
Abstract
Objective
The aim of this study was to assess the effect of moderate or severe renal impairment on the pharmacokinetic (PK) properties of milvexian.
Methods
This open-label, parallel-group study assessed the PK, safety, and tolerability of a single oral 60 mg dose of milvexian in participants with normal renal function (n = 8; estimated glomerular filtration rate [eGFR] ≥ 90 mL/min/1.73 m2) and participants with moderate (n = 8; eGFR ≥ 30 to ≤ 59 mL/min/1.73 m2) or severe (n = 8; eGFR < 30 mL/min/1.73 m2) renal impairment. Regression analysis was performed using linear regression of log-transformed PK parameters versus eGFR.
Results
Milvexian was well tolerated, with no deaths, serious adverse events, or serious bleeding reported. The maximum milvexian concentration (Cmax) was similar for all groups. Based on a regression analysis of milvexian concentration versus eGFR, participants with eGFR values of 30 and 15 mL/min/1.73 m2, respectively, had area under the curve (AUC) values that were 41% and 54% greater than in participants with normal renal function. Median time to maximum concentration (Tmax) was similar for the three groups (4.5–5.0 h). The half-life increased for participants with moderate (18.0 h) or severe (17.7 h) renal impairment compared with those with normal renal function (13.8 h).
Conclusion
A single dose of milvexian 60 mg was safe and well tolerated in participants with normal renal function and moderate or severe renal impairment. There was a similar increase in milvexian exposure between the moderate and severe renal groups.
Clinical Trials Registration
This study was registered with ClinicalTrials.gov (NCT03196206, first posted 22 June 2017).
Similar content being viewed by others

This open-label study assessed the pharmacokinetics, safety, and tolerability of milvexian, an oral small molecule inhibitor of factor XIa, in participants with normal renal function and participants with moderate or severe renal impairment. |
A single dose of milvexian 60 mg was safe and well tolerated in participants with normal renal function and in those with moderate or severe renal impairment. |
Modest increases in milvexian exposure were observed in participants with moderate and severe renal impairment, but these are not likely to be clinically relevant. |
1 Introduction
Patients with cardiovascular and thromboembolic diseases are at an increased risk of serious thrombotic events, which necessitates the use of antithrombotic therapies [1,2,3,4,5]. However, fears about an increased risk of bleeding events can lead to patients either not receiving antithrombotic therapy or being suboptimally treated with currently available agents [6,7,8]. Thus, there is a need for novel anticoagulants with an improved benefit/risk profile compared with current standards of care.
In the coagulation cascade, hemostasis and thrombosis are balanced through the regulation of blood factors, cellular components (e.g., platelets), and other coordinating proteins [9]. Thrombin plays a primary role in the coagulation cascade by activating platelet aggregation; Factors V, VIII, XI, and XIII; and forming fibrin [9, 10]. The zymogen Factor XI (FXI) is converted by thrombin to the activated protease Factor XIa (FXIa). FXIa leads to formation of a clot through direct activation of Factor IX, which in turn activates Factor X to convert prothrombin to thrombin, followed by the thrombin-mediated conversion of fibrinogen to fibrin [11, 12]. The downstream conversion of prothrombin to thrombin via FXIa is amplified through a positive feedback loop.
Modulation of FXIa may provide a novel mechanism for systemic anticoagulation with the potential to improve the benefit/risk profile of existing anticoagulants [13,14,15,16]. Based on findings from clinical and preclinical models, hemostasis is not solely dependent on the FXI pathway, and FXIa inhibitors have the potential to reduce thrombus formation [17,18,19,20]. Notably, spontaneous bleeding is rare in individuals with congenital FXI deficiencies and, for these individuals, mild bleeding after a serious injury or surgery is the only clinical manifestation of FXI deficiency [21,22,23]. Additionally, results from in vivo and clinical studies have shown that there is a reduced risk of adverse cardiovascular events and venous thromboembolism with FXI deficiency [13, 16, 24,25,26]. These findings support further investigation of FXIa inhibitors to prevent thrombotic events with a safer bleeding profile [11, 12, 27].
Milvexian (BMS-986177/JNJ-70033093) is a potentially first-in-class, oral, small-molecule that inhibits FXIa with high affinity and selectivity [28]. Milvexian is being developed to prevent thrombotic events in diverse patient populations. It has demonstrated antithrombotic activity while preserving hemostasis in preclinical models of arterial and venous thrombosis, and was generally safe and well tolerated in phase I studies in healthy participants and in individuals with hepatic impairment [29,30,31,32]. Milvexian is being investigated in an ongoing phase II study for the secondary prevention of major cardiovascular events in patients with acute ischemic stroke [33]. A separate study on the prevention of total venous thromboembolism events in patients undergoing total knee replacement surgery has been completed [34].
