic drug, was used to enhance p62-mediated autophagy and apoptosis of glioblastoma multiform (GBM) cells by regulating the Wnt/β-catenin pathway289 (Table 9).
Necroptosis signaling pathways in cancer
Universally, the occurrence of cancer is closely related to cell death. In addition to apoptosis, necrotic apoptosis is also related to the development of cancer. However, studies have shown that necroptosis plays a dual role in cancer progression and development.290 Necroptosis is a regulatory form of cell death, which is different from apoptosis, autophagy and necrosis by releasing inflammatory mediators as a mechanism to defend against viruses. However, the potential molecular mechanism of necroptosis is complex and has not been fully elucidated. Some studies believe that RIPK1 has no kinase activity in complex I. inhibiting RIPK1 activity by necrostatin-1 has no effect on TNF-induced NF-κB signaling pathway, but it can prevent the formation of complex IIB from inhibiting necroptosis.291,292 Therefore, the role of RIPK1 in cells can determine whether cancer cells survive or undergo necroptosis through targeted drugs.292 RIPK3 activation of MLKL is a key regulatory pathway in necroptosis. The upstream inducer DR, Toll-like receptor (TLR), or virus induces the activation of RIPK3 through RIPK1, toll-like receptor adaptor molecule 1 (TICAM1), and Z-DNA–binding protein 1 (ZBP1), respectively. Additionally, the adhesion receptor (AR) activates RIPK3 via an unknown adaptor protein or kinase.293 Necroptosis plays a dual role in the occurrence and development of tumor cells. On the one hand, necroptosis can inhibit the proliferation, migration, and invasion of tumor cells. On the other hand, necroptosis may be involved in the growth of early tumors. Recent studies suggest that tumor cells resistant to apoptosis may be sensitive to the necroptosis pathway,294,295 suggesting that the study of tumor cells’ necroptosis and its regulatory mechanism is expected to become a target for the treatment of tumors (Fig. 4 and Table 10).

Small-molecule compounds targeting necroptosis pathways in cancer. There are three main pathways to fight tumor by targeting necroptosis. In TNF-α After binding with TNFR1 on the plasma membrane, the downstream protein molecules TRADD, TRAF, cIAPs, LUBA and RIPK1 are recruited to form complex I and activate NF-κB to promote the survival of tumor cells. Then RIPK1 promotes the recruitment of pro-caspase-8 to produce activated Caspase-8 and form complex IIa. If caspase-8 is inhibited or there is no expression of caspase-8, RIP3 is recruited to form rip1-rip3 necrosome, causing ripk3 phosphorylation, MLKL is recruited to form complex IIb and induce necroptosis. In addition, TLR ligand can also mediate RIPK3-MLKL dependent necrosis through TICAM1. ZBP1 acts upstream of RIPK3 and interacts with RIPK3 through its RHIM domain to mediate necroptosis in response to viral infection
Targeting RIPK1/ RIPK3/MLKL
It is generally believed that the dysfunction of necroptosis is related to the occurrence and development of tumors. For example, RIP3 expression is significantly downregulated in patients with AML, an invasive hematopoietic malignancy known to block hematopoietic differentiation and cell death. The decrease of RIP3 reduces the death of hematopoietic cells, which is related to the occurrence of AML.296 Another study reported that the genetic defect of RIP3 transformed flt3-itd and runxeto-driven mouse bone marrow proliferation into AML by increasing the accumulation of leukemia-initiating cells.297 Therefore, the role of RIP3 in the pathogenesis of AML may be determined by the cellular environment. Furthermore, low expression of MLKL is associated with reduced overall survival in patients with colon cancer after surgery.298 A study analyzed that RIPK1 protein and mRNA levels were significantly upregulated in tumor samples from 40 patients with colorectal adenocarcinoma, while ripk3 and p-MLKL decreased in colorectal cancer, suggesting that necroptosis may be reduced and apoptosis may be increased in tumor cells.299 Similarly, studies have found that the expression of RIPK3 in colorectal cancer tissues is significantly lower than that in adjacent normal tissues, and the upregulation of RIPK3 can inhibit the proliferation, migration, and invasion of colorectal cancer cells.300 Interestingly, RIPK3 was found to be highly expressed in tumors in a mouse inflammatory bowel cancer (CAC) model, and further analysis of colon cancer tissue chips from 168 tumors and 103 non-tumor controls found that RIPK3 was highly expressed in human colorectal cancers. However, after knocking out the RIPK3 gene in mice, the number and size of tumors were significantly reduced, and the tumor load was also reduced. RIPK3 may be involved in early tumor growth through enteritis mouse model experiments.300 MLKL is also downregulated in pancreatic and cervical squamous cell carcinomas, where low levels of MLKL in plasma predict poor prognosis in pancreatic and ovarian cancers.301 Altogether, this information provides research directions for studying necrotic proteins in tumor development. Based on the above research results, current research believes that necroptosis is a double-edged sword, and the role of RIPK1/RIPK3/MLKL in various cancer tissues still needs to be confirmed by multi-center, prospective clinical controlled experiments.
Studies have shown that traditional necroptosis inducers or existing chemotherapeutic agents can necrotize many cancer cell lines. These cell lines cover almost all common cancer types, especially colorectal cancer cells and hematopoietic system tumors, which are more sensitive to necroptosis inducers.294 Necrostatin-1 (NEC-1) is a specific inhibitor of necroptosis by preventing the interaction between ripk1 and ripk3, which can specifically inhibit necroptosis without affecting normal cell function and apoptosis. A study studied the antitumor effect of NEC-1 through colitis associated cancer (CAC) mouse model and considered that NEC-1 can significantly inhibit tumor growth and development by inhibiting JNK/c-Jun signal pathway.302 At the same time, several necrostatin related compounds with necroptosis inhibitory activity, including NEC-3, NEC-4, NEC-5, NEC-7 and so on, have been gradually studied in depth. Screening necroptosis inducers is also a new strategy for drug resistance of tumor cells. Resibufagenin induces CRC cell necroptosis by inducing RIPK3 mediated activation of glycogen phosphorylase (PYGL), glutamine synthetase (GLUL), and glutamate dehydrogenase (GLUDL) so as to inhibit tumor growth.303 It shows that RIPK3 is not only a molecular switch of necrotic cells but also a hub to control the metabolic state of cells.304,305 Some complexes have been proved to induce necrotic apoptosis of tumor cells and play an antitumor role, but they are still limited to basic research. The polypeptide Su-X targeting survivin-XIAP complex can induce necrotic apoptosis of tumor cells. The necrotic apoptosis-related proteins (p-RIPK1, p-RIP3, and p-MLKL) in the cells treated with Su-X are significantly increased, suggesting that Su-X can inhibit the development of tumors by inducing necrotic apoptosis of tumor cells.306 Apoptosis inducers are largely related to cancer drug resistance. Drugs that kill tumor cells through non-apoptotic pathways can bypass the traditional drug resistance. RIPK1 and RIPK3 are the key molecules of cell death and survival pathway and important potential targets for tumor treatment.307 Reactivation of transcriptional reporter activity (RETRA) is a small molecule that can induce the expression of the p53 regulatory gene in the mutant (MT) p53 cells. It shows a necrotic form through the phosphorylation of RIPK1/RIPK3/MLKL so that the cervical cancer cell cycle stagnates in the S phase, p21 is upregulated, cyclin-d3 is downregulated, mitochondrial hyperpolarization increases the production of ROS, and finally selectively induces cervical cancer cell necroptosis regardless of p53 state, and it has no cytotoxic effect on normal human peripheral blood mononuclear cells (PBMC).308 The combined application of RETRA and necroptosis inhibitor Necrostatin-1 reversed the effect of RETRA and saved the death of cervical cancer cells.308 It is suggested that RETRA-induced necrosis and apoptosis can be used as a potential treatment for apoptosis-resistant cervical cancer. In the clinical use of cyclin D-CDK4 inhibitor (CDK4I) in HR+ breast cancer, the problem of CDK4 inhibitor resistance is usually caused by compensatory CDK2 activity.309 A therapeutic liposome peptide NP-ALT inhibits the tyrosine phosphorylation of p27kip1 (CDKN1B) and the activity of CDK4 and CDK2 by inducing ROS and RIPK1 dependent necroptosis in breast cancer cells and xenotransplantation models resistant to endocrine therapy. It provides a new treatment option for HR+ tumors resistant to endocrine and CDK4 inhibitors.309 In recent years, traditional Chinese medicine has had obvious curative effects in the treatment and remission of cancer, and there are relatively few adverse reactions. Major bioactive curcumin derived from the rhizome of Curcuma longa induces necroptosis and apoptosis by increasing cleaved caspase-3 and cleaved PARP, p-RIP3, and p-MLKL proteins and finally reduces the viability of tolerant prostate cancer pc-3act cells.310 Arctigenin, a mitochondrial complex I inhibitor, induces necroptosis in prostate cancer cells through ROS-mediated mitochondrial damage and increased CCN1 levels, ultimately increasing p-RIP3 and p-MLKL levels. Pretreatment with the necroptosis inhibitor necrostatin-1 restored their levels and prostate cancer cell viability.311 Ophiopogonin D’ (OPD’), a natural compound extracted from Ophiopon japonicus, induces significant necroptosis in androgen-dependent LNCaP cancer cells by activating and increasing Fas ligand (FasL)-dependent RIPK1 protein expression and exerts antitumor effects.312
In addition, the combination of FMRP protein with RIPK1 mRNA suggests that FMRP regulates the necroptosis pathway by monitoring the metabolism of RIPK1 mRNA. The use of FMR1 anti transcription therapy in CRC cell lines will upregulate RIPK1 and cause necrotic apoptosis of CRC cells.313 In conclusion, the susceptibility to necrotic apoptosis inducers may be of great significance for the clinical treatment of colorectal tumors. Interestingly, radiotherapy is one of the main methods of treating cancer. More than 50% of tumor patients receive radiotherapy during their disease treatment, of which 40% can be cured by radiotherapy.314 However, tumor recurrence is still one of the key factors of treatment failure.315 Radiation-induced necroptosis results in the formation of RIPK1/RIP3/MLKL necrotic bodies by upregulating the phosphorylation of RIPK1 and RIPK3.316 The blocking of necroptosis regulatory genes, especially MLKL, by low-dose chemical inhibitors or gene deletion can significantly inhibit the recurrence of tumors in and out of mice, and even reduce the tumorigenicity of mice.316 Detecting the increase of IL-8 in colorectal cancer cells after irradiation, puts forward a new way - RIPK1/RIP3/MLKL/JNK/IL-8 is involved in tumor re proliferation mediated by necroptosis cells,316 MLKL/JNK/IL-8 may become a potential target to block tumor regrowth and improve the efficacy of radiotherapy. This study suggests that radiotherapy for necrotic apoptosis may be an effective way to improve the effect of radiotherapy.
Targeting TLR4/TICAM1
Although RIPK3 and MLKL appear to be common players in necroptosis, but RIPK1 is not involved in TLRs-mediated necroptosis.317 TLRs are commonly expressed in innate immune cells and tumor cells.318 TLR ligands mediate RIPK3-MLKL-dependent necrosis through TICAM1(also known as TRIF). Interestingly, TLR4 has both tumor-promoting and tumor-suppressive effects,318 and targeting TLR4 activation or inhibition may be a potential therapeutic strategy for different types of tumors. Many studies have shown that the gene expression profile of peripheral blood mononuclear cells (PBMC) in blood samples that are more specific and more convenient to collect clinically shows unique gene expression characteristics in several cancers.319 A significant and unique gene expression feature was found in breast cancer patients,320 which showed the possibility of dividing breast cancer patients into subgroups.320 Sporadic thyroid cancer is the most common endocrine malignancy in which the risk of papillary thyroid cancer (PTC) is associated with single nucleotide polymorphisms (SNPs). One study showed that the TICAM1 (rs8120) gene in PTC was associated with SNPs.321 In addition, there was a significant interaction between TICAM1 (rs8120) and FOXE1 (rs10984377), a known susceptibility locus for thyroid cancer, suggesting that multiple RCD mechanisms and host factors may interact in complex ways to increase the risk of PTC and FTC.321 TLR4 is overexpressed in breast tumors, thereby promoting tumor progression and metastasis. The natural product Curcumin inhibits the survival of breast cancer cells by inhibiting the TLR4-dependent TICAM1 signaling pathway, reducing the level of interferon (IFN-α/β).322 Nobiletin (NOB), an O-methylated flavonoid, inhibits the growth of different types of prostate cancer cells to varying degrees by inhibiting TLR4/TICAM1/IRF3 and TLR9/IRF7 signaling pathways, depending on hormonal status and aggressiveness characteristics.323
Targeting ZBP1
As an interferon-induced z-nucleic acid sensor, ZBP1, also known as DAI/DLM-1, acts upstream of RIPK3 and interacts with RIPK3 through its Rhim (RIP homotype interaction motif) domain to mediate necrosis and apoptosis in response to viral infection or TLR signal transduction.324 The N-terminal domain (ND) of ZBP1 is important for the ZBP1-ZBP1 homologous interaction, and the RHIM domain of its C-terminal region interacts with RIPK3, triggering RIPK3-dependent necroptosis.325 Notably, the virus can not only directly bind to RIPK3 but also promote the binding of host protein ZBP1 to RIPK3, ultimately activating MLKL.322 Interestingly, in irradiated tumor cells, zbp1-mlkl necrotic cascade induces cytoplasmic DNA accumulation and then autonomously activates CGAs sting signal to drive persistent inflammation. Resection of caspase-8 can enhance the activation of the sting pathway and the antitumor effect of radiation by activating mlkl and improve the alternative radiotherapy strategy.326 Metastasis involves separating tumor cells from primary tumors and acquiring migration and invasion ability. These abilities are mediated by various events, including the loss of intercellular contact, the increase of adhesive transfer, and the inability to maintain normal cell polarity. Inhibition of ZBP1-mediated necroptosis promotes tumor growth in mouse colorectal cancer and melanoma models.327 Other similar studies have proved that the loss of ZBP1 function relieves the regulation of many mRNAs involved in cell movement and cell cycle regulation, resulting in phenotypic changes in breast cancer, which not only increases the growth capacity of metastatic cells but also promotes cell migration.328 In T47D and MDA231 human breast cancer cells, targeting imp1/zbp1 regulates the local expression of many cell movement-related mRNAs, such as those encoding E-cadherin, α/β- actin, and arp2/3 complex, so as to stabilize intercellular junction and focal adhesion and inhibit tumor cell invasion.329 Interestingly, another study had shown that the expression of ZBP1 was significantly increased in necrotic tumors. ZBP1 is a key regulator of tumor necrosis and apoptosis. Its deletion blocks tumor necrosis and apoptosis during tumor development and inhibits tumor metastasis in MVT-1 breast cancer model, providing a potential drug target for controlling tumor metastasis.199
Other targets
Cell FLICE (FADD-like IL-1β-converting enzyme) inhibitor protein (c-FLIP) is not only a major anti-apoptotic protein but also an important cytokine and chemoresistance factor that inhibits cytokine and chemotherapy-induced apoptosis. It has been reported that the expression level of c-FLIP is elevated in colorectal cancer,330 and the c-FLIP isomer is involved in switching apoptosis and necrotic cell death.331 The c-FLIP isomer in ribosomes determines whether cell death occurs ripk3 mediated bad apoptosis or caspase-dependent apoptosis. The defect of apoptosis signal transduction and the upregulation of drug transporters in cancer cells produce clinical drug resistance by significantly limiting the effectiveness of cancer chemotherapy. Overexpression of p-glycoprotein, Bcl-2 or Bcl-xL may be the main cause of clinical tumor drug resistance. Interestingly, drugs that induce non-apoptotic cell death can overcome cancer drug resistance. Shikonin, a naturally occurring naphthoquinone sensitive breast cancer cell line, and drug-resistant cell line, showed the same necroptosis, which proved that shikonin avoided the apoptosis resistance mediated by p-glycoprotein, Bcl-2 and Bcl-xL by inducing necroptosis of drug-resistant cancer cell lines, thereby inhibiting drug resistance.332
As a newly discovered mode of programmed death, necroptosis is closely related to the physiological process of many cases. At present, the research on necroptosis is mostly in the basic experimental stage. Necroptosis plays an opposite role in antitumor. On the one hand, it can inhibit the proliferation and migration of tumor cells; On the other hand, it can promote tumor growth and participate in early tumor formation. Further study on the molecular mechanism of necrotic apoptosis pathway and the relationship between upstream and downstream signal molecules of related signal pathways, exploring its role in different tumor modes and finding corresponding targeted drugs are one of the directions to improve the effect of tumor treatment in the future.
Pyroptosis signaling pathways in cancer
Pyroptosis is an inflammatory regulated cell death mediated by GSDM, which is mainly characterized by membrane perforation, cell swelling, cell content overflow, chromatin condensation, and DNA breakage.333,334 The human GSDM family includes six members, including GSDM-A, -B, -C, -D, -E, and DFNB59; all GSDM family members have N-terminal pore-forming domain, C-terminal self inhibitory domain, and ring domain connecting N-terminal and C-terminal domain. Among them, GSDMD and GSDME are the most complete to study the mechanism of inducing cell death. During the formation of the tumor and tumor microenvironment, pyroptosis has the dual effects of inhibiting and promoting its formation:335 on the one hand, inflammatory bodies released during pyroptosis can inhibit the proliferation and metastasis of tumor cells, and its mechanism is that NLRP3 inflammatory bodies produced by pyroptosis inhibit tumorigenesis by secreting inflammatory cytokines; On the other hand, the aggregation of inflammatory bodies contributes to the formation of tumor microenvironment and promotes tumor occurrence and development. Studies have shown that NLRP3 inflammasome plays an important role in the aggregation of myeloid-derived suppressor cells (MDSC) and the inhibition of antitumor T cell immune response after DC immunization.336 NLRP3 in tumor-associated macrophages drives the polarization of immunosuppressive CD4+ T cells in the tumor immune microenvironment of pancreatic ductal adenocarcinoma through IL-1.337 In addition, the production of IL-22 depends on the activation of NLRP3 inflammatory bodies and the subsequent release of IL-1 from immune cells. IL-22 is closely related to the development of a variety of tumors, such as lung cancer, skin cancer, breast cancer, and gastric cancer.338 A bioorthogonal chemistry system (BCS) selectively releases active GSDM to tumor cells. However, only 10–30% of tumor cells undergo pyroptosis to completely remove tumor grafts, and there is no tumor regression in immunodeficient mice.339 Therefore, how to use the new weapon of pyroptosis to develop new tumor treatment schemes, reduce the drug resistance of chemotherapy drugs and enhance the body’s immunity has become an urgent problem to be solved (Fig. 5 and Table 11).

Small-molecule compounds targeting pyroptosis pathways in cancer. There are two main pathways and two other pathways that exert antitumor activity by targeting pyroptosis. Pyroptosis pathway can be divided into classical pyroptosis pathway and non-classical pyroptosis pathway. The activation of classical pyroptosis pathway is initiated by PAMPs or damps. After NLRs or ALRs recognize specific stimuli, they start to assemble to form inflammatory bodies and process to form activated caspase-1. Caspase-1 cuts GSDMD, and the N-terminal of GSDMD is located and aggregated on the cell membrane to form pores. In addition, caspase-1 cleaves pro-IL-1β and pro-IL-18 to form mature IL-1β and IL-18, and the intracellular contents are secreted outside the membrane through the membrane pore. The nonclassical focal death pathway depends on the activation of caspase-4/Caspase-5/caspase-11. After stimulated by LPS in the cytoplasm, caspase-4/Caspase-5/caspase11 can directly bind to the conserved structure lipid A of LPS, cause oligomerization and activation, further cut GSDMD, cause the N-terminal of GSDMD to disintegrate and locate in the cell membrane to form membrane pores. In caspase-3-dependent cell death, GSDME is the reaction substrate of Caspase-3. GSDME can be cleaved by activated caspase-3 to generate its N-terminal fragment, which performs pyroptosis by penetrating the plasma membrane. Granzyme B can also participate in NK cell induced pyroptosis by cleaving GSDME. In addition, TNF-γ acts on TNFR, activates the N-terminal cleavage of GSDMC mediated by caspase-8, locates in the cell membrane, forms membrane pores, and finally induces pyroptosis. IFN-γ by acting on ifngr, causes the N-terminal of GSDMB to cleave and locate to the cell membrane to form membrane pores and induce pyroptosis. Granzyme A can also induce pyroptosis by cleaving GSDMB
Targeting Caspase-1/-4/-5/-11/GSDMD
Caspase-1-dependent cell pyroptosis, also known as the classical inflammatory corpuscle pathway, is based on the activation of inflammatory corpuscles. Caspase-4/-5/-11 dependent cell pyroptosis has nothing to do with the inflammatory corpuscle complex, so this pathway is also known as the non-classical inflammatory corpuscle pathway. There is a close connection between pyroptosis and apoptosis. Knockdown of GSDMD can block IL-1β secretion and convert pyroptosis into apoptosis, thereby promoting tumor cell killing.340 It has been studied to genetically engineer GSDMD by inserting other protease sites or Caspase-3/-7 cleavage sites between the N-terminal and C-terminal domains of GSDMD, which can also convert apoptosis into pyroptosis.341 Interestingly, the expression of GSDMD in gastric cancer cells was lower than that in adjacent normal tissue cells, which may promote the proliferation of cancer cells. GSDMD reduces the expression of Cyclin A2 and Cyclin-Dependent Kinase (CDK2) by inhibiting ERK1/2, STAT3, and PI3K/Akt in gastric cancer (GC) cells. Therefore, the decrease of GSDMD expression in GC cells increases the expression of the Cyclin/CDK complex as a substance that regulates the cell cycle, promotes the transition from the S phase to the G2 phase, and accelerates the proliferation of GC cells.342 However, in NSCLC, the expression level of GSDMD in tumor tissues is significantly higher than that in adjacent tissues and normal tissues.343 Silencing GSDMD in NSCLC cells or using GSDMD inhibitors can inhibit the progression of NSCLC to a certain extent, which indicates that the high expression of GSDMD may promote the progression of NSCLC.343 Studies have shown that treatment of endometrial cancer cell lines Ishikawa and HEC1A with the hydrogen-rich medium in vitro can upregulate the release of inflammatory mediator IL-1β and promote cell pyroptosis.344 The volume and weight of the tumor were significantly reduced compared with the untreated group when water was administered to the mice transplanted with endometrial cancer tumors by gavage.344 After the GSDMD gene was knocked out in endometrial cancer cells, and the above results showed no significant difference between the hydrogen-rich and non-hydrogen-rich groups.344 This indicates that the pyroptosis of endometrial cancer cells may be achieved through hydrogen-induced GSDMD-mediated IL-1β secretion, so hydrogen-rich-induced pyroptosis of endometrial cancer cells may become a new therapeutic method.
Due to the resistance to apoptosis, chemotherapy as the main treatment of advanced human esophageal squamous cell carcinoma (ESCC) has little effect. The upregulation of proline, glutamate, and leucine-rich protein-1 (PELP1) in advanced ESCC is highly correlated with cancer progression and poor prognosis. Metformin or pyroptosis inducer activates mir-497 by targeting the mir-497/PELP1 axis, inhibits PELP1 expression, increases the level of cleaved GSDMD, induces pyroptosis in ESCC, and improves the prognosis of ESCC.189 A new antitumor molecule 2-(alpha-naphthoyl) ethyltrimethylammonium Iodide (α-NETA) induces pyroptosis in different epithelial ovarian cancer (EOC) cell lines through GSDMD/ caspase-4 pathway and reduces the size of EOC tumor in mice. Knockout of caspase-4 or GSDMD seriously hindered the killing activity of α- NETA on EOC cells.345 Accumulating evidence suggests that abnormal expression of lncRNAs may regulate cancer cell proliferation and metastasis.346 LncRNA RP1-85F18.6 is highly expressed in colorectal cancer and can regulate ΔNp63 at the transcriptional and translational levels (its role is opposite to that of p53), thereby participating in the proliferation, invasion, survival, and metastasis of colorectal cancer cells, including inhibiting the apoptosis of tumor cells.347 It is worth noting that LncRNA RP1-85F18.6 is also involved in the pyroptosis process of colorectal cancer cells. Downregulation of LncRNA RP1-85F18.6 induces pyroptosis in CRC cells by silencing ΔNp63 and cleaving GSDMD.347 Studies have found that lncRNA GAS5 can promote inflammasome assembly and expression by interfering with glucocorticoid receptor expression, thereby upregulating the expression of IL-1β and IL18 inflammatory mediators mediated by caspase-1, promoting tumor cell pyroptosis, and promoting tumor cell pyroptosis.348 In both cell and animal experiments, ovarian tumor cell growth and migration were inhibited in a time-dependent manner; when lncRNA GAS5 was knocked out, as its expression decreased, the pyroptotic effect was weakened, and the tumor-inhibiting effect was also weakened.348 The above results indicate that lncRNA GAS5 can inhibit tumor progression by promoting the pyroptosis of ovarian cancer cells, and the reduction of GAS5 expression can lead to the occurrence of ovarian cancer.
Targeting Caspase-3/GSDME
In Caspase-3-dependent pyroptosis, GSDME is the reaction substrate of Caspase-3, and GSDME is involved in the regulation of secondary necrosis.349,350 Under the stimulation of apoptosis, GSDME can be cleaved by activated caspase-3 to generate its N-terminal fragment (gsdme NT), which performs pyroptosis by penetrating the plasma membrane.349,350 Granzyme is an exogenous serine protease released by cytotoxic T lymphocyte (CTL) and natural killer (NK) cells. After entering the target cells, it induces the apoptosis of the target cells by activating the apoptosis-related enzyme system. The human body contains five kinds of granzymes, namely granzymes A, B, H, K, and M. Granzyme participates in NK cell induced pyroptosis by cleaving GSDME.351 There is a two-way relationship between pyroptosis and tumor treatment. Activating chronic inflammation can promote tumor development while activating acute pyroptosis will lead to necrotic pyroptosis so as to inhibit tumor progression and achieve the effect of antitumor treatment. Some research results show that the pyroptosis caused by hypoxia in the tumor center can promote the development of tumor and reduce the survival rate of patients.352 Chronic inflammation caused by inflammatory mediators increases the risk of tumorigenesis. Epithelial pyroptosis releases HMGB1, which can promote the occurrence of colitis-related colorectal cancer by activating the ERK1/2 pathway.353 On the other hand, as inflammatory pyroptosis, pyroptosis can activate the immune system. Immune stimulants, including HMGB1, can induce the activation of dendritic cells and antitumor T cells. Tumor-infiltrating immune cells can induce pyroptosis of tumor cells.354 Chimeric antigen receptor gene-modified T (car-t) cells can induce GSDME-dependent pyroptosis in leukemia cells, and its mechanism involves the cleavage of activated GSDME in leukemia cells by granzyme B released by car-t cells. It is worth noting that the activity of car-t cells to induce target pyroptosis is determined by the number of perforin/granzyme B in car-t cells, rather than the number of perforin/granzyme B in existing CD8+ T cells.354 In addition, granzyme A has also been shown to induce pyroptosis of tumor cells by cutting GSDMB.355 The lysed GSDMB was introduced into mouse tumor cells, and the tumor could be effectively controlled by immune checkpoint therapy.355 The expression of GSDMB is induced by IFN - and the combination of IFN - and immune checkpoint blocking can greatly activate antitumor immunity. Therefore, the Granzyme family plays an important role in CTL-induced pyroptosis. The activation of pyroptosis in CTL can enhance cytotoxicity. At present, immune checkpoint inhibitors (ICI) have broad application prospects in clinical tumor treatment, but relevant data show that only 1/3 of patients respond to ICI.356 There is a synergistic effect between cell focal death and ICI. Inducing target cell focal death will promote anti-ICI tumors to obtain sensitivity to ICI.339 The mechanism is that the rupture of inflammatory cell membrane promotes the overflow of cell contents, triggers a strong inflammatory response and a large number of lymphocyte infiltration, and these significantly increased lymphocytes further induce caspase-3-dependent tumor pyroptosis, forming positive feedback to improve the antitumor effect.357 A special chimeric costimulatory converting receptor (CCCR), which is composed of the extracellular region of PD-1, the transmembrane and cytoplasmic region of NKG2D, and the cytoplasmic region of 41BB.358 CCCR modified NK92 cells showed enhanced activity against human lung cancer H1299 cells in vitro by extensively inducing pyroptosis. However, another study suggested that the inhibitory effect of antigen-specific CTL on the tumor was not related to pyroptosis.359 Therefore, pyrosis is a kind of pyroptosis in the form of immune stimulation, which can cooperate with ICI to improve the effect of immunotherapy.
One study found that GSDME was highly expressed in SH-SY5Y neuroblastoma, HeLa cervical cancer cells, and MeWo skin melanoma cells.350 GSDME positive SH-SY5Y cells showed pyroptosis characteristics after treatment with chemotherapeutic drugs topotecan, etoposide, cisplatin, or Irinotecan.350 After GSDME positive HeLa cells were treated with DOX or 5-fluorouracil (5-FU), the originally induced apoptosis was switched to CASP3 dependent pyroptosis.350 3′,5′-degraded chalcone (C10) activates caspase-3 by inducing PKCδ/JNK pathway, induces PARP and GSDME-dependent pyroptosis, increases the proportion of sub G1 PC3 cells, and selectively inhibits the proliferation of prostate cancer cells in vitro and in vivo.360 Dihydroartemisinin (DHA), a derivative of artemisinin extracted from the traditional Chinese medicine Artemisia annua, can increase the expression of natural dermal protein E (DFNA5) and melanoma 2 (AIM2) by activating Caspase-3, and finally, induce pyroptosis to inhibit the proliferation and tumorigenicity of breast cancer cells.361 Knockout of aim2 and DFNA5 could significantly enhance the resistance of breast cancer cells to DHA.361 It reveals the new anti-cancer mechanism of DHA and also brings a promising treatment strategy for breast cancer.
Other targets
So far, few studies have focused on other GSDM family members except GSDMD and GSDME. In an acidic environment, α-ketoglutarate (α-KG) is inhibited by MDH1 and transformed into L-2HG, which increases the level of ROS, leads to the oxidation of the death receptor DR6 located in plasma membrane, triggers its endocytosis, recruits caspase-8-mediated GSDMC cleavage, and finally induces pyroptosis.81 Treatment with α-KG derivative DMΑ-KG can further improve the level of ROS and make cancer cells that originally resisted pyroptosis more vulnerable to pyroptosis induced by α-KG.81 It reveals that GSDMC has potential clinical value in tumor treatment. A study found two new gene association sites (17q12 and 8q24.21) related to the risk of childhood acute lymphoblastic leukemia.362 The peak slice size related to acute lymphoblastic leukemia in 17q12 is about 200KB, and the pyrolytic substrate GSDMB is also expressed in this fragment.362 At the gene level, GSDMB may affect the site expression of 17q12, thus affecting the risk of acute lymphoblastic leukemia; the relationship between GSDMB as a substrate causing cell death and the pathogenesis, treatment, and prognosis of acute lymphoblastic leukemia needs to be further studied.362
Ferroptosis signaling pathways in cancer
ACSl4, LPCAT3, and ALOXs (especially ALOX15) pathways mediate the oxidation of polyunsaturated fatty acids, including arachidonic acid, which is necessary for the lipotoxicity of ferroptosis. The upregulation of ACSl4 is the sign of ferroptosis.293 On the contrary, some antioxidant systems, especially the XC system (including core component SLC7A11), GPX4, NFE2L2, and some heat shock proteins (such as HSPs), inhibit the ferroptosis lipid peroxidation process.293 The ultimate goal of clarifying the potential mechanism of ferroptosis is to obtain better cancer treatment options. Therefore, based on the molecular regulation mechanism of ferroptosis, it is worthwhile to specifically target the key regulators of ferroptosis for triggering ferroptosis. Some drugs or compounds have been found to induce ferroptosis of tumor cells, which can be divided into two categories according to the mechanism of action (Fig. 6 and Table 12).

Small-molecule compounds targeting ferroptosis pathways in cancer. There are two main pathways to exert antitumor activity by targeting ferroptosis. The intracellular antioxidant stress system mainly relies on GPX4 to remove excess lipid peroxide. GPX4 of the antioxidant system will reduce the lipid peroxide PL-OOH to the corresponding lipid alcohol PL-OH, so as to reduce the burden of lipid peroxidation and protect the cell membrane from damage. Cystine enters the cytoplasm through SLC7A11, transforms into cystine, and enters the GSH biosynthesis pathway. GSH is involved in the hydrolysis of PL-OOH by GPX4. The inhibition of GSH synthesis or the inactivation of GPX4 can make the excess PL-OOH in cells unable to be cleared, resulting in cell oxidative damage and the occurrence of ferroptosis. It is worth noting that in this process, BAP1, ATF3, Beclin1 and p53 will inhibit the function of SLC7A11. When intracellular iron is overloaded, a large number of free radicals can react with PUFA of cell membrane phospholipids under the catalysis of ester oxygenase and iron, and produce a large number of PL-OOH through the promotion of POR, a positive regulatory protein of iron death phospholipid peroxidation in tumor cells, resulting in ferroptosis. In addition to the direct pathway of PUFA mediated PL-OOH production, PUFA can also be incorporated into phospholipid membrane through ACSL4, esterified into PUFA COA, esterified into PL through LPCAT3, and then oxidized into toxic PL-OOH by lipoxygenases (LOXs). In addition, the extracellular Fe3+ is combined with transferrin, transported into the cell through TFR1 and reduced to Fe2+, and then stored in the intracellular LIP with the help of intracellular NRF2. Fe2+ can transfer electrons to produce free radicals or ROS with oxidation ability through Fenton reaction with peroxide, so as to promote the oxidation process of LOXs
Targeting ACSL4/LPCAT3/ALOX15
Lipid peroxidation (LP) reflects the process that biofilms, lipids, and other lipid-containing molecules related to PUFA are oxidized by oxides such as ROS to form lipid peroxidate (LPO). Current research shows that the accumulation of PUFA oxide is a sign of ferroptosis, and its accumulation process mainly involves deoxygenation inhibition, mainly referring to GPX4 and ferroptosis suppressor protein 1 (FSP1) and the enhancement of peroxidation catalyzed by iron and a series of enzymes.363 Arachidonic acid (AA) and adrenaline (ADA) can be esterified into acyl CoA derivatives by acyl CoA synthetase long-chain family member 4 (ACSl4) and then esterified into phosphatidyl ethanolamine (PE) by recombinant lysophosphatidyl-choline acyltransferase 3 (lpcat3), and then oxidized into toxic lipid hydroperoxides by LOXs. LOXs is an iron-containing enzyme and the most important lipid oxidase in ferroptosis.364 Therefore, the activation of ACSl4, lpcat3, and LOXs will lead to excessive lipid peroxidation and ferroptosis. ACSL member ACSl4 is an important contributor to the ferroptosis of tumor cells.365 ACSl4 tends to catalyze the conversion of arachidonic acid and adrenic acid in polyunsaturated fatty acids into arachidonyl coenzyme A and adrenoyl coenzyme A, respectively. These products can participate in the synthesis of negatively charged phospholipids such as phosphatidylethanolamine or phosphatidylinositol. Phosphatidylethanolamine is a key substrate for lipid peroxidation during ferroptosis, especially when GPX4 is inhibited. Therefore, ACSl4 gene knockout or functional inhibition can effectively prevent the occurrence of ferroptosis.8,366 A number of studies have suggested that ACSl4 can be used as a biomarker to predict whether tumor cells can successfully die of iron.8,365,367 ACSl4 mediated ferroptosis can inhibit the proliferation of glioma cells, so it has the potential to become a new target for glioma treatment.368 Interestingly, although ACSl4 is indispensable for ferroptosis induced by erastin or (1s, 3R) - rsl3, it cannot determine the final occurrence of p53-alox12 mediated ferroptosis.369 Whether this process needs the participation of other members of the ACSL family remains to be studied.
The incurable cancer is called pancreatic ductal adenocarcinoma (PDAC), driven by mutations in constitutively active KRAS.370,371 Oncogenic KRAS reprogrammes PDAC cells to a highly anti-apoptotic state.372 Resistance to apoptosis makes PDAC highly resistant to the mitochondrial pattern of apoptosis-regulated cell death.372 Artesunate (ART), an antimalarial drug, has the highest cytotoxicity in PDAC cell lines with constitutively active KRAS, specifically inducing ROS and lysosomal iron-dependent cell death.373 PDAC patients have increased sensitivity to Ras-driven ferroptosis.373 Combined treatment with iron ptosis inhibitor ferrostatin-1 can block art-induced lipid peroxidation and cell death and increase the long-term survival and proliferation of PDAC cells.373 Lycorine extracted from Amaryllidaceae general can reduce the expression level of GPX4, increase the expression level of ACSL4, increase the expression level of 5-HETE, 12-HETE, 15-HETE and MDA, reduce the ratio of GSH/GSSG, induce tumor cells to produce ferroptosis and inhibit the proliferation of renal cell carcinoma (RCC).278
Targeting SLC7A11/GPX4/NFE2L2
Erastin (ERA) is a typical ferroptosis inducer, which can directly inhibit the cystine/glutamate antiporter system xc− on the cell membrane, reducing the transport of extracellular cystine into cells.40 The deletion of cystine reduces intracellular glutathione synthesis, which indirectly inhibits downstream GPX4, accelerates the accumulation of lipid peroxides to lethal doses, and induces ferroptosis.43 Butionine-sulfoximine can block the synthesis of glutathione by inhibiting the activity of γ-glutamine cysteine synthase and, finally promote ferroptosis.43 Various drugs have been found to induce ferroptosis through a mechanism of glutathione depletion. In addition to the indirect inhibition of GPX4 by depletion of glutathione, it can also directly inhibit GPX4 and induce ferroptosis. Tumor cells such as diffuse large B-cell lymphoma and renal cell carcinoma have been found to be very sensitive to ferroptosis regulated by GPX4,43 and some drug-resistant tumor cells are also highly dependent on GPX4 to maintain their own survival.374 Therefore, GPX4 is a new target for some tumor therapy.