Patients with renal impairment are at heightened risk of bleeding when taking anticoagulant therapies [35]; however, it is unknown if this is purely due to increases in drug exposure, intrinsic features of this patient population, or a combination of both [36,37,38]. The increased risk of bleeding in patients with renal impairment has been observed even without anticoagulant therapies, with one study demonstrating a 1.5-fold increased risk of bleeding in patients with chronic kidney disease compared with those without chronic kidney disease [39]. Patients with chronic kidney disease demonstrate abnormalities in platelet physiology (e.g., α–granules) in addition to deregulation of arachidonic acid and prostaglandin metabolism that results in reduced adhesion and aggregation, increasing bleeding risk [40, 41]. Additionally, uremic patients exhibit higher concentrations of prostacyclin and increased nitric oxide generation by platelets, which both independently inhibit platelet aggregation and contribute to dysfunctional hemostasis and increased bleeding risk. Therefore, patients with renal impairment who use currently available antithrombotics may benefit from a drug with an improved safety profile, such as milvexian.
Based on results from a previous first-in-human study, renal excretion of milvexian is estimated to be below 20%, therefore renal impairment may not have a large impact on milvexian exposure [31]. However, patients with impaired renal function can display altered pharmacokinetic (PK) properties because of inhibition of several pathways of hepatic and intestinal metabolism and transport [42], and PK evaluation in certain patient populations with renal impairment is recommended by regulatory bodies [43, 44]. The future patient population for milvexian may include patients with end-stage renal disease (ESRD), necessitating investigation of the effect renal impairment has on milvexian PK properties.
This study assessed the effect of renal impairment on the PK and pharmacodynamic (PD) properties of milvexian as well as the safety and tolerability of milvexian in participants with normal renal function and moderately or severely impaired renal function.
2 Methods
2.1 Ethics
This study was conducted in accordance with Good Clinical Practice, as defined by the International Council for Harmonisation and in accordance with the ethical principles underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). The study was registered with ClinicalTrials.gov (NCT03196206, first posted 22 June 2017). The protocol, amendments, and participant informed consent received appropriate approval by the Independent Ethics Committee (IEX) and the Institutional Review Board (IRB) of IntegReview IRB (now Advarra; Columbia, MD, USA) prior to initiation of the study at the site. Prior to the beginning of the study, all participants provided written informed consent, including consent for any screening procedures conducted to establish participant eligibility for the study. The study was conducted at two clinical sites (Clinical Pharmacology of Miami LLC, Miami, FL, USA; and Orlando Clinical Research Center, Orlando, FL, USA) from 6 July 2017 to 4 March 2018.
2.2 Study Design
This was an open-label, parallel-group study to evaluate the PK, safety, and tolerability of a single dose of milvexian in participants with normal renal function and participants with moderate or severe renal impairment. As the study was nonrandomized, enrolled participants, including those not dosed, were assigned sequential participant numbers. Participants were matched by age, body weight, and sex. Participants underwent screening evaluations to determine eligibility within 21 days before study treatment administration. Eligible participants were enrolled in one of three renal function groups based on estimated glomerular filtration rate (eGFR). The eGFR was determined by the Modification of Diet in Renal Disease formula. Normal renal function was defined as an eGFR ≥ 90 mL/min/1.73 m2, moderate renal impairment was defined as an eGFR ≥ 30 to ≤ 59 mL/min/1.73 m2, and severe renal impairment was defined as an eGFR < 30 mL/min/1.73 m2. On Day 1, all participants in the three renal function groups received an oral 60 mg dose of milvexian after consumption of a standard meal. Selection of the 60 mg dose was based on in vivo preclinical pharmacology data from the rabbit electric arterial thrombosis model, differences in the affinity of milvexian for rabbit and human FXIa, and modeling results [45]. Simcyp PBPK simulator v15 was employed using a minimal physiologically based PK (PBPK) model developed for milvexian to estimate the potential increase in exposures when simulating varying degrees of renal impairment based on eGFR. The 60 mg dose is within the dose linear range of milvexian, and therefore the results from this study can be extrapolated to infer differences with renal impairment at any other dose of milvexian within the known dose-proportional range (20–200 mg) [31].
2.3 Participants
Eligible participants included men and women not of childbearing potential, aged 18–70 years, with a body mass index of 18.0–32.0 kg/m2 for participants with normal renal function or 18.0–38.0 kg/m2 for participants with moderate or severe renal impairment. In all three renal function groups, eligible participants were in good health as determined by no clinically significant deviation from normal in medical or surgical history, physical examination, electrocardiograms (ECGs), and clinical laboratory determinations, except for renal insufficiency, which was predefined by renal function group. Participants were ineligible if they had an indwelling catheter in preparation for dialysis, history of coagulopathy, gastrointestinal disease, or any major surgery within 4 weeks of study treatment administration (or planned within 2 weeks of study completion). Additionally, participants in the severe renal function group could not be on dialysis.