Due to its poor water solubility and effect, ERA hinders its further use in vivo. In order to enhance the water solubility of the scaffold, piperazine erastin analysis (PE) was obtained by introducing piperazine into the middle of the aniline ring of ERA.43 PE showed significant activity in the nude mouse tumor prevention model but had limited effect on the determined tumor growth.43 In order to improve the effect, AE was obtained by replacing piperazine with aldehyde based on PE, but it was accompanied by poor metabolic stability, and solubility.43 In order to achieve the best balance between reactivity, stability and solubility, the active carbonyl was transferred to the scaffold without electrophilic function, and the metabolically stable electrophilic ketone erastin analog imidazole ketone erastin (IKE) was obtained. Ike showed strong efficacy and selective lethality to BJ-derived tumor cells expressing oncogenic HRAS by inducing ferroptosis.375 Interestingly, the mechanism of action of a compound (MOA) is a set of target proteins and effector proteins necessary to produce its pharmacological effects in a specific cellular environment. MOA is of great significance in evaluating the efficacy and toxicity of compounds.376 A study found that altretamine is a new GPX4 inhibitor similar to sulfasalazine through the prediction-based MOA analysis of detecting mechanism of action by network dysregulation (demand) and combined with experiments, inhibiting the growth and development of diffuse large B-cell lymphoma (DLBCL) by inducing ferroptosis.377 FINO2, an endoperoxide containing 1,2-dioxane, induces ferroptosis in engineered cancer by playing a dual effect, directly oxidizing iron and indirectly inhibiting GPX4, showing a new mechanism of iron apoptosis inducer.378
Other targets
Excess iron can cause tissue damage and increase the risk of cancer. The most important mechanism of iron biotoxicity is the Fenton reaction of too much Fe2+ in cells, which leads to the accumulation of ROS and the production of a large number of hydroxyl radicals, resulting in the damage of cellular proteins, lipids, and DNA. Intervening in iron absorption and metabolism has become a method to treat tumors and other diseases. The application of iron-based nanoparticles can induce ferroptosis of tumor cells and inhibit tumor growth.379 In a word, tumor cells can increase the content of iron in cells by regulating iron metabolism, making cancer tissues more sensitive to ferroptosis. Studies have shown that intracellular iron overload can be prevented by knocking out the transferrin receptor (TFRC) on the cell surface, or the storage of iron in the inert pool can be increased by upregulating cytoplasmic ferritin to inhibit the occurrence of ferroptosis.380 Similarly, inhibiting the transcription factor iron-responsive element binding protein 2 (ireb2), which regulates iron metabolism, can reduce ferroptosis.40 In contrast, blocking intracellular iron output by knocking out solute carrier family 40 member 1 (slc40a1) will accelerate erastin-induced ferroptosis in neuroblastoma cells.381 In conclusion, the iron metabolism pathway and ferritin phagocytosis are the keys to regulating ferroptosis. In addition, mitochondrial lipids are also an important source of lipid peroxides during ferroptosis. Inhibition of mitochondrial tricarboxylic acid (TCA) cycle or functional mitochondrial electron transport chain (ETC) can reduce ferroptosis caused by cysteine deprivation.382 When cysteine is absent, cells will metabolize through glutamine decomposition, increase TCA in mitochondria, increase the production of lipid ROS, hyperpolarize mitochondrial membrane, and eventually collapse, thus inducing cell ferroptosis. In addition, mitochondrial fatty acid metabolism genes, including citrate synthase and recombinant acyl-coenzyme A synthetase long-chain family member 2 (acsf2), may be necessary genes for the occurrence of ferroptosis induced by erastin. In order to avoid the excessive accumulation of unfolded proteins in the endoplasmic reticulum, eukaryotic cells can activate a series of signal pathways to maintain endoplasmic reticulum homeostasis, which is called endoplasmic reticulum stress (ERs). Erastin can upregulate ERS response genes and induce the occurrence of ERS.383 Activating transcription factor 4 (ATF4) is the main signal transduction pathway of ERS involved in the activation of ferroptosis. It can increase cation transport regulators like protein 1 (CHAC1), promote the degradation of GSH, and induces the occurrence of ferroptosis. Puma, a regulator of apoptosis, is also downstream of ATF4 and upregulated in ERS induced by art, a ferroptosis inducer.383 There is evidence that ferrostatin-1 (fer-1) and liproxstatin-1 (lip-1) cannot reduce erastin-induced ERS by inhibiting lipid peroxidation.383
Other RCD signaling pathways in cancer
Targeting parthanatos through PARP1/AIFM1 signaling pathways in cancer
Parthanatos is a new form of regulated cell death different from apoptosis. Parthanatos is closely related to the occurrence and development of many diseases such as tumors. Because the abnormal activation of PARP-1 is a prerequisite for inducing parthanatos, parthanatos is also known as PARP-1-mediated apoptosis. In addition, AIF and macrophage MIF are also key factors in the occurrence of parthanatos. In this process, PARP-1 acts as a DNA damage receptor, and its enzyme activity is rapidly activated after DNA damage,384 resulting in a sharp increase of PARP-1 activity by nearly 500 times. The generation of polymer par is dispersed in the cytoplasm or the target protein is parylated, and the dynamic balance between the generation and degradation of par in cells is broken, resulting in different lengths of par,385 which induces the release of AIF from mitochondria. In addition, AIF can bind to nuclease MIF and activate MIF. AIF and MIF then translocate to the nucleus, resulting in nuclear shrinkage, chromatin agglutination, and large DNA fragments ranging from 15 to 50 KB, resulting in parthanatos386 (Fig. 7A and Table 13).

Small-molecule compounds targeting other pathways of RCD in cancer. Other main subroutines of RCD include parthanatos, entosis, NETosis and LCD. A When DNA is lost, PARP-1 is abnormally activated to produce a large amount of par. When the mitochondrial membrane is depolarized, the levels of ATP and NADPH decrease. AIF enters the nucleus from mitochondria, chromatin condenses and produces a large number of DNA fragments ranging from 15 KB to 50 KB, which induces the occurrence of parthanatos and promotes or inhibits tumorigenesis. B Entosis is an RCD form of "cannibalism" of cells. One cell engulfs and kills another cell, which is characterized by CIC structure. Cell adhesion and cytoskeleton rearrangement pathways (such as myosin, RhoA and ROCK) and other signaling molecules and regulatory factors (such as CDC42) play an important role in regulating the induction of entosis. It is worth noting that entosis can promote tumorigenesis through cell division and escape, or inhibit tumorigenesis through LCD and apoptosis. C The process of neutrophils secreting nets is called NETosis, which can promote tumor recurrence and metastasis. Overexpression of G-CSF and IL-8 in tumors can increase the number of neutrophils in blood, produce ROS and cause the formation of nets. In addition, NADPH oxidase can also directly produce ROS and promote the formation of nets. D When cells are exposed to lysosomal detergent, dipeptide methyl ester, lipid metabolites and ROS, lysosomes rupture and LCD is mediated by hydrolase or iron released by LMP, which inhibits the occurrence and development of tumors
Existing studies have shown that parthanatos is closely related to tumorigenesis and development.387 A study conducted microarray analysis on the expression of the PARP-1 gene in more than 8000 tumor samples.388 The results showed that the expression level of PARP-1 in breast cancer, ovarian cancer, endometrial cancer, lung cancer, skin cancer, and non-Hodgkin’s lymphoma was higher than that in the same amount of normal tissues, indicating that parthanatos was closely related to the above tumors. By constructing PARP-1 knockout mice, it was found that the risk of epithelial cancer in PARP-1 knockout mice was significantly reduced. The mechanism is that downregulating the level of PARP-1 protein can inhibit the activity of NF-κB and the expression of tumor-promoting related proteins regulated by NF-κB and inhibit the occurrence of parthanatos.387 In addition, PARP-1 knockout mice could significantly reduce the incidence of colorectal cancer induced by oxymethane (AOM) combined with dextran sulfate sodium (DSS). Mechanism study found that downregulating PARP-1 protein level can inhibit the occurrence of induced colorectal cancer by inhibiting the expression of cyclin D and transcription factor signal transducer and activator of transcription 3 (STAT3).389 The influence of parthanatos on tumorigenesis and development is mainly reflected in two aspects. On the one hand, in the process of rapid proliferation, radiotherapy, or chemotherapy, DNA is easy to be destroyed and leads to tumor cell apoptosis. One of the most important functions of PARP-1 is to participate in DNA repair, which is conducive to the survival of tumor cells. Therefore, the purpose of inducing tumor cell apoptosis can be achieved by inhibiting the activity of PARP-1. On the other hand, the occurrence of parthanatos mainly comes from the abnormal activation of PARP-1, so it can also lead to the occurrence of parthanatos in tumor cells by enhancing the activity of PARP-1 to inhibit the proliferation of tumor cells. Because PARP-1 is involved in many DNA repair pathways and the maintenance of genomic stability,390 the regulation of PARP-1 activity is an important means for the clinical treatment of related cancers.
Breast cancer proteins, especially BRCA1 and BRCA2, participate in homologous recombination repair (HRR). In clinical trials, PARP inhibitors are mainly concentrated in cancer patients with homologous recombination repair defects, including breast cancer and ovarian cancer patients with BRCA1 and BRCA2 mutations (gBRCA1/2m). At present, olaparib, niraparib, rucaparib, veliparib, and talazoparib inhibit the anticancer activity of parthanatos by inhibiting the catalytic activity of PARP-1 and PARP-2.391,392,393,394,395 β-Lapachone, a natural product obtained from the bark of the lapacho tree, induces parthanatos through the NQO1-dependent ROS-mediated RIPK1-PARP1-AIF pathway and promotes hepatocellular carcinoma cell death.396 The addition of a PARP-1-specific inhibitor blocked β-Lapachone-induced cell death.396 Deoxypodophyllotoxin (DPT), a natural active compound extracted from Anthriscus sylvestris, inhibits glioma growth by inducing excessive ROS, upregulating PARP-1 expression, and inducing nuclear translocation of AIF in xenograft glioma and glioma cells in vitro.397
Targeting Entosis through CDC42/RHOA/ROCK/Myosin signaling pathways in cancer
Entosis is an RCD form of "cannibalism" of cells.293 One cell engulfs and kills another cell, which is characterized by intracellular cell structure. After activation of entosis, similar cells are phagocytized and killed through LC3-related phagocytosis (LAP) and cathepsin B (CTSB) - mediated lysosomal degradation pathway. Cell adhesion and cytoskeleton rearrangement pathways (such as actin, myosin, RhoA, and rock) play an important role in controlling the induction of entosis. In addition to cell adhesion and cytoskeleton rearrangement pathways, other signaling molecules and regulatory factors (such as Cdc42) also participate in the regulation of entosis through different mechanisms (Fig. 7B and Table 13).
As early as more than 100 years ago, researchers observed an interesting cell sheath cell structure in human tumor tissue.398 At first, people speculated whether it was phagocytosis. Later, it was found that the internal cells forming this overlapping structure were living cells, and it was defined as cell in cell (CIC) structure.399 Due to the limitations of experimental conditions and instruments, the early research on CIC structure remained in the stage of phenomenon description. In recent years, scientists have made more in-depth research on the formation mechanism and biological function of CIC structure. Cell in cell structure refers to cell overlapping structure, which is mainly classified into the homotypic overlapping structure and heterotypic overlapping structure. CIC structure is a process in which one or more living cells exist in another cell to form a unique cell structure of cell sleeve cells and produce biological effects. It is common in tumor tissues and cells cultured in vitro. Usually, we call the cells inside the CIC structure effector cells and the cells outside the CIC structure target cells. It can be observed that the typical morphological feature of CIC structure is that after the effector cells enter the cytoplasm of the target cells, they are wrapped by the target cells, showing a "bird’s eye" shape, while the nucleus of the target cells is squeezed by the internal effector cells to form a "Crescent" shape.400 Entosis is a CIC structure formed between epithelial-derived cells (mainly tumor cells).401 Its mediated internal cell death is mainly lysosomal mediated caspase-3 (cysteine protease-3) - independent death.401 Some studies have found that it negatively regulates the formation of adhesion protein family molecule pcdh7 in entotic cell-in-cell structure and also found that the force ring plays a key role in the formation of entotic cell-in-cell structure.402 In mitotic surveillance, entosis selectively promotes aneuploid progeny cells to drill into adjacent cells to form cell-in-cell structure by activating the p53 signaling pathway and then is cleared to maintain the genomic stability of epithelial cells. This study also revealed the physiological function of entosis, found a new mechanism of abnormal cell clearance outside the regulation of the cell cycle, and revealed a new pathway for the p53 gene to maintain epithelial homeostasis and inhibit tumorigenesis at the cell level; On the one hand, it enriches the connotation of the existing mitotic surveillance mechanism; On the other hand, it expands the extension of entosis as a cell death mechanism involved in important biological processes of cancer.402
Targeting NETosis through NAPDH/ROS signaling pathways in cancer
NETosis is a form of RCD driven by NET, which is regulated by NADPH oxidase-mediated ROS production and histone citrullination.293 Neutrophil extracellular traps (NETs) are a form of inflammatory cell death found in 2004.403 They can trap bacteria, fungi, protozoa, and viruses. Nets are secreted by activated neutrophils and composed of DNA fibers, histones, and antibacterial proteins.404 They can fix pathogens and expose them to locally high lethal concentrations of effector proteins. The process of neutrophils secreting nets is called NETosis, which is the inflammatory cell death mode of neutrophils.405 NETosis involves a dynamic process of multiple signals and steps: the production of ROS, the nuclear migration of neutrophil elastase (NE), anti-myeloperoxidase antibody (MPO), histone modification, and chromatin degradation are the core mechanisms of the formation of nets.406,407 NETosis mediates histone citrullination, which eventually leads to chromatin deconcentration, nuclear membrane destruction, and chromatin fiber release (Fig. 7C and Table 13).
Nets are formed in the tumor microenvironment. The inducements of NETosis formation in malignant tumors include tumor cell colony-stimulating factor (G-CSF), and endothelial cell IL-8.408 Overexpression of G-CSF in tumors can increase the number of neutrophils in the blood, produce ROS and cause the formation of nets.406 Pancreatic cancer (PACA) cells can directly or indirectly induce the formation of nets.409 NETosis exists in animal models and tumor patients’ blood and tumor tissues. NETosis have both tumor-promoting and antitumor effects, which depend on the state of the immune system and the interaction of the tumor microenvironment.405,407 Studies have been devoted to the relationship between the formation of NETosis and tumorigenesis, progression, and metastasis and revealed the direct effect of NETosis on tumor cell proliferation through protease or activation signal.410 Tumor cells can induce the formation of NETosis in vivo and in vitro. Studies have shown that tumor cells can induce neutrophils to induce NETosis. Compared with the normal control group, neutrophils in the circulation of mouse models of chronic myeloid leukemia, breast cancer, and lung cancer were more likely to induce NETosis.411 The systemic effect of the tumor on the body leads to the increase of neutrophils activity forming NETosis.411 The close relationship between tumor cells and NETosis in the tumor microenvironment highlights the role of NETosis in tumor progression and metastasis. NETosis can awaken dormant tumor cells and promote tumor recurrence and metastasis. In addition, they can trap tumor cells in circulation and promote tumor proliferation and metastasis. The formation of NEtosis usually requires the activation of neutrophils and the production of ROS by NADPH oxidase. At present, it is still necessary to further study the possibility of NEtosis as a tumor treatment target and pharmacological interference with the formation of NEtosis. Although the relevant studies have achieved satisfactory results in the tumor model, the research on tumor patients is not satisfactory. It is necessary to focus further research on balancing the regulation and formation of NEtosis, taking NEtosis as a therapeutic target without affecting immune function.
Targeting lysosome-dependent cell death through LMP signaling pathways in cancer
Lysosome-dependent cell death (LCD), also known as lysosomal cell death, is a form of RCD mediated by hydrolase (cathepsin) or iron released by LMP, which is characterized by lysosomal rupture.293 When cells are exposed to lysosomal detergent, dipeptide methyl ester, lipid metabolites, and ROS, lysosomes rupture and then release a large number of hydrolases, leading to the occurrence of LCD. Cathepsin plays a major role in LCD. Blocking the expression or activity of cathepsin can reduce the occurrence of LCD. Lysosomal membrane permeabilization can also amplify cell death signal transduction in the case of apoptosis, autophagy-dependent cell death, and ferroptosis, which increases the complexity of the cell death pathway (Fig. 7D and Table 13).
At present, there are many mechanisms to explain lysosomal permeability. In view of the important role of lysosomal function in cancer cells, researchers have developed a variety of small molecular compounds for lysosomes, which can induce lysosomal membrane permeability or interfere with lysosomal function to kill tumor cells. For example, chloroquine can induce lysosomal membrane permeability to regulate lysosomal function, so as to restore the sensitivity of refractory non-small cell lung cancer cells to cisplatin;412 Azithromycin (AZM) can increase the expression of lysosomal galectin-3 spots, enhance the permeability of lysosomal membrane mediated by lansoprazole (LPZ), and significantly enhance the death of cancer cells induced by LPZ.413 These findings suggest that cancer cells that are not sensitive to traditional therapy may be effectively treated by using activated lysosomal cell death pathway. In addition, tumor cell lysosomes are more fragile than normal cells and are more prone to lysosomal membrane permeability and lysosomal-dependent cell death. Therefore, the intervention of lysosome-dependent cell death pathway may be an effective treatment strategy for many types of cancer. However, the current understanding of lysosome-dependent cell death is insufficient, and the related molecular targets and molecular mechanisms need to be further studied.
Cancer treatment through multiple RCD signaling pathways
Simultaneous regulation of two or more RCD subroutines will be a promising treatment strategy for cancer. Lu01-m, a natural product with structural diversity and a variety of biological activities, induces cytotoxic activity in a variety of human prostate cancer cells through a variety of RCD mechanisms. Lu01-m mainly induces G2 / M phase cell cycle arrest and DNA damage, and finally induces apoptosis, necroptosis, autophagy, and other inhibition of tumor colony formation and tumor cell migration.414 Green tea extract Polyphenon E® blocks cell cycle G0/G1 checkpoint in PNT1a cells, activates caspases and cleaves poly(ADP ribose) polymerase 1 after autophagy, enabling cells to undergo anoikis.415 Polyphenon E® significantly enlarges the endoplasmic reticulum in PC3 cells, strongly upregulates GADD153/CHOP, and activates Puma, ultimately inducing necroptosis in prostate cancer cells.415 These new targets and strategies sensitize anti-apoptotic cells to other death pathways. Nobiletin extracted from citrus fruits can increase PARP levels in a dose-dependent manner, induce DNA damage and lead to apoptosis.416 It can also reduce mitochondrial membrane potential, induce ROS production and autophagy, induce GSDMD/ GSDME mediated pyroptosis, regulate a variety of regulatory cell death pathways, and significantly inhibit the proliferation of human ovarian cancer cells (Hocc).416 GSDME is highly expressed in human lung cancer. Both paclitaxel and cisplatin can significantly induce apoptosis of lung cancer cells, but cisplatin can induce more persistent caspase-3/GSDME-dependent pyroptosis than paclitaxel, suggesting that cisplatin may have additional advantages in the treatment of lung cancer with high expression of GSDME.417 In addition, cisplatin induces apoptosis and ferroptosis in A549 and HCT116 cells by causing reduced glutathione depletion and glutathione peroxidase inactivation, revealing a new potential mechanism of traditional chemotherapeutic drugs.418 A novel molecule, BAY 87-2243 (‘BAY’), induces mPTP opening and Δψ depolarization by inhibiting mitochondrial respiratory chain complex I (CI), promoting autophagosome formation, mitosis, and the associated increase in ROS, ultimately inducing ferroptosis, resulting in melanoma cell death.419 Knockout of autophagy-related gene 5 (ATG5) or addition of the ferroptosis inhibitor ferrostatin-1 inhibited BAY-stimulated autophagosome formation, increased cellular ROS, and tumor cell death.419 DHA, a semi-synthetic derivative of artemisinin, mediates cell cycle arrest in head and neck cancer cells through forkhead box protein M1 (FoxM1), reduces the expression of angiogenic factors, changes the angiogenesis phenotype of cancer cells, induces ferroptosis and apoptosis of cancer cells, and has efficient and specific antitumor activity.420 Heme oxygenase-1 (HO-1) has an antitumor function in cancer cells but has a cytoprotective function in normal cells.421,422 Piperlongumine (PL), a natural alkaloid isolated from pepper, inactivates kelch like ECH related protein-1 (Keap1) through mercaptan modification, then activates nuclear factor erythroid 2-related factor 2 (Nrf2), upregulates the expression of HO-1, induces apoptosis of breast cancer cells, promotes the production of ROS, and induces ferroptosis.423 However, it has no effect on normal human breast epithelial cells and finally forms a mechanism of selective killing of cancer cells423 (Table 14).
Combination therapy of targeted small-molecule compounds in cancer
Combination therapy for synthetic lethality in cancer
Synthetic lethality is a genetic interaction between two genes in which mutations in a pair of genes result in cell death, while the mutation of either gene is not lethal.424 Some DNA double-strand break repair (DSBR) genes have synthetic lethal relationships with oncogenes or tumor suppressor genes, and these genetic abnormalities could be targeted to kill cancer cells selectively.425 It can be a new direction of targeted tumor therapy to search for a target gene that has a synthetic killing effect with oncogenes and exert synthetic lethality. Therefore, combination therapy based on synthetic lethality is a viable and promising therapeutic strategy for cancer treatment.
PARP inhibitors are the first clinically approved cancer drugs designed to exploit synthetic lethality.426 At present, PARP inhibitors therapy has been successfully used to treat advanced breast and ovarian cancer patients harboring BRCA1/2 mutations and exhibiting homologous recombination (HR) deficiency.427 But some of these patients showed signs of resistance that limit the efficacy of PARP inhibitor monotherapy. Therefore, various research groups adopted a combination therapy strategy to induce cancer cells to produce synthetic lethal effects. In BRCA-mutant TNBC patients,428 there was increased expression of the MYC gene, which reduced the survival rate. The combination of MYC inhibitor, dinaciclib, and PARP inhibitor, niraparib, induced a potent synthetic lethal effect on TNBC cells with MYC overexpression.429 Sirtuin2 (SIRT2) is a kind of epigenetic regulator, and its disorder is the main factor in inducing cancer.430 SIRT2 inhibitor and sorafenib combination treatment exhibited potent ability to induce MCF-7 cell apoptosis, as well potential synthetic lethality effect.431 Aurora-A overexpression has been observed in breast cancer and is associated with poor prognosis. Aurora-A inhibitor, MLN8237 as a single agent, was ineffective in prolonging patient survival, so a strategy of combined inhibition of Aurora-A and Haspin may cooperatively regress the viability of breast cancer cells in vitro and in vivo.432 Moreover, IMMU-132, an antibody conjugate drug, and PARP inhibitors could significantly inhibit breast cancer cell growth and were well tolerated regardless of BRCA1/2 status. Currently, this combination strategy is being tested in phase II clinical trials in patients with advanced or metastatic solid tumors (NCT03992131).433 In addition, CHEK1 inhibitor, LY2603618, and Aurora-A inhibitor, alisertib, had a synthetic lethal effect. The combined treatment could trigger apoptosis of ovarian cancer cells and enhance the therapeutic effect of chemotherapy drugs.434
Studies have shown that thyroid carcinoma cells with BRAF V600E mutation were resistant to PLX4032, a BRAF inhibitor, and the dual blocking of EGFR and BRAF continuously inhibited the ERK/Akt pathway and led to an increased level of apoptosis as well induction of synthetic lethality.435 In BRAF-mutant and RAS/RAF wild-type CRC cells, double blocking of NEDD8 and EGFR pathways led to cell growth arrest and increased apoptosis induction.436 Sun et al. reported the novel synthetic lethal partners, Src and PARP1, that exerted a pronounced anticancer effect on HCC cells and could suppress the resistance to PARP1 inhibition.437 Moreover, a combination of sorafenib and selumetinib was also an effective therapy in HCC cells having high p-ERK levels. The efficacy and safety of the mixture in advanced HCC were demonstrated in phase I/II clinical trial (NCT01029418).438 In an FLT3(ITD)-positive AML cells, FLT3 inhibitor, AC220 combined with PARP1 inhibitor significantly inhibited proliferating leukemia stem cells and delayed disease onset.439 The combination of APG-2575 and ibrutinib could effectively inhibit the DLBCL cells with high expression of Bcl-2 and had a synergistic antitumor effect, which could improve the clinical treatment of lymphoma to a certain extent.440 Besides, a phase II clinical trial (NCT04494503) tested the safety, pharmacokinetic, pharmacodynamic, and efficacy of APG-2575 combined with ibrutinib in patients with relapsed/refractory CLL and small lymphocytic lymphoma (SLL). Additionally, Pan et al. had found that Bcl-2 inhibition and p53 activation could overcome apoptosis resistance and induce synthetic lethality in AML models.441 In phase II clinical trial, the safety and early efficacy of idasanutlin in combination with ABT-199 in young patients with neuroblastoma, AML, and ALL were evaluated (NCT04029688). There are many combined treatment strategies for synthetic lethality, and we have selected several representative examples for discussion and summary in Table 15.442,443,444
Combination therapy for synergistic effects in cancer
Synergistic therapy has been widely applied in clinical treatment as a strategy to improve the efficacy of anticancer therapy. The appropriate drug combinations could acquire better curative effects with fewer adverse reactions and produce synergistic effects by acting on multiple pathogenesis of the disease.445 The combination of natural active compounds with therapeutic drugs should be a promising therapeutic strategy that can overcome various clinical drug deficiencies and improve the in vivo efficacy of drugs.446 This group combined piperine with celecoxib to synergistically exert an antiproliferation effect on colon cancer cells. Besides, the co-treatment could cause mitochondrial dysfunction and the activation of caspase to trigger apoptosis.447 Tetrandrine as a bisbenzylisoquinoline alkaloid extracted from Stephania tetrandrine S. Moore, when combined with protein kinase A inhibitor H89, the therapeutic efficacy enhanced notably.448 It was found that tetrandrine and H89 could synergistically inhibit tumor cell growth and induce apoptosis and autophagy by regulating ROS-induced PKA and ERK signaling pathways. In addition, it showed that c-Myc amplified cancer cells were more sensitive to tetrandrine/H89 combined therapy treatment.448 Additionally, co-treatment of curcumin analog PGV-1 and citrus flavonoid compound diosmin enhanced the cytotoxic effect on 4T1 cancer cells. Targeted regulation of cyclin-dependent kinase 1 (CDK1), KIF11, and AURKA proteins blocked the cell cycle and increased the number of mitotic catastrophes, resulting in senescence and death of 4T1 cancer cells.449
Chemotherapy is a principal treatment for cancer, but chemotherapy targets not only tumor cells but also healthy cells, leading to a variety of adverse effects.450 Therefore, combining chemotherapy drugs with other anticancer compounds could reduce side effects and synergistically increase the antitumor activity of drugs. In this work, combined treatment of curcumin and 5-FU could significantly increase the apoptosis rate of cancer cells, prolong the survival of immunodeficient mice, as well reduce the toxicity and adverse effects of 5-FU.451 Likewise, 5-FU had a synergistic effect with natural product withaferin-A in inhibiting proliferation. This combination could modulate endoplasmic reticulum (ER) stress, suppress the β-catenin pathway, induce cell arrest at the G2M phase, and thus induce CRC cells’ autophagy and apoptosis.452 Recently, flubendazole and 5-FU had been found to synergically inhibit cell proliferation and promote cell death by targeting p53 protein and activating ferroptosis.453 As we all know, dietary polyphenols could be used as a chemical sensitizer to enhance drug efficacy and reduce chemotherapy resistance. It was shown that the dietary flavonoid fisetin and paclitaxel possessed a synergistic effect on A549 NSCLC cells by activating mitotic catastrophe and promoting autophagic cell death of cancer cells which was indicated as a novel chemotherapy approach for NSCLC treatment.454 In addition, it was reported that arsenic trioxide (ATO) could induce apoptosis of breast cancer cells at high concentrations, but it was easy to cause side effects.455 Therefore, this group used the hTERT inhibitor BIBR1532, combined with ATO, to make cells sensitive at a low concentration of ATO, which could synergistically inhibit the survival, proliferation, and accelerated apoptosis of breast cancer cells.455
Targeted drug combination therapy is a prospective clinical therapeutic strategy that could induce apoptosis and autophagic cell death by regulating some targeted proteins and signaling pathways. WEE1 was overexpressed in some tumors, and inhibition or downregulation of WEE1 could lead to mitotic catastrophe. WEE1 inhibitors play a key role in the treatment of tumors.456 Combined inhibition of BET and WEE1 could synergistically attenuate the growth of NSCLC cells.457 Besides, the study had shown that BET inhibitor increased WEE1 inhibitor AZD1775-induced DNA double-strand breaks and cytotoxicity. The blockade of BET protein BRD4 would downregulate the nonhomologous end-joining (NHEJ) activity. When combined with the WEE1 inhibitor, it also diminished myelin transcription factor 1 (MYT1) expression, thereby promoting mitotic catastrophe.457 Moreover, PARP inhibitor rucaparib and PI3K inhibitor BKM120 had shown synergetic antitumor effects on GBM cell lines. BKM120 could reduce HR repair molecule expression, thus increasing the level of rucaparib-induced apoptosis, which could significantly improve the antitumor efficacy.458 A recent study found that the combination of CBL0137 and HDAC inhibitor panobinostat could synergistically suppress the growth of MYCN-amplified neuroblastoma cancer cells, accompanied by the induction of IFN response and the inhibition of DNA damage repair.459 Additionally, Ponatinib, a tyrosine kinase inhibitor (TKI), combined with asciminib could produce a synergistic apoptosis-induced effect in BCR-ABL1 mutant CML cell lines and murine Ba/F3 cells.460 AC220 (Quizartinib), an FLT3 receptor tyrosine kinase inhibitor, was used for AML treatment in clinical. In order to improve its anticancer effect, it is necessary to explore the potential synergistic effect of AC220 and other small molecules. A present study found that when treated in combination with the autophagy inhibitor TAK-165, AC220 could induce cancer cell death by activating chaperone-mediated autophagy.461 These results suggest that targeted autophagy should be regarded as an effective strategy to enhance the efficacy of cancer therapy. A study manifested that combined treatment with Fin56, a ferroptosis inducer, and Torin 2, an mTOR inhibitor, could synergistically inhibit the viability of bladder cancer cells, as well as induce ferroptosis and autophagy-dependent cell death through the glutathione peroxidase 4 (GPX4) protein degradation increased.462 Moreover, metformin combined with sulfasalazine, a ferroptosis inducer, had a synergistic effect on activating ferroptosis and repressing breast cancer cell proliferation463 (Table 16).
Combination therapy for reduced drug resistance in cancer
Primary and acquired drug resistance is one of the most important signs of cancer. Some new combination therapy strategies focus on the development of new targeted therapies. Chemotherapy, as a common tumor treatment strategy, leads to high mortality due to the frequent occurrence of drug resistance during treatment. Bcl-2 inhibitors have been shown to reverse chemoresistance and enhance sensitivity to other chemotherapeutic drugs in cancer.464 The combination of resveratrol (RSV) and anticancer drug docetaxel (DTX) stimulates cell apoptosis by inhibiting Bcl-2 and increasing Bax, induces necroptosis, reverses DTX resistance and synergistically enhances the anticancer effect of DTX.465 Acquired drug resistance is usually due to the reactivation of MEK-ERK1/2 pathway caused by NRAS mutation, increased BRAF copy number, and abnormal BRAF splicing.466,467,468 The efficacy of BRAFi and/or MEKI is associated with tumor T cell infiltration. The loss of CD8 + T cells and the inflow of tumor-associated macrophages are associated with acquired drug resistance to metastatic melanoma.469 The development of resistance to BRAFi+MEKi in metastatic melanoma remains a major clinical challenge. A study in BRAFi+MEKi-resistant melanoma by using etoposide to re-induce pyroptosis, inhibition of the ERK1/2 pathway, and cleavage of GSDME-induced pyroptosis produced a more potent antitumor immune response, overcoming treatment resistance.470 It is suggested that targeting this regulated cell death pathway represents a potential strategy for salvage treatment of patients with BRAFi + MEKI resistant melanoma. Cetuximab, an EGFR inhibitor, is commonly used as a targeted therapy for patients with breast cancer. Because long-term use is easy to leads to anti-EGFR treatment resistance, cetuximab resistance is the main clinical drug resistance problem in the treatment of EGFR overexpression cancer. The level of mir-155-3p is upregulated in breast cancer cells.471 GSDME is the direct binding target of mir-155-5p.472 Cetuximab combined with mir-155-5p antagomir can promote the pyroposis of EGFR overexpressed breast cancer cells, induce apoptosis, enhance the antitumor effect of cetuximab, and provide an alternative treatment for breast cancer patients by upregulating GSDME-N and cleaved caspase-1.472 Colorectal cancer (CRC) is the third most commonly diagnosed cancer in the world. Despite the continuous improvement of the treatment level of colorectal cancer, there are still a considerable number of advanced patients with postoperative metastasis or patients with metastasis at the initial diagnosis, and most of them involve the liver, lung, peritoneum, and distant lymph nodes.314 In addition to chemotherapy, there are different targeted therapies for metastatic colorectal cancer (mCRC). Anti EGFR antibodies (such as cetuximab or panimab) combined with chemotherapy is an effective treatment for patients with RAS wild-type MCRC only.473 Since downstream KRAS mutations exist in about 50% of CRC, the effectiveness of EGFR inhibitor combination therapy is usually limited by intrinsic drug resistance. The combination of β-elemene and cetuximab can induce ferroptosis and inhibit the migration of CRC cells mutated in KRAS by inducing ROS accumulation, consuming GSH, lipid peroxidation, upregulating HO-1 and transferrin, downregulating GPX4, SLC7A11, FTH1 and SLC40A1.474 The above effects were eliminated after the use of ferroptosis inhibitor.474 By inducing ferroptosis, it is expected to provide a prospective strategy for CRC patients with KRAS mutation. In addition, the combination of Andrographis and 5-FU, the active component from Andrographis paniculata, in xenograft animal models and CRC cell lines, inhibits CRC in a dose-dependent manner by activating ferroptosis and inhibiting β- catenin/Wnt signaling pathway, which is more effective than 5-FU or Andrographis alone.475 Apoptosis has been shown to be involved in the development of sorafenib resistance in HCC. Nuclear factor erythroid 2-related factor 2 (Nrf2) overexpression inhibits apoptosis and contributes to the chemical resistance of some cancers.476 However, Nrf2 plays a dual role in the treatment of cancer, depending on the type and stage of cancer. P62-keap1-nrf2 pathway plays an important role in protecting HCC cells from ferroptosis by upregulating ROS-related genes.477 Alkaloid trigonelline reverses drug resistance by inhibiting Nrf2 in vitro and tumor xenotransplantation model, thus increasing the anti HCC activity of erastin and sorafenib477 (Table 17).
Novel therapeutic strategies targeting RCD subroutines in cancer
At present, with the development of molecular biology, the molecular typing of most cancers has gradually become clear, and the small molecule targeted therapeutic drugs under its guidance is also undergoing phase I-III clinical research. However, the progress of targeted therapy is slow, and some cancers are not sensitive to endocrine therapy and molecular targeted therapy. However, the existing traditional chemotherapy has some problems, such as strong side effects and drug resistance. Therefore, optimizing the existing clinical treatment methods and looking for new and effective cancer treatment methods have become the current research hotspot.478
Some drugs can effectively induce cancer cells to produce RCD, but they can not be directly used in clinical cancer treatment because of their poor water solubility, nephrotoxicity, and other toxic side effects. Recently, studies have shown that exosomes and nanosystems play a great role in the treatment of cancer. In addition, nanoparticles have unique advantages in disease treatment because of their good hydrophilicity and targeting. Nanoparticles combined with drugs or physical methods targeting ferroptosis are a new and potential treatment strategy. Ferritin nanoparticles, known as erastin and rapamycin binding, are a nanodrug inspired by Abraxane. When ferritin nanoparticles are swallowed by cancer cells, the nanosystem can release erastin and rapamycin to inhibit systeam Xc− and autophagy, respectively, resulting in ferroptosis in tumor cells.479 In addition, the organic metal network (mon-p53) wrapped with p53 plasmid combined with iron can also induce the occurrence of ferroptosis by releasing iron and p53 plasmid to inhibit systeam Xc−. In mice with cancer, mon-p53 treatment not only inhibited tumor growth but also prolong the life of mice.480 In addition, a novel cascaded copper-based metal-organic framework therapeutic nanocatalyst using HKUST-1 amplifies the treatment of hepatocellular carcinoma by integrating the cyclooxygenase-2 (COX-2) inhibitor meloxicam and the chemotherapeutic drug sorafenib.481 In recent years, a variety of regulatory cell death forms have been found and characterized by their corresponding molecular mechanisms. These mechanisms can stimulate autophagy-dependent cell death, apoptosis, necroptosis, pyroptosis, ferroptosis, and so on, providing a new choice for cancer treatment research. In order to effectively improve the intracellular iron level, promote the ferroptosis induction pathway, establish an effective ferroptosis/pyroptosis mediated system, and create an efficient ferroptosis/pyroptosis double induction nano delivery system TF-LipoMof@PL.482 This system is loaded with the metal organic skeleton (MOF) of the pH-sensitive lipid layer modified by transferrin.482 The iron in the cell is enriched by the iron-containing MOF. The modified transferrin on the lipid layer further promotes the endocytosis of MOF.482 The upregulated TfR expression on the surface of various tumor cells not only provides a natural advantage for more effective iron-mediated iron endocytosis, but also provides a target for therapeutic drug delivery.483 PL is loaded as a ferroptosis inducer in an iron-containing MOF coated with a dope pH-sensitive lipid layer decorated with transferrin. This system can induce Fenton reaction, provide H2O2 for the double induction system, increase ROS in tumor cells, cause ferroptosis and pyroptosis in cells, and achieve an effective anticancer effect.482 In recent years, a large number of studies have found that exosomes have the advantages of low immunogenicity, high biocompatibility, and high efficiency, and have good advantages in drug delivery. A research team constructed a folic acid (FA)-labeled Erastin-loaded exosome preparation (erastin@FA-exo) to target folate receptor-overexpressing TNBC cells. Compared with free Erastin, Erastin@FAexo increased the uptake rate of Erastin in MDA-MB-231 cells and induced ferroptosis by inhibiting Systeam Xc−.484
Concluding remarks and future perspectives
Of note, the dynamic balance between cell survival and death is not only a necessary condition to maintain the homeostasis of the organisms, but one of the important conditions for the growth and development. When excessive cell proliferation or normal cell death is inhibited, the incidence of malignant tumors increases greatly. Thus, the two significant characteristics of malignant tumors are uncontrolled cell proliferation and escaping programmed cell death.485 Tumor has many phenotypes, which are the basis of tumor cell growth and metastasis. Resistance to programmed cell death (PCD) is one of its important phenotypes.486,487 In the traditional sense, cell death is divided into regulatory and non-regulatory types. With the deepening of research, more and more regulatory cell death modes have been found and named. In this review, we summarize 9 subroutines of regulated cell death (RCD), including autophagy-dependent cell death, apoptosis, necroptosis, pyroptosis, ferroptosis, parthanatos, entosis, netotic cell death and lysosome-dependent cell death.488 Autophagy-dependent cell death played the Jannus role in the occurrence and development of tumors. In the early stage of tumor occurrence, autophagy-dependent cell death exerted a preventive effect in controlling or killing cancer cells, while in the formed tumor cells, autophagy-dependent cell death could maintain the survival of cancer cells and promote development. Apoptosis has been recognized as a key intracellular process that maintain organism homeostasis and promote survival. Inhibition or resistance of apoptosis often leads to the occurrence of tumors. In recent years, in order to reduce toxicity, reduce the risk of disease progression, promote individualized treatment of tumors and improve the prognosis of patients, researchers have done a lot of research in the direction of tumor treatment by targeting RCD. In this review, we summarize those 104 drugs treat tumors by mainly targeting apoptosis, 65 drugs treat tumors by mainly targeting autophagy dependent cell death, 12 drugs treat tumors by mainly targeting pyroptosis, 8 drugs treat tumors by mainly targeting ferroptosis, and 10 drugs treat tumors by mainly targeting other targets of regulated cell death (RCD). Long-term activation or inhibition of a single RCD subroutine often leads to drug resistance. We summarized 8 antitumor drugs that target multiple RCD subroutines and 36 combination therapies that target RCD.