2.4 Safety Assessments
Safety and tolerability were assessed based on medical review of adverse event (AE) reports and the results of vital sign measurements, ECG measurements, physical examinations, and clinical laboratory tests.
2.5 Pharmacokinetic and Pharmacodynamic Assessments
The PK properties of milvexian were derived from plasma concentration versus time and urinary excretion data. Assessed parameters included maximum observed plasma concentration (Cmax), time of Cmax (Tmax), area under the plasma concentration-time curve (AUC) from time zero to time of the last quantifiable concentration (AUCt), AUC from time zero extrapolated to infinite time (AUC∞), terminal plasma half-life (T½), apparent total body clearance (CLT/F), fraction of unbound drug (fu), Cmax of free drug (Cmax fu), AUC from time zero to time of the last quantifiable concentration of free drug (AUCt fu), AUC from time zero extrapolated to infinite time of free drug (AUC∞ fu), apparent total body clearance of free drug (CLT/F fu), total amount recovered in urine (Urt), percentage dose of milvexian total amount recovered in urine (%Urt), renal clearance (CLR), and protein binding.
Activated partial thromboplastin time (aPTT) and FXI clotting activity were examined as exploratory PD biomarkers.
2.6 Clinical Laboratory Evaluations
Plasma samples were analyzed for milvexian concentration and milvexian protein binding using a validated liquid chromatography with tandem mass spectrometry (LC-MS/MS) assay at ICON Laboratory Services, Inc. (Whitesboro, NY, USA). Samples were analyzed using Analyst version 1.4.2 (Applied Biosystems, Framingham, MA, USA). LC-MS/MS assays had a lower limit of quantification of 1.0 ng/mL and an upper limit of quantification of 1000 ng/mL. Individual participant PK parameter values were derived by noncompartmental methods using a validated PK analysis program. The cumulative milvexian Urt was calculated as the summation of the product of the concentration of the analyte with the volume of urine collected over a collection interval. To determine CLR, the cumulative amount of milvexian excreted in urine was divided by the plasma AUC over the same time interval, data permitting. The designated protein-binding blood sample was analyzed for milvexian protein binding by LC-MS/MS. Validated assays were performed at Labcorp Colorado Coagulation to measure aPTT and FXI clotting activity (Englewood, CO, USA).
2.7 Sample Collection
Blood and/or urine samples were obtained at screening and on Days −1 and 4 for clinical laboratory evaluations in all three renal function groups. Additional blood and/or urine samples were obtained from participants in the moderate and severe renal impairment groups on Day 2.
Details on the timing of sample collection for PK and PD assessments are shown in Online Resource 1.
2.8 Statistical Analyses
The population for safety analysis included all participants who received one dose of milvexian. The evaluable PK population included all participants who received milvexian and from whom valid PK parameter data were obtained. The PD population included all participants who received milvexian and had any available PD biomarker data. Determination of the study sample size was not based on statistical power considerations. However, it was calculated that data from eight healthy participants and eight participants with various levels of impaired renal function would provide 80% probability for the 90% confidence interval (CI) of the geometric mean ratio (GMR) for milvexian to be within (78.58%, 127.25%), (88.69%, 112.75%), and (88.69%, 112.75%) of the GMR point estimate for Cmax, AUCt, and AUC∞, respectively.
All milvexian PK data were summarized using descriptive statistics. A regression analysis was performed using linear regression of log-transformed Cmax, AUCt, and AUC∞ versus eGFR or creatinine clearance (CrCL; estimated using the Cockcroft–Gault equation). In the regression analysis, the dependent variables were the log-transformed PK parameters, and baseline eGFR or CrCL values were the independent variables. Predicted values for each of the PK parameters and associated 90% CIs were calculated for eGFR and CrCL equal to 15, 30, and 90 (mL/min/1.73 m2 for eGFR and mL/min for CrCL), representing the lower bound cut-off for severe renal impairment, moderate renal impairment, and normal renal function, respectively. In addition, GMRs of each PK parameter’s predicted values were calculated for eGFR and CrCL values of 30: 90 (moderate renal impairment: normal renal function) and 15: 90 (severe renal impairment: normal renal function). All statistical analyses and calculations were performed using SAS® software version 9.3 (SAS Institute Inc., Cary, NC, USA).
3 Results
3.1 Participants
A total of 43 participants were enrolled in this study, of whom 24 participants (55.8%) entered the treatment period, received one dose of study treatment, and completed the study. Of the patients who did not enter the treatment period, the majority (n = 18) no longer met the study eligibility criteria; the remaining participant withdrew consent. Eight participants were enrolled in each renal function group (normal renal function, eGFR ≥ 90 mL/min/1.73 m2; moderate renal impairment, eGFR ≥ 30 to ≤ 59 mL/min/1.73 m2; and severe renal impairment, eGFR < 30 mL/min/1.73 m2). All 24 participants were included in the safety, PK, and PD analyses. Table 1 outlines the demographics and characteristics of the participants enrolled in each renal function group.