Compared with ACD, RCD is controlled by specific signal transduction pathways and can be regulated by genetic signals or drug intervention. Loss of control over single or mixed types of regulatory cell death can lead to a variety of human diseases, including cancer. Different lethal subroutines in RCD will affect the progress of cancer and the response to treatment. In the early stage of the disease, cancer cells may have the characteristics of anti-cancer treatment because of the mutations that destroy the RCD pathway, and avoiding RCD is one of the important signs of cancer. Through the simultaneous regulation of multiple RCD signal pathways by a drug or gene, the drug resistance of cancer cells to a specific type of RCD can be avoided, so as to achieve the purpose of treatment. Further in-depth studies of the complexity of RCD in the body, especially in malignant tumors, is not only conducive to deepen a better understanding of intracellular signal molecules and the maintenance of homeostasis, but of great significance for the confirmation of therapeutic targets, the development of new small-molecule drugs and the intricate mechanisms of drug resistance. Based on the current research results, focusing on the crosstalk between different RCD pathways may be a new direction of cancer treatment research in the future.
References
Zheng, R. et al. Cancer incidence and mortality in China, 2016. J. Natl Cancer Center. 2, 1–9 (2022).
Cheng, T. et al. The International Epidemiology of Lung Cancer: latest trends, disparities, and tumor characteristics. J. Thorac. Oncol. 11, 1653–1671 (2016).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Christgen, S., Tweedell, R. & Kanneganti, T. Programming inflammatory cell death for therapy. Pharmacol. Therapeut. 232, 108010 (2022).
Liao, M. et al. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: a revisited perspective from molecular mechanisms to targeted therapies. J. Hematol. Oncol. 15, 44 (2022).
Fairlie, W. D., Tran, S. & Lee, E. F. Crosstalk between apoptosis and autophagy signaling pathways. Int. Rev. Cell Mol. Biol. 352, 115–158 (2020).
Nagata, S. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 36, 489–517 (2018).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Pistritto, G. et al. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging 8, 603–619 (2016).
Inoue-Yamauchi, A. et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat. Commun. 8, 16078 (2017).
Derakhshan, A., Chen, Z., & Van Waes, C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin. Cancer Res. 23, 1379–1387 (2017).
Xiang, H. et al. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm. Sin. B 10, 569–581 (2020).
Cui, J., Shen, H. M. & Lim, L. H. K. The role of autophagy in liver cancer: crosstalk in signaling pathways and potential therapeutic targets. Pharmaceuticals 13, 432 (2020).
Mokarram, P. et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 13, 781–819 (2017).
Zhang, J. et al. Mechanisms of autophagy and relevant small-molecule compounds for targeted cancer therapy. Cell. Mol. Life. Sci. 75, 1803–1826 (2018).
Yoshida, G. J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J. Hematol. Oncol. 10, 67 (2017).
Song, S. L. et al. Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 232, 2977–2984 (2017).
Su, W. et al. Identification of autophagic target RAB13 with small-molecule inhibitor in low-grade glioma via integrated multi-omics approaches coupled with virtual screening of traditional Chinese medicine databases. Cell Prolif. 54, e13135 (2021).
Guo, Z. et al. Ampelopsin inhibits human glioma through inducing apoptosis and autophagy dependent on ROS generation and JNK pathway. Biomed. Pharmacother. 116, 108524 (2019).
Sethi, A. et al. An updated patent review of galectin-1 and galectin-3 inhibitors and their potential therapeutic applications (2016-present). Expert Opin. Ther. Pat. 31, 709–721 (2021).
Zhang, N. et al. Shikonin induces colorectal carcinoma cells apoptosis and autophagy by targeting galectin-1/JNK signaling axis. Int. J. Biol. Sci. 16, 147–161 (2020).
Wu, X. et al. Dihydroartemisinin modulates apoptosis and autophagy in multiple myeloma through the P38/MAPK and Wnt/β-catenin signaling pathways. Oxid. Med. Cell. Longev. 2020, 6096391 (2020).
Tian, S. et al. F1012-2 inhibits the growth of triple negative breast cancer through induction of cell cycle arrest, apoptosis, and autophagy. Phytother. Res. 32, 908–922 (2018).
Ofengeim, D. & Yuan, J. Regulation of RIPK1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14, 727–736 (2013).
Chen, J. et al. Molecular insights into the mechanism of necroptosis: the necrosome as a potential therapeutic target. Cells 8, 1486 (2019).
Dondelinger, Y. et al. NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015).
Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 10, 1729 (2019).
Lafont, E. et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018).
Dillon, C. et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 1, 401–407 (2012).
Wang, L., Du, F. & Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
Newton, K. et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574, 428–431 (2019).
Mompeán, M. et al. The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 173, 1244–1253.e1210 (2018).
Amin, P. et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. NatI. Acad. Sci. USA 115, E5944–E5953 (2018).
Ding, J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
Feng, S., Fox, D. & Man, S. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J. Mol. Biol. 430, 3068–3080 (2018).
Zhang, Y. et al. Plasma membrane changes during programmed cell deaths. Cell Res 28, 9–21 (2018).
Franchi, L., Muñoz-Planillo, R. & Núñez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 (2012).
Chen, G. & Nuñez, G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 (2010).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Masaldan, S. et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 14, 100–115 (2018).
Sterling, J. et al. Iron importers Zip8 and Zip14 are expressed in retina and regulated by retinal iron levels. Exp. Eye Res. 155, 15–23 (2017).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Harraz, M. M., Dawson, T. M. & Dawson, V. L. Advances in neuronal cell death 2007. Stroke 39, 286–288 (2008).
Wang, Y., Luo, W. & Wang, Y. PARP-1 and its associated nucleases in DNA damage response. DNA Repair. 81, 102651 (2019).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417 (2020).
Cao, K. & Tait, S. W. G. Apoptosis and cancer: force awakens, phantom menace, or both? Int. Rev. Cell Mol. Biol. 337, 135–52 (2018).
Pfeffer, C. M. & Singh, A. T. K. Apoptosis: a target for anticancer therapy. Int. J. Mol. Sci. 19, 448 (2018).
Yuan, X. et al. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 37, 733–748 (2018).
Villa-Morales, M. & Fernandez-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 16, 85–101 (2012).
Diaz Arguello, O. A. & Haisma, H. J. Apoptosis-inducing TNF superfamily ligands for cancer therapy. Cancers 13, 1543 (2021).
Lim, B. et al. Targeting TRAIL in the treatment of cancer: new developments. Expert Opin. Ther. Targets 19, 1171–1185 (2015).
Shishodia, G. et al. Tetrandrine (TET) induces death receptors Apo trail R1 (DR4) and Apo trail R2 (DR5) and sensitizes prostate cancer cells to TRAIL-induced apoptosis. Mol. Cancer Ther. 17, 1217–1228 (2018).
Kline, C. L. et al. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci. Signal. 9, ra18 (2016).
Graves, P. R. et al. Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem. Biol. 14, 1020–1029 (2019).
Phillips, D. C. et al. Hexavalent TRAIL fusion protein eftozanermin alfa optimally clusters apoptosis-inducing TRAIL receptors to induce on-target antitumor activity in solid tumors. Cancer Res. 81, 3402–3414 (2021).
Sp, N. et al. Tannic acid promotes TRAIL-induced extrinsic apoptosis by regulating mitochondrial ROS in human embryonic carcinoma. Cells Cells 9, 282 (2020).
Sang Eun, H. et al. Scutellarein induces Fas-mediated extrinsic apoptosis and G2/M cell cycle arrest in Hep3B hepatocellular carcinoma cells. Nutrients 11, 263 (2019).
Chiu, C. F. et al. The novel camptothecin derivative, CPT211, induces cell cycle arrest and apoptosis in models of human breast cancer. Biomed. Pharmacother. 128, 110309 (2020).
Kim, S. M. et al. Apigetrin induces extrinsic apoptosis, autophagy and G2/M phase cell cycle arrest through PI3K/AKT/mTOR pathway in AGS human gastric cancer cell. J. Nutr. Biochem. 83, 108427 (2020).
Hsu, F. T., Chiang, I. T. & Wang, W. S. Induction of apoptosis through extrinsic/intrinsic pathways and suppression of ERK/NF-κB signalling participate in anti-glioblastoma of imipramine. J. Cell. Mol. Med. 24, 3982–4000 (2020).
Wu, J. & Wood, G. S. Analysis of the effect of gentian violet on apoptosis and proliferation in cutaneous T-cell lymphoma in an in vitro study. JAMA Dermatol. 154, 1191–1198 (2018).
Rajan, P. K. et al. The role of histone acetylation-/methylation-mediated apoptotic gene regulation in hepatocellular carcinoma. Int. J. Mol. Sci. 21, 8894 (2020).
Ramaiah, M. J., Tangutur, A. D. & Manyam, R. R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 277, 119504 (2021).
Chidambaram, A. et al. Design, synthesis, and characterization of α, β-unsaturated carboxylic acid, and its urea based derivatives that explores novel epigenetic modulators in human non-small cell lung cancer A549 cell line. J. Cell. Physiol. 233, 5293–5309 (2018).
Wang, Y. et al. YIPF2 promotes chemotherapeutic agent-mediated apoptosis via enhancing TNFRSF10B recycling to plasma membrane in non-small cell lung cancer cells. Cell Death Dis. 11, 242 (2020).
Xiao, R. et al. Antagonist of cIAP1/2 and XIAP enhances anti-tumor immunity when combined with radiation and PD-1 blockade in a syngeneic model of head and neck cancer. Oncoimmunology 7, e1471440 (2018).
Deng, Z. et al. Ruthenium complexes with phenylterpyridine derivatives target cell membrane and trigger death receptors-mediated apoptosis in cancer cells. Biomaterials 129, 111–126 (2017).
Farghadani, R. et al. In vivo acute toxicity evaluation and in vitro molecular mechanism study of antiproliferative activity of a novel indole Schiff base beta-diiminato manganese(III) complex in hormone-dependent and triple negative breast cancer cells. Peerj 7, e7686 (2019).
Lee, K. C. et al. Induction apoptosis of erinacine A in human colorectal cancer cells involving the expression of TNFR, Fas, and Fas ligand via the JNK/p300/p50 signaling pathway with histone acetylation. Front. Pharmacol. 10, 1174 (2019).
Dyari, H. R. E. et al. A novel synthetic analogue of ω-3 17,18-epoxyeicosatetraenoic acid activates TNF receptor-1/ASK1/JNK signaling to promote apoptosis in human breast cancer cells. FASEB J. 31, 5246–5257 (2017).
Nascimento, F. R. et al. Dibenzoylmethane derivative inhibits melanoma cancer in vitro and in vivo through induction of intrinsic and extrinsic apoptotic pathways. Chem. Biol. Interact. 351, 109734 (2022).
Kang, T. H. et al. Natural compound licochalcone B induced extrinsic and intrinsic apoptosis in human skin melanoma (A375) and squamous cell carcinoma (A431) cells. Phytother. Res. 31, 1858–1867 (2017).
Tung, S. Y. et al. Apoptotic mechanisms of gastric cancer cells induced by isolated erinacine S through epigenetic histone H3 methylation of FasL and TRAIL. Food Funct. 12, 3455–3468 (2021).
Chang, K. F. et al. Cedrol suppresses glioblastoma progression by triggering DNA damage and blocking nuclear translocation of the androgen receptor. Cancer Lett. 495, 180–190 (2020).
Kim, H. J. et al. C5, a cassaine diterpenoid amine, induces apoptosis via the extrinsic pathways in human lung cancer cells and human lymphoma cells. Int. J. Mol. Sci. 21, 1298 (2020).
Yang, D. L. et al. Demethylzeylasteral (T-96) initiates extrinsic apoptosis against prostate cancer cells by inducing ROS-mediated ER stress and suppressing autophagic flux. Biol. Res. 54, 27 (2021).
Basu, A. The interplay between apoptosis and cellular senescence: Bcl-2 family proteins as targets for cancer therapy. Pharmacol. Therapeut. 230, 107943 (2022).
Campbell, K. J. & Tait, S. W. G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 8, 180002 (2018).
Adams, J. M. & Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27–36 (2018).
Zhang, J.-Y. et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 31, 980–997 (2021).
Yamaguchi, R., Lartigue, L. & Perkins, G. Targeting Mcl-1 and other Bcl-2 family member proteins in cancer therapy. Pharmacol. Ther. 195, 13–20 (2019).
Ohgino, K. et al. Intracellular levels of reactive oxygen species correlate with ABT-263 sensitivity in non-small-cell lung cancer cells. Cancer Sci. 111, 3793–3801 (2020).
Yang, I. H. et al. ABT-263 exhibits apoptosis-inducing potential in oral cancer cells by targeting C/EBP-homologous protein. Cell. Oncol. 42, 357–368 (2019).
Lochmann, T. L. et al. Venetoclax is effective in small-cell lung cancers with high BCL-2 expression. Clin. Cancer Res. 24, 360–369 (2018).
Song, S. et al. Targeting cancer stem cells with a pan-BCL-2 inhibitor in preclinical and clinical settings in patients with gastroesophageal carcinoma. Gut 70, 2238–2248 (2021).
Kurppa, K. J. et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37, 104–122.e112 (2020).
Wei, H. et al. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci. 108, 478–487 (2017).
Shin, J. W. et al. BCI induces apoptosis via generation of reactive oxygen species and activation of intrinsic mitochondrial pathway in H1299 lung cancer cells. Sci. China Life Sci. 61, 1243–1253 (2018).
Zhu, P. J. et al. Myeloid cell leukemin-1 inhibitors: a growing arsenal for cancer therapy. Drug. Discov. Today 25, 1873–1882 (2020).
Hird, A. W. & Tron, A. E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol. Therapeut. 198, 59–67 (2019).
Zhu, P. J. et al. Discovery of 3,5-dimethyl-4-sulfonyl-1H-pyrrole-based myeloid cell leukemia 1 inhibitors with high affinity, selectivity, and oral bioavailability. J. Med. Chem. 64, 11330–11353 (2021).
Tron, A. E. et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 9, 5341 (2018).
Jin, S. et al. Xanthones from the bark of garcinia xanthochymus and the mechanism of induced apoptosis in human hepatocellular carcinoma HepG2 cells via the mitochondrial pathway. Int. J. Mol. Sci. 20, 4803 (2019).
Han, J. M., Kim, H. L. & Jung, H. J. Ampelopsin inhibits cell proliferation and induces apoptosis in HL60 and K562 leukemia cells by downregulating AKT and NF-κB signaling pathways. Int J. Mol. Sci. 22, 4265 (2021).
Anaya-Eugenio, G. D. et al. A pentamethoxylated flavone from Glycosmis ovoidea promotes apoptosis through the intrinsic pathway and inhibits migration of MCF-7 breast cancer cells. Phytother. Res. 35, 1634–1645 (2021).
Shin, M. K. et al. In vivo and in vitro effects of tracheloside on colorectal cancer cell proliferation and metastasis. Antioxidants 10, 513 (2021).
Gamage, C. D. B. et al. Deoxypodophyllotoxin exerts anti-cancer effects on colorectal cancer cells through induction of apoptosis and suppression of tumorigenesis. Int. J. Mol. Sci. 20, 2612 (2019).
Fu, D. J. et al. Molecular diversity of trimethoxyphenyl-1,2,3-triazole hybrids as novel colchicine site tubulin polymerization inhibitors. Eur. J. Med. Chem. 165, 309–322 (2019).
Hou, Q. Q. et al. Discovery of novel steroidal-chalcone hybrids with potent and selective activity against triple-negative breast cancer. Bioorg. Med. Chem. 28, 115763 (2020).
Ndagi, U., Mhlongo, N., & Soliman, M. E. Metal complexes in cancer therapy - an update from drug design perspective. Drug Des. Dev. Ther. 11, 599–616 (2017).
Huang, X. et al. Pt(IV) complexes conjugating with chalcone analogue as inhibitors of microtubule polymerization exhibited selective inhibition in human cancer cells. Eur. J. Med. Chem. 146, 435–450 (2018).
Pöthig, A. & Casini, A. Recent developments of supramolecular metal-based structures for applications in cancer therapy and imaging. Theranostics 9, 3150–3169 (2019).
Tang, B. et al. An iridium (III) complex as potent anticancer agent induces apoptosis and autophagy in B16 cells through inhibition of the AKT/mTOR pathway. Eur. J. Med. Chem. 145, 302–314 (2018).
Qi, Y. Y. et al. Two new Cu(II) dipeptide complexes based on 5-methyl-2-(2'-pyridyl)benzimidazole as potential antimicrobial and anticancer drugs: Special exploration of their possible anticancer mechanism. Eur. J. Med. Chem. 154, 220–232 (2018).
Liu, G. et al. Structure-activity relationship studies on Bax activator SMBA1 for the treatment of ER-positive and triple-negative breast cancer. Eur. J. Med. Chem. 178, 589–605 (2019).
Feng, S. et al. HG30, a tetrahydroanthraquinone compound isolated from the roots of Prismatomeris connate, induces apoptosis in human non-small cell lung cancer cells. Biomed. Pharmacother. 100, 124–131 (2018).
Hong, S. H. et al. Anti-proliferative and pro-apoptotic effects of licochalcone A through ROS-mediated cell cycle arrest and apoptosis in human bladder cancer cells. Int. J. Mol. Sci. 20, 3820 (2019).
El-Kashef, H. et al. Synthesis of a novel series of (Z)-3,5-disubstituted thiazolidine-2,4-diones as promising anti-breast cancer agents. Bioorg. Chem. 96, 103569 (2020).
Zhu, H. et al. Synthesis of chalcone derivatives: inducing apoptosis of HepG2 cells via regulating reactive oxygen species and mitochondrial pathway. Front. Pharmacol. 10, 1341 (2019).
Liu, R. et al. Discovery and development of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives as Bcl-2/Mcl-1 inhibitors. Bioorg. Chem. 88, 102938 (2019).
Si, L. et al. Potent effects of dioscin against pancreatic cancer via miR-149-3P-mediated inhibition of the Akt1 signalling pathway. Br. J. Pharmacol. 174, 553–568 (2017).
Pan, Z. et al. Cinobufagin induces cell cycle arrest at the S phase and promotes apoptosis in nasopharyngeal carcinoma cells. Biomed. Pharmacother. 122, 109763 (2020).
Santucci, R. et al. Cytochrome c: an extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 136, 1237–1246 (2019).
González-Arzola, K. et al. New moonlighting functions of mitochondrial cytochrome c in the cytoplasm and nucleus. FEBS Lett. 593, 3101–3119 (2019).
Bock, F. J. & Tait, S. W. G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100 (2020).
Prudent, J. & McBride, H. M. The mitochondria-endoplasmic reticulum contact sites: a signalling platform for cell death. Curr. Opin. Cell Biol. 47, 52–63 (2017).
Charan, M. et al. Macrophage migration inhibitory factor inhibition as a novel therapeutic approach against triple-negative breast cancer. Cell Death Dis. 11, 774 (2020).
Xiao, S. et al. Novel ginsenoside derivatives have shown their effects on PC-3 cells by inducing G1-phase arrest and reactive oxygen species-mediate cell apoptosis. Bioorg. Chem. 112, 104864 (2021).