3.2 Safety and Tolerability
Administration of a single dose of milvexian 60 mg was generally safe and well tolerated across the three renal function groups. There were no deaths or serious AEs (SAEs) leading to discontinuation during the study. One SAE was reported in one participant during the screening period, and this participant was not assigned to a renal function group and did not receive milvexian. The incidence of AEs was low and was equally distributed across renal function groups. Three participants (12.5%) reported one AE following administration of milvexian. Two participants (8.3%) reported headache, and one participant (4.2%) reported somnolence. An overall summary of AEs is presented in Table 2. All AEs were mild in intensity and considered by the investigator to be related to study treatment. No treatment was required for AEs, and all AEs resolved without sequelae. No bleeding events were reported. In addition, there were no clinically relevant findings or trends in clinical laboratory test, ECG, vital sign, or physical examination results.
3.3 Pharmacokinetics
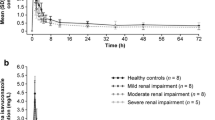
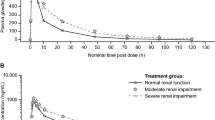
Mean milvexian plasma concentration-time profiles are shown in Fig. 1, and PK parameters are summarized in Table 3. Mean concentrations of milvexian were higher in participants with moderate or severe renal impairment compared with participants with normal renal function following a single dose of milvexian. A regression analysis indicated that Cmax was similar for all renal function groups, while AUCt and AUC∞ increased with decreasing eGFR and CrCL. From the slopes estimated in the eGFR-based regression analysis, on average, participants with eGFR values of 30 and 15 mL/min/1.73 m2 would have a 39% and 50% increase in AUCt and a 41% and 54% increase in AUC∞, respectively, compared with participants with normal renal function (eGFR 90 mL/min/1.73 m2) (Fig. 2). From the estimated slopes in the CrCL-based regression analysis, on average, participants with CrCL values of 30 and 15 mL/min would have a 38% and 49% increase in AUCt and a 40% and 52% increase in AUC∞, respectively, compared with a participant with normal renal function (CrCL 90 mL/min) (Fig. 2).

Mean (± SD) milvexian plasma concentration versus time profile. SD standard deviation.

Predicted values of milvexian PK parameters based on renal impairment from the regression analysis of eGFRa and CrCLb in a moderate and b severe renal impairment. aPredicted values for each PK parameter and associated 90% CI for eGFR (mL/min/1.73 m2) equal to 15, 30, and 90 were obtained from the linear regression model. GMRs of each PK parameter predicted values were calculated for eGFR values of 30: 90 (moderate renal impairment: normal renal function) and 15: 90 (severe renal impairment: normal renal function). bPredicted values for each PK parameter and associated 90% CI for CrCL (mL/min) equal to 15, 30, and 90 were obtained from the linear regression model. GMRs of each PK parameter predicted values were calculated for CrCL values of 30: 90 (moderate renal impairment: normal renal function) and 15: 90 (severe renal impairment: normal renal function). PK pharmacokinetic, eGFR estimated glomerular filtration rate, CrCL creatinine clearance, GMR geometric mean ratio, CI confidence interval, Cmax maximum observed concentration, AUCt area under the plasma concentration-time curve from time zero to time of the last quantifiable concentration, AUC∞ area under the plasma concentration-time curve from time zero extrapolated to infinite time
Median Tmax was similar across all renal function groups. As a result of decreasing CLT/F with decreasing renal function (15.3, 11.6, and 8.10 L/h in participants with normal renal function, moderate renal impairment, and severe renal impairment, respectively), the T½ was slightly longer for the moderate (17.7 h) and severe (18.0 h) renal impairment groups compared with the normal renal function group (13.8 h). The CLR was lower for both the moderate and severe renal impairment groups compared with the normal renal function group. Correspondingly, excretion in the urine, as measured by %Urt, was lower in the moderate and severe renal impairment groups compared with those with normal renal function. Protein binding was similar for all renal function groups and the unbound parameters showed similar results across the different renal function groups.
3.4 Pharmacodynamics
Mean baseline (predose Day 1) aPTT values were similar across renal function groups and ranged from 23.9 to 25.3 s. Administration of a single oral dose of milvexian resulted in a concentration-dependent prolongation of aPTT that was similar across all renal function groups (Fig. 3). Mean peak aPTT values were achieved at 5, 5, and 6 h postdose, corresponding to percentage changes from baseline of 127%, 97%, and 100% for the normal renal function, moderate renal impairment, and severe renal impairment groups, respectively.