Armentano, B. et al. 5-(Carbamoylmethylene)-oxazolidin-2-ones as a promising class of heterocycles inducing apoptosis triggered by increased ROS levels and mitochondrial dysfunction in breast and cervical cancer. Biomedicines 8, 35 (2020).
Güzelcan, E. A. et al. Synthesis of new derivatives of boehmeriasin A and their biological evaluation in liver cancer. Eur. J. Med. Chem. 166, 243–255 (2019).
Mohamed, M. F., Saddiq, A. A. & Abdelhamid, I. A. Attacking the mitochondria of colorectal carcinoma by novel 2-cyanoacrylamides linked to ethyl 1,3-diphenylpyrazole-4-carboxylates moiety as a new trend for chemotherapy. Bioorg. Chem. 103, 104195 (2020).
Deng, X. et al. Drp1-mediated mitochondrial fission contributes to baicalein-induced apoptosis and autophagy in lung cancer via activation of AMPK signaling pathway. Int. J. Biol. Sci. 16, 1403–1416 (2020).
Lincet, H. & Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 34, 3751–3759 (2015).
Liu, W. et al. Xanthohumol inhibits colorectal cancer cells via downregulation of Hexokinases II-mediated glycolysis. Int. J. Biol. Sci. 15, 2497–2508 (2019).
Liu, W. Y. et al. Lycorine induces mitochondria-dependent apoptosis in hepatoblastoma HepG2 cells through ROCK1 activation. Front. Pharmacol. 10, 651 (2019).
Liao, W. et al. Curcumin inhibited growth of human melanoma A375 cells via inciting oxidative stress. Biomed. Pharmacother. 95, 1177–1186 (2017).
Yang, N. et al. Emodin induced SREBP1-dependent and SREBP1-independent apoptosis in hepatocellular carcinoma cells. Front. Pharmacol. 10, 709 (2019).
Xu, Z. F. et al. Oroxyloside inhibits human glioma progression by suppressing proliferation, metastasis and inducing apoptosis related pathways. Biomed. Pharmacother. 97, 1564–1574 (2018).
Cahill, K. E., Morshed, R. A. & Yamini, B. Nuclear factor-kappa B in glioblastoma: insights into regulators and targeted therapy. Neuro Oncol. 18, 329–339 (2016).
Patel, M. et al. NF-kappa B pathways in the development and progression of colorectal cancer. Transl. Res. 197, 43–56 (2018).
Bosman, M. C. J., Schuringa, J. J. & Vellenga, E. Constitutive NF-kappa B activation in AML: causes and treatment strategies. Crit. Rev. Oncol. Hematol. 98, 35–44 (2016).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Nair, J. S., Musi, E. & Schwartz, G. K. Selinexor (KPT-330) induces tumor suppression through nuclear sequestration of IκB and downregulation of survivin. Clin. Cancer Res. 23, 4301–4311 (2017).
Abdul Razak, A. R. et al. First-in-class, first-in-human phase i study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J. Clin. Oncol. 34, 4142–4150 (2016).
Liu, X. et al. Puerarin Inhibits proliferation and induces apoptosis by upregulation of miR-16 in bladder cancer cell line T24. Oncol. Res. 26, 1227–1234 (2018).
Bukowski, K., Kciuk, M. & Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 21, 3233 (2020).
Labbozzetta, M., Notarbartolo, M. & Poma, P. Nuclear factor-kappa B signaling inhibitors revert multidrug-resistance in breast cancer cells. Int. J. Mol. Sci. 21, 3070 (2020).
Abdin, S. M. et al. Nuclear factor-κB signaling inhibitors revert multidrug-resistance in breast cancer cells. Chem. Biol. Interact. 340, 109450 (2021).
Kryczka, J. et al. Molecular mechanisms of chemoresistance induced by cisplatin in NSCLC cancer therapy. Int. J. Mol. Sci. 22, 8885 (2021).
Du, Y. et al. Isorhamnetin enhances the radiosensitivity of A549 cells through interleukin-13 and the NF-κB signaling pathway. Front. Pharmacol. 11, 610772 (2020).
Shrestha, S. et al. Aurantoside C targets and induces apoptosis in triple negative breast cancer cells. Mar. Drugs 16, 361 (2018).
Qiu, J. et al. Hyperoside induces breast cancer cells apoptosis via ROS-mediated NF-κB signaling pathway. Int. J. Mol. Sci. 21, 131 (2019).
Zhu, P. et al. Arnicolide D exerts anti-melanoma effects and inhibits the NF-κB pathway. Phytomedicine 64, 153065 (2019).
Chen, J. et al. Glaucocalyxin A induces cell cycle arrest and apoptosis via inhibiting NF-κB/p65 signaling pathway in melanoma cells. Life Sci. 271, 119185 (2021).
Ruibin, J. et al. Cardamonin induces G2/M phase arrest and apoptosis through inhibition of NF-κB and mTOR pathways in ovarian cancer. Aging 12, 25730–25743 (2020).
Hu, M. et al. Ginsenoside Rk1 induces apoptosis and downregulates the expression of PD-L1 by targeting the NF-κB pathway in lung adenocarcinoma. Food Funct. 11, 456–471 (2020).
Shen, M. et al. Betulinic acid induces ROS-dependent apoptosis and S-phase arrest by inhibiting the NF-κB pathway in human multiple myeloma. Oxidative Med. Cell. Longev. 2019, 5083158 (2019).
Aubrey, B. J. et al. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 25, 104–113 (2018).
Boutelle, A. M. & Attardi, L. D. p53 and tumor suppression: it takes a network. Trends Cell Biol. 31, 298–310 (2021).
Duffy, M. J., Synnott, N. C. & Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 83, 258–265 (2017).
Sabapathy, K. & Lane, D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 15, 13–30 (2018).
Blandino, G. & Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 37, 30 (2018).
Wang, S. M. et al. Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb. Perspect. Med 7, a026245 (2017).
Martinez-Zapien, D. et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 529, 541–545 (2016).
Yu, D. H. et al. Targeting MDMX for cancer therapy: rationale, strategies, and challenges. Front. Oncol. 10, 1389 (2020).
Liu, Y. et al. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 176, 92–104 (2019).
Zhang, J. et al. A small-molecule inhibitor of MDMX suppresses cervical cancer cells via the inhibition of E6-E6AP-p53 axis. Pharmacol. Res. 177, 106128 (2022).
Soares, J. et al. DIMP53-1: a novel small-molecule dual inhibitor of p53-MDM2/X interactions with multifunctional p53-dependent anticancer properties. Mol. Oncol. 11, 612–627 (2017).
Rozenberg, J. M. et al. Dual role of p73 in cancer microenvironment and DNA damage response. Cells 10, 3516 (2021).
Jiang, L. et al. Protoporphyrin IX is a dual inhibitor of p53/MDM2 and p53/MDM4 interactions and induces apoptosis in B-cell chronic lymphocytic leukemia cells. Cell Death Disco. 5, 77 (2019).
Yi, H. et al. A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J. Exp. Clin. Cancer Res. 37, 97 (2018).
Lu, W. et al. Selective targeting p53(WT) lung cancer cells harboring homozygous p53 Arg72 by an inhibitor of CypA. Oncogene 36, 4719–4731 (2017).
Gu, L. et al. Inhibition of MDM2 by a rhein-derived compound AQ-101 suppresses cancer development in SCID mice. Mol. Cancer Ther. 17, 497–507 (2018).
Ramraj, S. K. et al. Novel ovarian cancer maintenance therapy targeted at mortalin and mutant p53. Int. J. Cancer 147, 1086–1097 (2020).
Sari, A. N. et al. Identification and characterization of mortaparib(Plus)-a novel triazole derivative that targets mortalin-p53 interaction and inhibits cancer-cell proliferation by wild-type p53-dependent and -independent mechanisms. Cancers 13, 835 (2021).
Elwakeel, A. et al. Mutant p53(L194F) harboring luminal-A breast cancer cells are refractory to apoptosis and cell cycle arrest in response to mortaparib(Plus), a multimodal small molecule inhibitor. Cancers 13, 3043 (2021).
Tadele, D. S. et al. A cell competition-based small molecule screen identifies a novel compound that induces dual c-Myc depletion and p53 activation. J. Biol. Chem. 296, 100179 (2021).
Devor, E. J. et al. The synthetic curcumin analog HO-3867 rescues suppression of PLAC1 expression in ovarian cancer cells. Pharmaceutical 14, 942 (2021).
Sato, H. et al. Andrographolide induces degradation of mutant p53 via activation of Hsp70. Int. J. Oncol. 53, 761–770 (2018).
Petsri, K. et al. Renieramycin T induces lung cancer cell apoptosis by targeting Mcl-1 degradation: a new insight in the mechanism of action. Mar. Drugs 17, 301 (2019).
Son, Y. et al. Protopine isolated from Nandina domestica induces apoptosis and autophagy in colon cancer cells by stabilizing p53. Phytother. Res. 33, 1689–1696 (2019).
Lin, S. Q. et al. Actinomycin V suppresses human non-small-cell lung carcinoma A549 cells by inducing G2/M phase arrest and apoptosis via the p53-dependent pathway. Mar. Drugs. 17, 572 (2019).
Rashmi, K. C. et al. A new pyrrole based small molecule from Tinospora cordifolia induces apoptosis in MDA-MB-231 breast cancer cells via ROS mediated mitochondrial damage and restoration of p53 activity. Chem. Biol. Interact. 299, 120–130 (2019).
Reddy, S. T. et al. Synthesis of some novel methyl β-orsellinate based 3, 5-disubstituted isoxazoles and their anti-proliferative activity: identification of potent leads active against MCF-7 breast cancer cell. Bioorg. Chem. 105, 104374 (2020).
Liu, Y. et al. A novel indolizine derivative induces apoptosis through the mitochondria p53 pathway in HepG2 cells. Front. Pharmacol. 10, 762 (2019).
Feng, Y. et al. Resveratrol derivative, trans-3, 5, 4'-trimethoxystilbene sensitizes osteosarcoma cells to apoptosis via ROS-induced caspases activation. Oxid. Med. Cell. Longev. 2021, 8840692 (2021).
Dabiri, Y. et al. p53-dependent anti-proliferative and pro-apoptotic effects of a gold(I) N-heterocyclic carbene (NHC) complex in colorectal cancer cells. Front. Oncol. 9, 438 (2019).
Ma, J. et al. Bromocoumarinplatin, targeting simultaneously mitochondria and nuclei with p53 apoptosis pathway to overcome cisplatin resistance. Bioorg. Chem. 99, 103768 (2020).
Lin, X. et al. Diplatin, a novel and low-toxicity anti-lung cancer platinum complex, activation of cell death in tumors via a ROS/JNK/p53-dependent pathway, and a low rate of acquired treatment resistance. Front. Pharmacol. 10, 982 (2019).
Lu, M. L., Wang, Y. & Zhan, X. Q. The MAPK pathway-based drug therapeutic targets in pituitary adenomas. Front. Endocrinol. 10, 330 (2019).
Yue, J. & López, J. M. Understanding MAPK signaling pathways in apoptosis. Int. J. Mol. Sci. 21, 2346 (2020).
Pranteda, A. et al. The p38 MAPK signaling activation in colorectal cancer upon therapeutic treatments. Int. J. Mol. Sci. 21, 2773 (2020).
Lee, C. W. et al. Cudraflavone C Induces Apoptosis of A375.S2 Melanoma Cells through Mitochondrial ROS Production and MAPK Activation. Int. J. Mol. Sci. 18, 1508 (2017).
Hsieh, M. J. et al. Celastrol, a plant-derived triterpene, induces cisplatin-resistance nasopharyngeal carcinoma cancer cell apoptosis though ERK1/2 and p38 MAPK signaling pathway. Phytomedicine 58, 152805 (2019).
Lin, F. Z. et al. Celastrol induces vincristine multidrug resistance oral cancer cell apoptosis by targeting JNK1/2 signaling pathway. Phytomedicine 54, 1–8 (2019).
Jiang, X. Y. et al. Diallyl trisulfide suppresses tumor growth through the attenuation of Nrf2/Akt and activation of p38/JNK and potentiates cisplatin efficacy in gastric cancer treatment. Acta Pharm. Sin. 38, 1048–1058 (2017).
Hu, S. C. et al. Liposomal avicequinone-B formulations: aqueous solubility, physicochemical properties and apoptotic effects on cutaneous squamous cell carcinoma cells. Phytomedicine 58, 152870 (2019).
Wang, L. et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 450, 22–31 (2019).
Kee, J. Y. et al. Gomisin a suppresses colorectal lung metastasis by inducing AMPK/p38-mediated apoptosis and decreasing metastatic abilities of colorectal cancer cells. Front. Pharmacol. 9, 986 (2018).
Sui, X. B. et al. Metformin: a novel but controversial drug in cancer prevention and treatment. Mol. Pharm. 12, 3783–3791 (2015).
Lu, C. C. et al. Metformin triggers the intrinsic apoptotic response in human AGS gastric adenocarcinoma cells by activating AMPK and suppressing mTOR/AKT signaling. Int. J. Oncol. 54, 1271–1281 (2019).
Liao, M. et al. Main active components of Si-Miao-Yong-An decoction (SMYAD) attenuate autophagy and apoptosis via the PDE5A-AKT and TLR4-NOX4 pathways in isoproterenol (ISO)-induced heart failure models. Pharmacol. Res. 176, 106077 (2022).
Fattahi, S. et al. PI3K/AKT/mTOR signaling in gastric cancer: epigenetics and beyond. Life Sci. 262, 118513 (2020).
Hu, X. et al. Dual PI3K/mTOR inhibitor PKI-402 suppresses the growth of ovarian cancer cells by degradation of Mcl-1 through autophagy. Biomed. Pharmacother. 129, 110397 (2020).
Fan, J. et al. 1,7-Bis(4-hydroxyphenyl)-1,4-heptadien-3-one induces lung cancer cell apoptosis via the PI3K/Akt and ERK1/2 pathways. J. Cell. Physiol. 234, 6336–6349 (2019).
Liu, M. M. et al. Apigenin 7-O-glucoside promotes cell apoptosis through the PTEN/PI3K/AKT pathway and inhibits cell migration in cervical cancer HeLa cells. Food Chem. Toxicol. 146, 111843 (2020).
Trejo-Solis, C. et al. Crosstalk of the Wnt/β-catenin signaling pathway in the induction of apoptosis on cancer cells. Pharmaceuticals 14, 871 (2021).
Baik, J. Y. et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat. Commun. 12, 2666–2666 (2021).
Or, C. R. et al. Obatoclax, a Pan-BCL-2 inhibitor, downregulates survivin to induce apoptosis in human colorectal carcinoma cells via suppressing WNT/β-catenin signaling. Int. J. Mol. Sci. 21, 1773 (2020).
Fathi, N. et al. STAT3 and apoptosis challenges in cancer. Int. J. Biol. Macromol. 117, 993–1001 (2018).
Cao, Y. et al. Mitochondrial ROS accumulation inhibiting JAK2/STAT3 pathway is a critical modulator of CYT997-induced autophagy and apoptosis in gastric cancer. J. Exp. Clin. Cancer Res. 39, 119 (2020).
Oh, H. N. et al. JAK2 regulation by licochalcone H inhibits the cell growth and induces apoptosis in oral squamous cell carcinoma. Phytomedicine 52, 60–69 (2019).
She, S. et al. Combined inhibition of JAK1/2 and DNMT1 by newly identified small-molecule compounds synergistically suppresses the survival and proliferation of cervical cancer cells. Cell Death Dis. 11, 724 (2020).
Amaravadi, R. K., Kimmelman, A. C. & Debnath, J. Targeting autophagy in cancer: recent advances and future directions. Cancer Disco. 9, 1167–1181 (2019).
Yun, C. W. et al. The dual role of autophagy in cancer development and a therapeutic strategy for cancer by targeting autophagy. Int. J. Mol. Sci. 22, 179 (2021).
Turco, E., Fracchiolla, D. & Martens, S. Recruitment and activation of the ULK1/Atg1 kinase complex in selective autophagy. J. Mol. Biol. 432, 123–134 (2020).
Zachari, M. & Ganley, I. G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 61, 585–596 (2017).
Papinski, D. & Kraft, C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J. Mol. Biol. 428, 1725–1741 (2016).
Liu, L. et al. A review of ULK1-mediated autophagy in drug resistance of cancer. Cancers 12, 352 (2020).
Egan, D. F. et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 59, 285–297 (2015).
Dower, C. M. et al. Targeted inhibition of ULK1 promotes apoptosis and suppresses tumor growth and metastasis in neuroblastoma. Mol. Cancer Ther. 17, 2365–2376 (2018).
Chen, Y. et al. Dual targeting of NUAK1 and ULK1 using the multitargeted inhibitor MRT68921 exerts potent antitumor activities. Cell Death Dis. 11, 712 (2020).
Sun, D. et al. Discovery of 5-bromo-4-phenoxy-N-phenylpyrimidin-2-amine derivatives as novel ULK1 inhibitors that block autophagy and induce apoptosis in non-small cell lung cancer. Eur. J. Med. Chem. 208, 112782 (2020).
Yao, C. et al. Rocaglamide enhances NK cell-mediated killing of non-small cell lung cancer cells by inhibiting autophagy. Autophagy 14, 1831–1844 (2018).
Chen, M. et al. The apple dihydrochalcone phloretin suppresses growth and improves chemosensitivity of breast cancer cells via inhibition of cytoprotective autophagy. Food Funct. 12, 177–190 (2021).
Jang, J. E. et al. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 13, 761–762 (2017).
Ouyang, L. et al. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy 13, 777–778 (2017).
Zhang, L. et al. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem. Sci. 8, 2687–2701 (2017).
Liu, J. et al. Blocking AMPK/ULK1-dependent autophagy promoted apoptosis and suppressed colon cancer growth. Cancer Cell Int. 19, 336 (2019).
Hu, Y. et al. The disulfiram/copper complex induces autophagic cell death in colorectal cancer by targeting ULK1. Front. Pharmacol. 12, 752825 (2021).
Xu, Z. et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 104, 575–587 (2020).
Johnson, S. C., Rabinovitch, P. S. & Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 493, 338–345 (2013).
Rabanal-Ruiz, Y., Otten, E. G. & Korolchuk, V. I. mTORC1 as the main gateway to autophagy. Essays Biochem. 61, 565–584 (2017).
Popova, N. V. & Jücker, M. The role of mTOR signaling as a therapeutic target in cancer. Int. J. Mol. Sci. 22, 1743 (2021).
Nuñez-Olvera, S. I. et al. Autophagy machinery as a promising therapeutic target in endometrial cancer. Front. Oncol. 9, 1326 (2019).
Erazo, T. et al. The new antitumor drug ABTL0812 inhibits the Akt/mTORC1 axis by upregulating tribbles-3 pseudokinase. Clin. Cancer Res. 22, 2508–2519 (2016).
Xu, S. et al. Impact on autophagy and ultraviolet B induced responses of treatment with the MTOR inhibitors rapamycin, everolimus, torin 1, and pp242 in human keratinocytes. Oxid. Med. Cell. Longev. 2017, 5930639 (2017).
Shi, D. et al. Autophagy induced by cardamonin is associated with mTORC1 inhibition in SKOV3 cells. Pharmacol. Rep. 70, 908–916 (2018).
Ai, T. et al. N-(1-Benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides: antiproliferative activity and effects on mTORC1 and autophagy. ACS Med. Chem. Lett. 8, 90–95 (2017).
Liu, M. Y. et al. Ginsenoside Rg5 inhibits human osteosarcoma cell proliferation and induces cell apoptosis through PI3K/Akt/mTORC1-related LC3 autophagy pathway. Oxid. Med. Cell. Longev. 2021, 5040326 (2021).