Mean (± SD) aPTT versus time profile. SD standard deviation, aPTT activated partial thromboplastin time
Mean baseline (predose Day 1) FXI clotting activity values were similar across renal function groups and ranged from 105.4 to 115.7%. Administration of a single oral dose of milvexian resulted in a concentration-related decrease in FXI clotting activity (Fig. 4). The magnitude of decrease appeared to be similar across renal function groups. Mean nadir FXI clotting activity values were achieved at 5, 5, and 7 h postdose for participants with normal renal function, and moderate and severe renal impairment corresponding to percentage changes from baseline of 39.4%, 31.9%, and 31.8%, respectively.

Mean (± SD) FXI clotting activity versus time profile. SD standard deviation, FXI Factor XI
4 Discussion
This study evaluated the PK, safety, and tolerability of a single, 60 mg dose of milvexian in participants with normal renal function and participants with moderate or severe renal impairment. Milvexian is a potentially first-in-class, oral, small-molecule FXIa inhibitor being developed to prevent and treat thrombotic events across diverse patient populations [28]. FXIa inhibitors have the potential to improve the benefit/risk profile of existing anticoagulants by reducing thrombus formation without causing an increase in bleeding [13,14,15,16,17,18,19,20]. There is additional unmet need for safer anticoagulants in renal impairment patients with concomitant cardiovascular disease as these patients have a higher intrinsic risk of bleeding [39], and this risk may further increase with currently available anticoagulants [36,37,38].
Administration of a single dose of 60 mg milvexian was generally safe and well tolerated in participants with normal renal function and participants with moderate or severe renal impairment. The incidence of AEs was low and was equally distributed independent of renal function. Of note, no bleeding events were reported. Bleeding risk in patients receiving currently available anticoagulants is variable [46], but patients with renal impairment are at a higher risk of bleeding events generally and may benefit from dose adjustments of anticoagulants [47, 48]. Safety and tolerability results from the current study of milvexian add to evidence from a phase I study in healthy volunteers as well as a phase I study in participants with hepatic impairment that also demonstrated the safety and tolerability of milvexian [31, 32].
Previous studies demonstrated low renal excretion of milvexian (< 20%), which suggested that renal impairment may not have a significant impact on exposure [31]. Results from the regression analysis in the current study indicate that Cmax and Tmax will be similar across all renal function groups, with an increase in overall exposure in the moderate and severe renal impairment groups of approximately 40% and 50%, respectively. The corresponding predicted increase in milvexian drug exposure in patients with renal impairment is similar to results reported for other anticoagulants [46]; for example, rivaroxaban has shown a 56% increase in exposure in patients undergoing dialysis compared with healthy participants [49]. Similarly, apixaban has shown a 44% increase in exposure in patients with severe renal impairment (CrCL of 15 mL/min) compared with healthy participants [50]. Overall, the magnitude of increases in milvexian exposure may not be clinically relevant in patients with renal impairment, pending assessment of exposure–response.
Administration of a single dose of milvexian resulted in a concentration-dependent aPTT prolongation that was similar across the three renal function groups. Similarly, administration of milvexian led to a concentration-related decrease in FXI clotting activity across the three renal function groups. Results from the PD assessments suggest that renal function does not alter the PK/PD relationship with either aPTT or FXI clotting activity.
A limitation of this study is that the PK data were obtained from a small sample of participants. However, as noted earlier, the sample size for this study was based on the precision estimate of the GMRs of specific PK measurements of milvexian for a group with renal impairment versus a healthy group. Despite a modest sample size, regression analyses from the current study, which included eight healthy participants and 16 participants with various levels of impaired renal function, provided 90% CIs within the specified range. In addition, baseline characteristics were generally balanced across groups for age, sex, race, and body mass index, and as such would not contribute to any significant changes in PK across renal groups. Future studies could include participants with ESRD to further investigate the impact of renal impairment on the PK and PD properties of milvexian.
5 Conclusions
The results of this study demonstrated that a single dose of 60 mg milvexian was generally safe and well tolerated in participants with normal renal function and participants with moderate or severe renal impairment. The PK analysis indicated that Cmax and Tmax were similar across renal function groups, and the PD analysis suggested that concentration-dependent prolongation of aPTT and concentration-dependent decreases in FXI clotting activity were similar across renal function groups. Although there was a modest increase in exposure, as measured by AUC, in the renal impairment groups, these values are not likely to be clinically relevant.
References
Cavallari I, Patti G. Efficacy and safety of oral anticoagulation in elderly patients with atrial fibrillation. Anatol J Cardiol. 2018;19(1):67–71. https://doi.org/10.14744/AnatolJCardiol.2017.8256.
Kapil N, Datta YH, Alakbarova N, Bershad E, Selim M, Liebeskind DS, et al. Antiplatelet and anticoagulant therapies for prevention of ischemic stroke. Clin Appl Thromb Hemost. 2017;23(4):301–18. https://doi.org/10.1177/1076029616660762.