Milošević, Z. et al. Potential of the dual mTOR kinase inhibitor AZD2014 to overcome paclitaxel resistance in anaplastic thyroid carcinoma. Cell. Oncol. 41, 409–426 (2018).
Wang, J. et al. Novel PI3K/Akt/mTOR signaling inhibitor, W922, prevents colorectal cancer growth via the regulation of autophagy. Int. J. Oncol. 58, 70–82 (2021).
Chen, J. N. et al. Synthesis and in vitro anti-bladder cancer activity evaluation of quinazolinyl-arylurea derivatives. Eur. J. Med. Chem. 205, 112661 (2020).
Lv, C. et al. The antitumor natural product tanshinone IIA inhibits protein kinase C and acts synergistically with 17-AAG. Cell Death Dis. 9, 165 (2018).
Xia, T. et al. 20(S)-Ginsenoside Rh2 displays efficacy against T-cell acute lymphoblastic leukemia through the PI3K/Akt/mTOR signal pathway. J. Ginseng. Res. 44, 725–737 (2020).
Yuan, J. M. et al. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 13, 19 (2020).
Sooro, M. A., Zhang, N. & Zhang, P. Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int. J. Cancer 143, 2116–2125 (2018).
Wan, B. et al. Alpha, 2'-dihydroxy-4,4'-dimethoxydihydrochalcone inhibits cell proliferation, invasion, and migration in gastric cancer in part via autophagy. Biomed. Pharmacother. 98, 709–718 (2018).
Hung, A. C. et al. The synthetic β-nitrostyrene derivative CYT-Rx20 induces breast cancer cell death and autophagy via ROS-mediated MEK/ERK pathway. Cancer Lett. 371, 251–261 (2016).
Wang, J. et al. Morusin induces apoptosis and autophagy via JNK, ERK and PI3K/Akt signaling in human lung carcinoma cells. Chem. Biol. Interact. 331, 109279 (2020).
Zhao, Y. et al. 8-C-(E-phenylethenyl)quercetin from onion/beef soup induces autophagic cell death in colon cancer cells through ERK activation. Mol. Nutr. Food Res. 61, 1600437 (2017).
Wang, J. et al. Tetrandrine sensitizes nasopharyngeal carcinoma cells to irradiation by inducing autophagy and inhibiting MEK/ERK pathway. Cancer Med. 9, 7268–7278 (2020).
Shi, Y., Norberg, E. & Vakifahmetoglu-Norberg, H. Mutant p53 as a regulator and target of autophagy. Front. Oncol. 10, 607149 (2020).
Foggetti, G. et al. Gambogic acid counteracts mutant p53 stability by inducing autophagy. BBA Mol. Cell Res 1864, 382–392 (2017).
Zhou, X., Hao, Q. & Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell. Biol. 11, 293–305 (2019).
Allende-Vega, N. & Villalba, M. Metabolic stress controls mutant p53 R248Q stability in acute myeloid leukemia cells. Sci. Rep. 9, 5637 (2019).
Foggetti, G. et al. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 39, BSR20181345 (2019).
Siqueira, E. D. S. et al. Trans-chalcone induces death by autophagy mediated by p53 up-regulation and β-catenin down-regulation on human hepatocellular carcinoma HuH7.5 cell line. Phytomedicine 80, 153373 (2021).
Wang, N. et al. β-asarone inhibited cell growth and promoted autophagy via P53/Bcl-2/Bclin-1 and P53/AMPK/mTOR pathways in Human Glioma U251 cells. J. Cell. Physiol. 233, 2434–2443 (2018).
Cha, Y. E. et al. 6-azauridine induces autophagy-mediated cell death via a p53- and AMPK-dependent pathway. Int. J. Mol. Sci. 22, 2947 (2021).
Kowalski, S. et al. New oxidovanadium(IV) coordination complex containing 2-methylnitrilotriacetate ligands induces cell cycle arrest and autophagy in human pancreatic ductal adenocarcinoma cell lines. Int. J. Mol. Sci. 20, 261 (2019).
Patra, T. et al. A combination of AZD5363 and FH5363 induces lethal autophagy in transformed hepatocytes. Cell Death Dis. 11, 540 (2020).
Islam, M. A., Sooro, M. A. & Zhang, P. Autophagic regulation of p62 is critical for cancer therapy. Int. J. Mol. Sci. 19, 1405 (2018).
Tao, M. et al. p62 as a therapeutic target for tumor. Eur. J. Med Chem. 193, 112231 (2020).
Chen, Y. et al. p62/SQSTM1, a central but unexploited target: advances in its physiological/pathogenic functions and small molecular modulators. J. Med. Chem. 63, 10135–10157 (2020).
Deitersen, J. et al. High-throughput screening for natural compound-based autophagy modulators reveals novel chemotherapeutic mode of action for arzanol. Cell Death Dis. 12, 560 (2021).
Robke, L. et al. Phenotypic identification of a novel autophagy inhibitor chemotype targeting lipid kinase VPS34. Angew. Chem. Int. Ed. Engl. 56, 8153–8157 (2017).
Chen, X. L. et al. DCZ5248, a novel dual inhibitor of Hsp90 and autophagy, exerts antitumor activity against colon cancer. Acta Pharm. Sin. 42, 132–141 (2021).
Shen, Q. et al. Synthesis and evaluation of tetrahydroquinolin-2(1H)-one derivatives as novel anti-pancreatic cancer agents via targeting autophagy. Eur. J. Med. Chem. 170, 28–44 (2019).
Quintana, M. et al. Identification of benzo[cd]indol-2(1H)-ones as novel Atg4B inhibitors via a structure-based virtual screening and a novel AlphaScreen assay. Eur. J. Med. Chem. 178, 648–666 (2019).
Chen, Y. H. et al. Diverse roles of FOXO family members in gastric cancer. World J. Gastrointest. Oncol. 13, 1367–1382 (2021).
Farhan, M. et al. The role of FOXOs and autophagy in cancer and metastasis-Implications in therapeutic development. Med. Res. Rev. 40, 2089–2113 (2020).
Zhang, J. et al. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 11, 629–642 (2015).
Körholz, K. et al. Broad-spectrum HDAC inhibitors promote autophagy through FOXO transcription factors in neuroblastoma. Cells 10, 1001 (2021).
Xiao, Q. et al. Histone deacetylase inhibitors promote epithelial-mesenchymal transition in Hepatocellular Carcinoma via AMPK-FOXO1-ULK1 signaling axis-mediated autophagy. Theranostics 10, 10245–10261 (2020).
Hu, H. et al. Inhibition of autophagy by YC-1 promotes gefitinib induced apoptosis by targeting FOXO1 in gefitinib-resistant NSCLC cells. Eur. J. Pharmacol. 908, 174346 (2021).
Ding, R. et al. WX20120108, a novel IAP antagonist, induces tumor cell autophagy via activating ROS-FOXO pathway. Acta Pharm. Sin. 40, 1466–1479 (2019).
Gao, Y. et al. FOXO3 inhibits human gastric adenocarcinoma (AGS) cell growth by promoting autophagy in an acidic microenvironment. Cell. Physiol. Biochem. 49, 335–348 (2018).
Verzella, D. et al. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 11, 210 (2020).
Zhu, Y. et al. Olanzapine induced autophagy through suppression of NF-κB activation in human glioma cells. CNS Neurosci. Ther. 25, 911–921 (2019).
Heng, Y. et al. Camptothecin inhibits neddylation to activate the protective autophagy through NF-κB/AMPK/mTOR/ULK1 Axis in human esophageal cancer cells. Front. Oncol. 11, 671180 (2021).
Hill, S. M., Wrobel, L. & Rubinsztein, D. C. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 26, 617–629 (2019).
Sun, T. et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat. Commun. 6, 7215 (2015).
Boutouja, F. et al. Regulation of the tumor-suppressor BECLIN 1 by distinct ubiquitination cascades. Int. J. Mol. Sci. 18, 2541 (2017).
Huang, P. J. et al. Potential of antiviral drug oseltamivir for the treatment of liver cancer. Int. J. Oncol. 59, 109 (2021).
Jiang, J. et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy 16, 106–122 (2020).
Du, Y. et al. Ferroptosis is involved in the anti-tumor effect of lycorine in renal cell carcinoma cells. Oncol. Lett. 22, 781–781 (2021).
Wang, Y. F. et al. Baicalein induces beclin 1- and extracellular signal-regulated kinase-dependent autophagy in ovarian cancer cells. Am. J. Chin. Med. 45, 123–136 (2017).
Chen, H. Y. et al. Isoliquiritigenin induces autophagy and inhibits ovarian cancer cell growth. Int. J. Mol. Sci. 18, 2025 (2017).
Wang, Y. et al. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 37, 63 (2018).
Sun, D. et al. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 51, e12402 (2018).
Zhu, S. et al. Inhibiting eukaryotic elongation factor 2 kinase: an update on pharmacological small-molecule compounds in cancer. J. Med. Chem. 64, 8870–8883 (2021).
Teng, J. F. et al. Polyphyllin VI, a saponin from Trillium tschonoskii Maxim. induces apoptotic and autophagic cell death via the ROS triggered mTOR signaling pathway in non-small cell lung cancer. Pharmacol. Res. 147, 104396 (2019).
Kim, T. W. & Lee, H. G. Apigenin induces autophagy and cell death by targeting EZH2 under hypoxia conditions in gastric cancer cells. Int. J. Mol. Sci. 22, 13455 (2021).
Tsai, C. H. et al. Docosahexaenoic acid promotes the formation of autophagosomes in MCF-7 breast cancer cells through oxidative stress-induced growth inhibitor 1 mediated activation of AMPK/mTOR pathway. Food Chem. Toxicol. 154, 112318 (2021).
D'Onofrio, N. et al. Colorectal cancer apoptosis induced by dietary δ-valerobetaine involves PINK1/parkin dependent-mitophagy and SIRT3. Int. J. Mol. Sci. 22, 8117 (2021).
Yao, Z. Q. et al. A novel small-molecule activator of Sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis. 9, 767 (2018).
Chu, C. W. et al. Thioridazine enhances P62-mediated autophagy and apoptosis through Wnt/β-catenin signaling pathway in glioma cells. Int. J. Mol. Sci. 20, 473 (2019).
Wang, Z. et al. Using drugs to target necroptosis: dual roles in disease therapy. Histol. Histopathol. 33, 773–789 (2018).
Zhou, W. & Yuan, J. Necroptosis in health and diseases. Semin. Cell. Dev. Biol. 35, 14–23 (2014).
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005).
Tang, D. et al. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019).
Su, Z. et al. Cancer therapy in the necroptosis era. Cell Death Differ. 23, 748–756 (2016).
He, G. et al. Regression of apoptosis-resistant colorectal tumors by induction of necroptosis in mice. J. Exp. Med. 214, 1655–1662 (2017).
Nugues, A. et al. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 5, e1384 (2014).
Höckendorf, U. et al. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer Cell 30, 75–91 (2016).
Li, X. et al. Association of mixed lineage kinase domain-like protein expression with prognosis in patients with colon cancer. Technol. Cancer Res. Treat. 16, 428–434 (2017).
Zeng, F. et al. RIPK1 binds MCU to mediate induction of mitochondrial Ca uptake and promotes colorectal oncogenesis. Cancer Res. 78, 2876–2885 (2018).
X, F. et al. Receptor-interacting protein kinase 3 is a predictor of survival and plays. Neoplasma 62, 592–601 (2015).
Seldon, C. et al. Chromodomain-helicase-DNA binding protein 5, 7 and pronecrotic mixed lineage kinase domain-like protein serve as potential prognostic biomarkers in patients with resected pancreatic adenocarcinomas. World J. Gastrointest. Oncol. 8, 358–365 (2016).
Liu, Z. et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am. J. Cancer Res. 5, 3174–3185 (2015).
Han, Q. et al. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J. Transl. Med. 16, 201 (2018).
Zhang, D.-W. et al. Multiple death pathways in TNF-treated fibroblasts: RIP3- and RIPK1-dependent and independent routes. Cell Res. 21, 368–371 (2011).
Vandenabeele, P. et al. The role of the kinases RIPK1 and RIP3 in TNF-induced necrosis. Sci. Signal. 3, 115 (2010).
Harshman, S. et al. Proteomic characterization of circulating extracellular vesicles identifies novel serum myeloma associated markers. J. Proteom. 136, 89–98 (2016).
Dannappel, M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014).
Mohanty, S. et al. RETRA induces necroptosis in cervical cancer cells through RIPK1, RIPK3, MLKL and increased ROS production. Eur. J. Pharmacol. 920, 174840 (2022).
Jilishitz, I. et al. NP-ALT, a liposomal:peptide drug, blocks p27Kip1 phosphorylation to induce oxidative stress, necroptosis, and regression in therapy-resistant breast cancer cells. Mol. Cancer Res. 19, 1929–1945 (2021).
Lee, Y.-J., Park, K.-S. & Lee, S.-H. Curcumin targets both apoptosis and necroptosis in acidity-tolerant prostate carcinoma cells. BioMed. Res. Int. 2021, 8859181–8859181 (2021).
Lee, Y.-J. et al. Arctigenin induces necroptosis through mitochondrial dysfunction with CCN1 upregulation in prostate cancer cells under lactic acidosis. Mol. Cell. Biochem. 467, 45–56 (2020).
Lu, Z. et al. Ophiopogonin D' induces RIPK1-dependent necroptosis in androgen-dependent LNCaP prostate cancer cells. Int. J. Oncol. 56, 439–447 (2020).
Di Grazia, A. et al. The fragile X mental retardation protein regulates RIPK1 and colorectal cancer resistance to necroptosis. Cell. Mol. Gastroenterol. Hepatol. 11, 639–658 (2021).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clin. 70, 313 (2020).
H, X. et al. Exosome-transmitted PSMA3 and PSMA3-AS1 promote proteasome inhibitor resistance. Clin. Cancer Res. 25, 1923–1935 (2019).
Li, B. et al. piRNA-823 delivered by multiple myeloma-derived extracellular vesicles promoted tumorigenesis through re-educating endothelial cells in the tumor environment. Oncogene 38, 5227–5238 (2019).
Wallach, D. et al. Programmed necrosis in inflammation: toward identification of the effector molecules. Science 352, aaf2154 (2016).
Zhao, S. et al. Toll-like receptors and prostate cancer. Front. Immunol. 5, 352 (2014).
Baine, M. J. et al. Transcriptional profiling of peripheral blood mononuclear cells in pancreatic cancer patients identifies novel genes with potential diagnostic utility. PLOS One 6, e17014 (2011).
Suzuki, E. et al. Gene expression profile of peripheral blood mononuclear cells may contribute to the identification and immunological classification of breast cancer patients. Breast Cancer 26, 282–289 (2019).
Sigurdson, A. et al. Selected single-nucleotide polymorphisms in FOXE1, SERPINA5, FTO, EVPL, TICAM1 and SCARB1 are associated with papillary and follicular thyroid cancer risk: replication study in a German population. Carcinogenesis 37, 677–684 (2016).
Güney Eskiler, G. et al. Inhibition of TLR4/TRIF/IRF3 signaling pathway by curcumin in breast cancer cells. J. Pharm. Pharm. Sci. Publ. Can. Soc. Pharm. Sci. Soc. canadienne des. Sci. pharmaceutiques 22, 281–291 (2019).
Deveci Ozkan, A. et al. Anti-inflammatory effects of nobiletin on TLR4/TRIF/IRF3 and TLR9/IRF7 signaling pathways in prostate cancer cells. Immunopharmacol. Immunotoxicol. 42, 93–100 (2020).
Kaiser, W. J. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 (2013).
Yang, D. et al. ZBP1 mediates interferon-induced necroptosis. Cell. Mol. Immunol. 17, 356–368 (2020).
Yang, Y. et al. ZBP1-MLKL necroptotic signaling potentiates radiation-induced antitumor immunity via intratumoral STING pathway activation. Sci. Adv. 7, eabf6290 (2021).
Karki, R. et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 37, 109858 (2021).
Gu, W., Pan, F. & Singer, R. Blocking beta-catenin binding to the ZBP1 promoter represses ZBP1 expression, leading to increased proliferation and migration of metastatic breast-cancer cells. J. Cell Sci. 122, 1895–1905 (2009).
Gu, W. et al. Regulation of local expression of cell adhesion and motility-related mRNAs in breast cancer cells by IMP1/ZBP1. J. Cell Sci. 125, 81–91 (2012).
GJ, U. et al. Overexpression of FLIPL is an independent marker of poor prognosis in colorectal. Clin. Cancer Res. 13, 5070–5075 (2007).
Safa, A. R. Roles of c-FLIP in apoptosis, necroptosis, and autophagy. J. Carcinog. Mutagen 6, 003 (2013).
Han, W. et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol. Cancer Ther. 6, 1641–1649 (2007).
Tsuchiya, K. Switching from apoptosis to pyroptosis: gasdermin-elicited inflammation and antitumor immunity. Int. J. Mol. Sci. 22, 426 (2021).
Zhang, Z., Zhang, Y. & Lieberman, J. Lighting a fire: can we harness pyroptosis to ignite antitumor immunity? Cancer Immunol. Res. 9, 2–7 (2021).
Rathinam, V. & Fitzgerald, K. Inflammasome complexes: emerging mechanisms and effector functions. Cell 165, 792–800 (2016).
van Deventer, H. et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 70, 10161–10169 (2010).
Daley, D. et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 214, 1711–1724 (2017).
Voigt, C. et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc. Natl Acad. Sci. USA 114, 12994–12999 (2017).
Wang, Q. et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426 (2020).
Micaroni, M. et al. Rab6a/a' are important Golgi regulators of pro-inflammatory TNF secretion in macrophages. PLOS One 8, e57034 (2013).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Wang, W. et al. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J. Dig. Dis. 19, 74–83 (2018).
Gao, J. et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol. Rep. 40, 1971–1984 (2018).
Yang, Y. et al. Hydrogen inhibits endometrial cancer growth via a ROS/NLRP3/caspase-1/GSDMD-mediated pyroptotic pathway. BMC Cancer 20, 28 (2020).
Qiao, L. et al. NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. FASEB J. 33, 12760–12767 (2019).
Deng, H. et al. Long non-coding RNAs: New biomarkers for prognosis and diagnosis of colon cancer. Tumour Biol. 39, 1010428317706332 (2017).
Ma, Y. et al. Biological functions and clinical significance of the newly identified long non-coding RNA RP1-85F18.6 in colorectal cancer. Oncol. Rep. 40, 2648–2658 (2018).
Li, J. et al. LncRNA GAS5 suppresses ovarian cancer by inducing inflammasome formation. Biosci. Rep. 38, BSR20171150 (2018).
Rogers, C. et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8, 14128–14128 (2017).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020).
Hou, J. et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 22, 1264–1275 (2020).
Tan, G. et al. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J. Hematol. Oncol. 13, 149 (2020).
Liu, Y. et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 5, eaax7969 (2020).
Zhou, Z. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, eaaz7548 (2020).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558 (2018).
Gao, Y. et al. Methotrexate-loaded tumour-cell-derived microvesicles can relieve biliary obstruction in patients with extrahepatic cholangiocarcinoma. Nat. Biomed. Eng. 4, 743–753 (2020).
Lu, C. et al. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol. Immunol. 122, 200–206 (2020).
Jaime-Sánchez, P. et al. Antigen-specific primed cytotoxic T cells eliminate tumour cells in vivo and prevent tumour development, regardless of the presence of anti-apoptotic mutations conferring drug resistance. Cell Death Differ. 25, 1536–1548 (2018).
Zhang, Y. et al. A novel 3',5'-diprenylated chalcone induces concurrent apoptosis and GSDME-dependent pyroptosis through activating PKCδ/JNK signal in prostate cancer. Aging 12, 9103–9124 (2020).
Li, Y. et al. Dihydroartemisinin induces pyroptosis by promoting the AIM2/caspase-3/DFNA5 axis in breast cancer cells. Chem. Biol. Interact. 340, 109434 (2021).
Wiemels, J. et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat. Commun. 9, 286 (2018).
Maiorino, M., Conrad, M. & Ursini, F. GPX4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid. Redox Signal. 29, 61–74 (2018).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Yuan, H. et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 478, 1338–1343 (2016).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Feng, J. et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharm. Sin. 42, 160–170 (2021).
Cheng, J. et al. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol. Rep. 43, 147–158 (2020).
Chu, B. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591 (2019).
Almoguera, C. et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53, 549–554 (1988).
Eser, S. et al. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 111, 817–822 (2014).
Arlt, A., Muerkoster, S. S. & Schafer, H. Targeting apoptosis pathways in pancreatic cancer. Cancer Lett. 332, 346–358 (2013).
Eling, N. et al. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2, 517–532 (2015).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017).
Larraufie, M.-H. et al. Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg. Med. Chem. Lett. 25, 4787–4792 (2015).
Scannell, J. W. et al. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug. Disc. 11, 191–200 (2012).
Woo, J. H. et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 162, 441–451 (2015).
Gaschler, M. M. et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515 (2018).
Wang, S. et al. Iron and magnetic: new research direction of the ferroptosis-based cancer therapy. Am. J. Cancer Res. 8, 1933–1946 (2018).
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Cell Chem. Bio. 15, 234–245 (2008).
Geng, N. et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharm. Sci. 22, 3826–3836 (2018).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363 e353 (2019).
Lee, Y.-S. et al. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J. Cell. Biochem. 120, 928–939 (2019).
Gupta, S. et al. Differential sensitivity of naive and subsets of memory CD4+ and CD8+ T cells to hydrogen peroxide-induced apoptosis. Genes. Immun. 8, 560–569 (2007).
Murata, M. M. et al. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 30, 2584–2597 (2019).
Baek, S. H. et al. Induction of mitochondrial dysfunction by poly(ADP-ribose) polymer: implication for neuronal cell death. Mol. Cells 36, 258–266 (2013).
Pazzaglia, S. & Pioli, C. Multifaceted role of PARP-1 in DNA repair and inflammation: pathological and therapeutic implications in cancer and non-cancer diseases. Cells 9, 41 (2019).
Galia, A. et al. PARP-1 protein expression in glioblastoma multiforme. Eur. J. Histochem. 56, e9 (2012).
Dorsam, B. et al. PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proc. Natl Acad. Sci. USA 115, E4061–E4070 (2018).
Yang, D. et al. Knockdown of macrophage migration inhibitory factor (MIF), a novel target to protect neurons from parthanatos induced by simulated post-spinal cord injury oxidative stress. Biochem. Biophys. Res. Commun. 523, 719–725 (2020).
Fong, P. C. et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134 (2009).
Mateo, J. et al. An adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparib. Target Oncol. 11, 401–415 (2016).
Sandhu, S. K. et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 14, 882–892 (2013).
Nishikawa, T. et al. Phase 1 dose-escalation study of single-agent veliparib in Japanese patients with advanced solid tumors. Cancer Sci. 108, 1834–1842 (2017).
de Bono, J. et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Disco. 7, 620–629 (2017).
Park, E. J. et al. β-Lapachone induces programmed necrosis through the RIPK1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis. 5, e1230–e1230 (2014).
Ma, D. et al. Deoxypodophyllotoxin triggers parthanatos in glioma cells via induction of excessive ROS. Cancer Lett. 371, 194–204 (2016).
Humble, J. G., Jayne, W. H. W. & Pulvertaft, R. J. V. Biological interaction between lymphocytes and. other cells. Brit. J. Haemat. 2, 283–294 (1956).
Tauber, A. I. Metchnikoff and the phagocytosis theory. Nat. Rev. Mol. Cell Biol. 4, 897–901 (2003).
Borensztejn, K. et al. Classification of cell-in-cell structures: different phenomena with similar appearance. Cells 10, 2569 (2021).
Overholtzer, M. et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131, 966–979 (2007).
Liang, J. et al. p53-dependent elimination of aneuploid mitotic offspring by entosis. Cell Death Differ. 28, 799–813 (2021).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Brinkmann, V. Neutrophil extracellular traps in the second decade. J. Innate Immun. 10, 414–421 (2018).
M, S. Neutrophil extracellular traps as a drug target to counteract chronic and acute. Curr. Pharm. Biotechnol. 19, 1196–1202 (2018).
Demers, M. et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl Acad. Sci. USA 109, 13076–13081 (2012).
Grayson, P. C. & Kaplan, M. J. At the bench: neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J. Leukoc. Biol. 99, 253–264 (2016).
Gupta, A. K. et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 584, 3193–3197 (2010).
Abdol Razak, N., Elaskalani, O. & Metharom, P. Pancreatic cancer-induced neutrophil extracellular traps: a potential contributor to cancer-associated thrombosis. Int. J. Mol. Sci. 18, 487 (2017).
Sangaletti, S. et al. Defective stromal remodeling and neutrophil extracellular traps in lymphoid tissues favor the transition from autoimmunity to lymphoma. Cancer Disco. 4, 110–129 (2014).
Demers, M. & Wagner, D. D. Neutrophil extracellular traps: a new link to cancer-associated thrombosis and potential implications for tumor progression. Oncoimmunology 2, e22946 (2013).
Circu, M. et al. Modulating lysosomal function through lysosome membrane permeabilization or autophagy suppression restores sensitivity to cisplatin in refractory non-small-cell lung cancer cells. PLOS One 12, e0184922–e0184922 (2017).
Takeda, A. et al. Macrolide antibiotics enhance the antitumor effect of lansoprazole resulting in lysosomal membrane permeabilization-associated cell death. Int. J. Oncol. 57, 1280–1292 (2020).
Lin, H.-Y. et al. The anti-proliferative activity of secondary metabolite from the marine Streptomyces sp. against prostate cancer cells. Life 11, 1414 (2021).
Rizzi, F. et al. Polyphenon E®, a standardized green tea extract, induces endoplasmic reticulum stress, leading to death of immortalized PNT1a cells by anoikis and tumorigenic PC3 by necroptosis. Carcinogenesis 35, 828–839 (2014).
Zhang, R. et al. Nobiletin triggers reactive oxygen species-mediated pyroptosis through regulating autophagy in ovarian cancer cells. J. Agric. Food Chem. 68, 1326–1336 (2020).
Zhang, C.-c et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 24, 312–325 (2019).
Guo, J. et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res. Treat. 50, 445–460 (2018).
Basit, F. et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 8, e2716–e2716 (2017).
Lin, R. et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 381, 165–175 (2016).
Bae, J. et al. Berberine protects 6-hydroxydopamine-induced human dopaminergic neuronal cell death through the induction of heme oxygenase-1. Mol. Cells 35, 151–157 (2013).
Lee, W.-Y. et al. The induction of heme oxygenase-1 suppresses heat shock protein 90 and the proliferation of human breast cancer cells through its byproduct carbon monoxide. Toxicol. Appl. Pharmacol. 274, 55–62 (2014).
Lee, H.-N. et al. Heme oxygenase-1 determines the differential response of breast cancer and normal cells to piperlongumine. Mol. Cells 38, 327–335 (2015).
Topatana, W. et al. Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J. Hematol. Oncol. 13, 118 (2020).
Li, S. et al. Development of synthetic lethality in cancer: molecular and cellular classification. Signal Transduct. Target Ther. 5, 241 (2020).
Lord, C. J. & Ashworth, A. PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158 (2017).
Turk, A. A. & Wisinski, K. B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 124, 2498–2506 (2018).
Liao, M. et al. Small-molecule drug discovery in triple negative breast cancer: current situation and future directions. J. Med. Chem. 64, 2382–2418 (2021).
Carey, J. P. W. et al. Synthetic lethality of PARP inhibitors in combination with MYC blockade is independent of BRCA status in triple-negative breast cancer. Cancer Res. 78, 742–757 (2018).
Wang, Y. et al. SIRT2: controversy and multiple roles in disease and physiology. Ageing Res. Rev. 55, 100961 (2019).
Wang, H. L. et al. Potential synthetic lethality for breast cancer: a selective sirtuin 2 inhibitor combined with a multiple kinase inhibitor sorafenib. Pharmacol. Res. 77, 106050 (2021).
Chen, A. et al. CRISPR/Cas9 screening identifies a kinetochore-microtubule dependent mechanism for Aurora-A inhibitor resistance in breast cancer. Cancer Commun 41, 121–139 (2021).
Cardillo, T. M. et al. Synthetic lethality exploitation by an anti-trop-2-SN-38 antibody-drug conjugate, IMMU-132, plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin. Cancer Res. 23, 3405–3415 (2017).
Alcaraz-Sanabria, A. et al. Synthetic lethality interaction between aurora kinases and CHEK1 inhibitors in ovarian cancer. Mol. Cancer Ther. 16, 2552–2562 (2017).
Notarangelo, T. et al. Dual EGFR and BRAF blockade overcomes resistance to vemurafenib in BRAF mutated thyroid carcinoma cells. Cancer Cell Int. 17, 86 (2017).
Invrea, F. et al. Synthetic lethality screening highlights colorectal cancer vulnerability to concomitant blockade of NEDD8 and EGFR pathways. Cancers 13, 3805 (2021).
Sun, C. et al. Inhibiting Src-mediated PARP1 tyrosine phosphorylation confers synthetic lethality to PARP1 inhibition in HCC. Cancer Lett. 526, 180–192 (2022).
Wang, C. et al. Phospho-ERK is a biomarker of response to a synthetic lethal drug combination of sorafenib and MEK inhibition in liver cancer. J. Hepatol. 69, 1057–1065 (2018).
Maifrede, S. et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood 132, 67–77 (2018).
Luo, Q. et al. A novel BCL-2 inhibitor APG-2575 exerts synthetic lethality with BTK or MDM2-p53 inhibitor in diffuse large B-cell lymphoma. Oncol. Res. 28, 331–344 (2020).
Pan, R. et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell 32, 748–760 (2017).
Zhang, P. et al. Targeting DNA damage repair functions of two histone deacetylases, HDAC8 and SIRT6, sensitizes acute myeloid leukemia to NAMPT inhibition. Clin. Cancer Res. 27, 2352–2366 (2021).
Zhang, Y. et al. Combined HDAC and bromodomain protein inhibition reprograms tumor cell metabolism and elicits synthetic lethality in glioblastoma. Clin. Cancer Res. 24, 3941–3954 (2018).
Lai, S. W. et al. Targeted PARP inhibition combined with FGFR1 blockade is synthetically lethal to malignant. Cells Patients Pancreat. Cancer Cells 9, 911 (2020).
Yu, W. D. et al. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. 452, 66–70 (2019).
Patra, S. et al. Chemotherapeutic efficacy of curcumin and resveratrol against cancer: chemoprevention, chemoprotection, drug synergism and clinical pharmacokinetics. Semin. Cancer Biol. 73, 310–320 (2021).
Srivastava, S. et al. Piperine and Celecoxib synergistically inhibit colon cancer cell proliferation via modulating Wnt/β-catenin signaling pathway. Phytomedicine 84, 153484 (2021).
Yu, M. et al. Combination therapy with protein kinase inhibitor H89 and Tetrandrine elicits enhanced synergistic antitumor efficacy. J. Exp. Clin. Cancer Res. 37, 114 (2018).
Musyayyadah, H. et al. The growth suppression activity of diosmin and PGV-1 co-treatment on 4T1 breast cancer targets mitotic regulatory proteins. Asian Pac. J. Cancer Prev. 22, 2929–2938 (2021).
Vodenkova, S. et al. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Therapeut. 206, 107447 (2020).
Zheng, X. et al. Low curcumin concentration enhances the anticancer effect of 5-fluorouracil against colorectal cancer. Phytomedicine 85, 153547 (2021).
Alnuqaydan, A. M. et al. Synergistic antitumor effect of 5-fluorouracil and withaferin-A induces endoplasmic reticulum stress-mediated autophagy and apoptosis in colorectal cancer cells. Am. J. Cancer Res. 10, 799–815 (2020).
Zhou, X. et al. Flubendazole, FDA-approved anthelmintic, elicits valid antitumor effects by targeting P53 and promoting ferroptosis in castration-resistant prostate cancer. Pharmacol. Res. 164, 105305 (2021).
Klimaszewska-Wisniewska, A. et al. Paclitaxel and the dietary flavonoid fisetin: a synergistic combination that induces mitotic catastrophe and autophagic cell death in A549 non-small cell lung cancer cells. Cancer Cell Int. 16, 10 (2016).
Nasrollahzadeh, A. et al. Arsenic trioxide and BIBR1532 synergistically inhibit breast cancer cell proliferation through attenuation of NF-κB signaling pathway. Life Sci. 257, 118060 (2020).
Bukhari, A. B. et al. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Invest. 129, 1329–1344 (2019).
Takashima, Y. et al. Bromodomain and extraterminal domain inhibition synergizes with WEE1-inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int. J. Cancer 146, 1114–1124 (2020).
Zhang, S. et al. BKM120 sensitizes glioblastoma to the PARP inhibitor rucaparib by suppressing homologous recombination repair. Cell Death Dis. 12, 546 (2021).
Xiao, L. et al. Dual targeting of chromatin stability by the curaxin CBL0137 and histone deacetylase inhibitor panobinostat shows significant preclinical efficacy in neuroblastoma. Clin. Cancer Res. 27, 4338–4352 (2021).
Gleixner, K. V. et al. Asciminib and ponatinib exert synergistic anti-neoplastic effects on CML cells expressing BCR-ABL1 (T315I)-compound mutations. Am. J. Cancer Res. 11, 4470–4484 (2021).
Ouchida, A. T. et al. Synergistic effect of a novel autophagy inhibitor and Quizartinib enhances cancer cell death. Cell Death Dis. 9, 138 (2018).
Sun, Y. et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis. 12, 1028 (2021).
Yang, J. et al. Metformin induces ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J. Exp. Clin. Cancer Res. 40, 206 (2021).
Bray, K. et al. Bcl-2 modulation to activate apoptosis in prostate cancer. Mol. Cancer Res. 7, 1487–1496 (2009).
Lee, S.-H. & Lee, Y.-J. Synergistic anticancer activity of resveratrol in combination with docetaxel in prostate carcinoma cells. Nutr. Res. Pract. 15, 12–25 (2021).
Hartsough, E., Shao, Y. & Aplin, A. E. Resistance to RAF inhibitors revisited. J. Invest. Dermatol. 134, 319–325 (2014).
Moriceau, G. et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 27, 240–256 (2015).
Wagle, N. et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Disco. 4, 61–68 (2014).
Song, C. et al. Recurrent tumor cell-intrinsic and -extrinsic alterations during MAPKi-induced melanoma regression and early adaptation. Cancer Disco. 7, 1248–1265 (2017).
Erkes, D. A. et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Disco. 10, 254–269 (2020).
Liu, J. H. et al. MicroRNA-155regulates the proliferation and metastasis of human breast cancers by targeting MAPK7. J. Buon. 24, 1075–1080 (2019).
Xu, W. et al. Downregulation of miR-155-5p enhances the anti-tumor effect of cetuximab on triple-negative breast cancer cells via inducing cell apoptosis and pyroptosis. Aging 13, 228–240 (2021).
Wu, C. C. et al. Tumor sidedness and efficacy of first-line therapy in patients with RAS/BRAF wild-type metastatic colorectal cancer: a network meta-analysis. Crit. Rev. Oncol. Hematol. 145, 102823 (2020).
Chen, P. et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 10, 5107–5119 (2020).
Sharma, P. et al. Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of β-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 41, 1385–1394 (2020).
Ren, D. et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl Acad. Sci. USA 108, 1433–1438 (2011).
Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016).
Nurgali, K., Jagoe, R. T. & Abalo, R. Editorial: adverse effects of cancer chemotherapy: anything new to improve tolerance and reduce sequelae? Front. Pharmacol. 9, 245–245 (2018).
Li, Y. et al. Nanoparticle ferritin-bound erastin and rapamycin: a nanodrug combining autophagy and ferroptosis for anticancer therapy. Biomater. Sci. 7, 3779–3787 (2019).
Zheng, D. W. et al. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. 17, 284–291 (2017).
Tian, H. et al. A cascaded copper-based nanocatalyst by modulating glutathione and cyclooxygenase-2 for hepatocellular carcinoma therapy. J. Colloid Interface Sci. 607, 1516–1526 (2022).
Xu, R. et al. Ferroptosis/pyroptosis dual-inductive combinational anti-cancer therapy achieved by transferrin decorated nanoMOF. Nanoscale Horiz. 6, 348–356 (2021).
Feng, H. et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 30, 3411–3423.e3417 (2020).
Yu, M. et al. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci. 110, 3173–3182 (2019).
Hanahan, D. & Weinberg, R. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Riggi, N., Aguet, M. & Stamenkovic, I. Cancer metastasis: a reappraisal of its underlying mechanisms and their relevance to treatment. Ann. Rev. Paleopathol. Mech. Dis. 13, 117–140 (2018).
Fang, Y. et al. Pyroptosis: a new frontier in cancer. Biomed. Pharmacother. 121, 109595 (2020).
Kreuzaler, P. & Watson, C. J. Killing a cancer: what are the alternatives? Nat. Rev. Cancer 12, 411–424 (2012).
Shao, J. et al. Luteoloside inhibits proliferation and promotes intrinsic and extrinsic pathway-mediated apoptosis involving MAPK and mTOR signaling pathways in human cervical cancer cells. Int. J. Mol. Sci. 19, 1664 (2018).
Ren, H. et al. Design, synthesis, and characterization of an orally active dual-specific ULK1/2 autophagy inhibitor that synergizes with the PARP inhibitor olaparib for the treatment of triple-negative breast cancer. J. Med. Chem. 63, 14609–14625 (2020).
Acknowledgements
The study was supported by the Fundamental Research Funds for the National Natural Science Foundation of China (82003879, 81773889, 82073998), Science and Technology Department of Sichuan Province (2022JDRC0045), Central Universities (YJ201880), Youth Talent Promotion Project of China Association for Science and Technology (CACM-2020-QNRC1-01) and 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC21061). Parts of images generated from BioRender.com.
Author information
Authors and Affiliations
Contributions
B.H., Y.C., and L.F. conceived this study. F.P., M.L. and R.Q. wrote the manuscript. M.L., R.Q. and S.Z. made the figures and tables. P.C. performed literature searching. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, F., Liao, M., Qin, R. et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Sig Transduct Target Ther 7, 286 (2022). https://doi.org/10.1038/s41392-022-01110-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01110-y
This article is cited by
-
Melanoma biology and treatment: a review of novel regulated cell death-based approaches
Cancer Cell International (2024)
-
Targeting ferroptosis for leukemia therapy: exploring novel strategies from its mechanisms and role in leukemia based on nanotechnology
European Journal of Medical Research (2024)
-
Dysregulated CREB3 cleavage at the nuclear membrane induces karyoptosis-mediated cell death
Experimental & Molecular Medicine (2024)
-
Deep learning enables the discovery of a novel cuproptosis-inducing molecule for the inhibition of hepatocellular carcinoma
Acta Pharmacologica Sinica (2024)
-
Cross-talk between disulfidptosis and immune check point genes defines the tumor microenvironment for the prediction of prognosis and immunotherapies in glioblastoma
Scientific Reports (2024)