Gurbel PA, Fox KAA, Tantry US, Ten Cate H, Weitz JI. Combination antiplatelet and oral anticoagulant therapy in patients with coronary and peripheral artery disease. Circulation. 2019;139(18):2170–85. https://doi.org/10.1161/CIRCULATIONAHA.118.033580.
Eisen A, Giugliano RP, Braunwald E. Updates on acute coronary syndrome: a review. JAMA Cardiol. 2016;1(6):718–30. https://doi.org/10.1001/jamacardio.2016.2049.
Faxon DP. Use of antiplatelet agents and anticoagulants for cardiovascular disease: current standards and best practices. Rev Cardiovasc Med. 2005;6(Suppl 4):S3-14.
Bracey A, Shatila W, Wilson J. Bleeding in patients receiving non-vitamin K oral anticoagulants: clinical trial evidence. Ther Adv Cardiovasc Dis. 2018;12(12):361–80. https://doi.org/10.1177/1753944718801554.
Shpak M, Ramakrishnan A, Nadasdy Z, Cowperthwaite M, Fanale C. Higher incidence of ischemic stroke in patients taking novel oral anticoagulants. Stroke. 2018;49(12):2851–6. https://doi.org/10.1161/STROKEAHA.118.022636.
Naqvi IA, Kamal AK, Rehman H. Multiple versus fewer antiplatelet agents for preventing early recurrence after ischaemic stroke or transient ischaemic attack. Cochrane Database Syst Rev. 2020;(8):CD009716. https://doi.org/10.1002/14651858.CD009716.pub2
Ten Cate H, Hackeng TM, de Frutos PG. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost. 2017;117(7):1265–71. https://doi.org/10.1160/TH17-02-0079.
Brass LF. Thrombin and platelet activation. Chest. 2003;124(3 Suppl):18S-25S. https://doi.org/10.1378/chest.124.3_suppl.18s.
Lowenberg EC, Meijers JC, Monia BP, Levi M. Coagulation factor XI as a novel target for antithrombotic treatment. J Thromb Haemost. 2010;8(11):2349–57. https://doi.org/10.1111/j.1538-7836.2010.04031.x.
Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115(13):2569–77. https://doi.org/10.1182/blood-2009-09-199182.
Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111(8):4113–7. https://doi.org/10.1182/blood-2007-10-120139.
Yang DT, Flanders MM, Kim H, Rodgers GM. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol. 2006;126(3):411–5. https://doi.org/10.1309/QC259F09UNMKVP0R.
Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol. 2016;36(7):1316–22. https://doi.org/10.1161/ATVBAHA.116.306925.
Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105(2):269–73. https://doi.org/10.1160/TH10-05-0307.
Pollack CV Jr, Kurz MA, Hayward NJ. EP-7041, a factor XIa inhibitor as a potential antithrombotic strategy in extracorporeal membrane oxygenation: a brief report. Crit Care Explor. 2020;2(9): e0196. https://doi.org/10.1097/CCE.0000000000000196.
Sakimoto S, Hagio T, Yonetomi Y, Ono T, Koyama S, Hashimoto A, et al. ONO-8610539, an injectable small-molecule inhibitor of blood coagulation factor XIa, improves cerebral ischemic injuries associated with photothrombotic occlusion of rabbit middle cerebral artery. Presented at the International Stroke Conference; 21–24 February 2017; Houston, TX, USA.
Thomas D, Kanefendt FS, Schwers S, Unger S, Yassen A, Boxnick S. First evaluation of the safety, pharmacokinetics and pharmacodynamics of BAY 2433334 a small molecule targeting coagulation factor XIa in healthy young male participants. Presented at the International Society on Thrombosis and Haemostasis (ISTH) 2020 Virtual Congress; 12–14 July 2020.
Weitz JI, Bauersachs R, Becker B, Berkowitz SD, Freitas MCS, Lassen MR, et al. Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: the FOXTROT randomized clinical trial. JAMA. 2020;323(2):130–9. https://doi.org/10.1001/jama.2019.20687.
Rosenthal RL, Dreskin OH, Rosenthal N. Plasma thromboplastin antecedent (PTA) deficiency; clinical, coagulation, therapeutic and hereditary aspects of a new hemophilia-like disease. Blood. 1955;10(2):120–31.
Franchini M, Veneri D, Lippi G. Inherited factor XI deficiency: a concise review. Hematology. 2006;11(5):307–9. https://doi.org/10.1080/10245330600921964.
Lee SE, Choi YJ, Chi SI, Kim HJ, Seo KS. Factor XI deficiency and orthognathic surgery: a case report on anesthesia management. J Dent Anesth Pain Med. 2015;15(1):25–9. https://doi.org/10.17245/jdapm.2015.15.1.25.
Minnema MC, Friederich PW, Levi M, von dem Borne PA, Mosnier LO, Meijers JC, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101(1):10–4. https://doi.org/10.1172/JCI781.
Wang X, Smith PL, Hsu MY, Gailani D, Schumacher WA, Ogletree ML, et al. Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. J Thromb Haemost. 2006;4(9):1982–8. https://doi.org/10.1111/j.1538-7836.2006.02093.x.
Preis M, Hirsch J, Kotler A, Zoabi A, Stein N, Rennert G, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129(9):1210–5. https://doi.org/10.1182/blood-2016-09-742262.
Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res. 2016;141(Suppl 2):S8–11. https://doi.org/10.1016/S0049-3848(16)30354-1.
Dilger AK, Pabbisetty KB, Corte JR, De Lucca I, Fang T, Yang W, et al. Discovery of milvexian, a high-affinity, orally bioavailable inhibitor of factor XIa in clinical studies for antithrombotic therapy. J Med Chem. 2022;65(3):1770–85. https://doi.org/10.1021/acs.jmedchem.1c00613.
Wong PC, Crain EJ, Dilger AK, Exler RR, Ewing WR, Gordon D, et al. Small-molecule factor XIa inhibitor, BMS-986177/JNJ-70033093, prevents and treats arterial thrombosis in rabbits at doses that preserve hemostasis. Poster PB0121. Presented at the International Society on Thrombosis and Haemostasis (ISTH) 2020 Virtual Congress; 12–14 July 2020.
Wang X, Qiu L, Du F, Shukla N, Nawrocki A, Chintala M. Antithrombotic effects of a novel small molecule FXIa inhibitor BMS-986177/JNJ-70033093 in a rabbit AV-shunt model of thrombosis. Poster PB0179. Presented at the International Society on Thrombosis and Haemostasis (ISTH) 2020 Virtual Congress; 12–14 July 2020.
Perera V, Wang Z, Luettgen J, Li D, DeSouza M, Cerra M, et al. First-in-human study of milvexian, an oral, direct, small molecule factor XIa inhibitor. Clin Transl Sci. 2021;15(2):330–42. https://doi.org/10.1111/cts.13148.
Perera V, Abelian G, Li D, Wang Z, Zhang L, Lubin S, et al. Single-dose pharmacokinetics of milvexian in participants with mild or moderate hepatic impairment compared with healthy participants. Clin Pharmacokinet. 2022. https://doi.org/10.1007/s40262-022-01110-9.
ClinicalTrials.gov. NCT03766581. A study on BMS-986177 for the prevention of stroke in patients receiving aspirin and clopidogrel (AXIOMATIC-SSP). https://clinicaltrials.gov/ct2/show/NCT03766581 Accessed 1 Sep 2020.
Weitz JI, Strony J, Ageno W, Gailani D, Hylek EM, Lassen MR, et al. Milvexian for the prevention of venous thromboembolism. N Engl J Med. 2021;385(23):2161–72. https://doi.org/10.1056/NEJMoa2113194.
Corpataux N, Spirito A, Gragnano F, Vaisnora L, Galea R, Svab S, et al. Validation of high bleeding risk criteria and definition as proposed by the academic research consortium for high bleeding risk. Eur Heart J. 2020;41(38):3743–9. https://doi.org/10.1093/eurheartj/ehaa671.
Jackevicius CA, Lu L, Ghaznavi Z, Warner AL. Bleeding risk of direct oral anticoagulants in patients with heart failure and atrial fibrillation. Circ Cardiovasc Qual Outcomes. 2021;14(2):e007230. https://doi.org/10.1161/CIRCOUTCOMES.120.007230.
Vio R, Proietti R, Rigato M, Calo LA. Clinical evidence for the choice of the direct oral anticoagulant in patients with atrial fibrillation according to creatinine clearance. Pharmaceuticals (Basel). 2021;14(3):279. https://doi.org/10.3390/ph14030279.
Alhousani M, Malik SU, Abu-Hashyeh A, Poznanski NJ, Al-Hasan S, Roth DF, et al. Using oral anticoagulants among chronic kidney disease patients to prevent recurrent venous thromboembolism: a systematic review and meta-analysis. Thromb Res. 2021;198:103–14. https://doi.org/10.1016/j.thromres.2020.11.036.
Ocak G, Rookmaaker MB, Algra A, de Borst GJ, Doevendans PA, Kappelle LJ, et al. Chronic kidney disease and bleeding risk in patients at high cardiovascular risk: a cohort study. J Thromb Haemost. 2018;16(1):65–73. https://doi.org/10.1111/jth.13904.
Lutz J, Menke J, Sollinger D, Schinzel H, Thurmel K. Haemostasis in chronic kidney disease. Nephrol Dial Transplant. 2014;29(1):29–40. https://doi.org/10.1093/ndt/gft209.
Hedges SJ, Dehoney SB, Hooper JS, Amanzadeh J, Busti AJ. Evidence-based treatment recommendations for uremic bleeding. Nat Clin Pract Nephrol. 2007;3(3):138–53. https://doi.org/10.1038/ncpneph0421.
Dreisbach AW, Lertora JJ. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4(8):1065–74. https://doi.org/10.1517/17425255.4.8.1065.
European Medicines Agency. Committee for Medicine Products for Human Use (CHMP). Note for Guidance on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Renal Function. https://www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-evaluation-pharmacokinetics-medical-products-patients-impaired-renal-function_en.pdf. Accessed 24 Mar 21.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for Industry. Pharmacokinetics in Patients with Impaired Renal Function—Study Design, Data Analysis, and Impact on Dosing and Labeling. https://www.fda.gov/media/71334/download. Accessed 24 Mar 2021.
Wong PC, Crain EJ, Bozarth JM, Wu Y, Dilger AK, Wexler RR, et al. Milvexian, an orally bioavailable, small-molecule, reversible, direct inhibitor of factor XIa: in vitro studies and in vivo evaluation in experimental thrombosis in rabbits. J Thromb Haemost. 2022;20(2):399–408. https://doi.org/10.1111/jth.15588.
Weir MR, Kreutz R. Influence of renal function on the pharmacokinetics, pharmacodynamics, efficacy, and safety of non-vitamin K antagonist oral anticoagulants. Mayo Clin Proc. 2018;93(10):1503–19. https://doi.org/10.1016/j.mayocp.2018.06.018.
Fox KA, Piccini JP, Wojdyla D, Becker RC, Halperin JL, Nessel CC, et al. Prevention of stroke and systemic embolism with rivaroxaban compared with warfarin in patients with non-valvular atrial fibrillation and moderate renal impairment. Eur Heart J. 2011;32(19):2387–94. https://doi.org/10.1093/eurheartj/ehr342.
Zeitouni M, Giczewska A, Lopes RD, Wojdyla DM, Christersson C, Siegbahn A, et al. Clinical and pharmacological effects of apixaban dose adjustment in the ARISTOTLE trial. J Am Coll Cardiol. 2020;75(10):1145–55. https://doi.org/10.1016/j.jacc.2019.12.060.
Dias C, Moore KT, Murphy J, Ariyawansa J, Smith W, Mills RM, et al. Pharmacokinetics, pharmacodynamics, and safety of single-dose rivaroxaban in chronic hemodialysis. Am J Nephrol. 2016;43(4):229–36. https://doi.org/10.1159/000445328.
Chang M, Yu Z, Shenker A, Wang J, Pursley J, Byon W, et al. Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of apixaban. J Clin Pharmacol. 2016;56(5):637–45. https://doi.org/10.1002/jcph.633.
Acknowledgements
The authors thank Eric Lawitz, MD, and Thomas Marbury, MD, for their contributions to this study. Medical writing support was provided by Alana Simorellis, PhD, of Cello Health Communications/MedErgy, and was funded by Bristol Myers Squibb and Janssen Global Services, LLC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
This study was conducted in accordance with Good Clinical Practice, as defined by the International Council for Harmonisation and in accordance with the ethical principles underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). The study was registered with ClinicalTrials.gov (NCT03196206, first posted 22 June 2017). The protocol, amendments, and participant informed consent received appropriate approval by the IEX and the IRB of IntegReview IRB (now Advarra; Columbia, MD, USA) prior to initiation of the study at the site. The study was conducted at two clinical sites (Clinical Pharmacology of Miami LLC, Miami, FL, USA; and Orlando Clinical Research Center, Orlando, FL, USA) from 6 July 2017 to 4 March 2018.
Consent to participate
Prior to the beginning of the study, all participants provided written informed consent, including consent for any screening procedures conducted to establish participant eligibility for the study.
Consent for publication
Not applicable.
Availability of data and materials
The data that support the findings of this study are not publicly available due to privacy or ethical restrictions. Please contact the corresponding author, Vidya Perera, for additional information.
Code availability
Not applicable.
Conflict of interest
VP, GA, DL, ZW, SL, AB, and BM are full-time employees of Bristol Myers Squibb. LZ is a full-time employee of Janssen.
Funding
This study was sponsored by Bristol Myers Squibb.
Author contributions
VP, GA, DL, ZW, LZ, SL, AB, and BM contributed to the study design and concept, data analysis and review, or data interpretation; critically reviewed the manuscript for intellectual content; and approved the final manuscript for publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Perera, V., Abelian, G., Li, D. et al. Single-Dose Pharmacokinetics of Milvexian in Participants with Normal Renal Function and Participants with Moderate or Severe Renal Impairment. Clin Pharmacokinet 61, 1405–1416 (2022). https://doi.org/10.1007/s40262-022-01150-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01150-1