
Abstract
The alteration of structural connectivity in fluvial networks is important for the genetic dynamics of aquatic species. Exploring the effects of network fragmentation through genetic analysis is crucial to assess the conservation status of riverine species. In this study, we investigated the genetic consequences of the altered connectivity of brown trout in the Deva–Cares catchment (northern Spain). We investigated (1) genetic diversity, (2) genetic differentiation and genetic structure, (3) migration rates and effective population size and (4) genetic differentiation and riverscape characteristics. Analysis of the genetic variation among 197 individuals from the 13 study sites revealed a high degree of genetic differentiation (FST = 0.181). Below-barrier study sites had higher genetic diversity and lower FST values, while headwater and above-barrier study sites had lower genetic diversity and higher FST values. Most of the genetic groups identified were separated by one or more impermeable barriers. We reported an abrupt decrease in genetic diversity and effective population size in upper course tributaries and isolated reaches. Likewise, a downstream-biased gene flow was found, and it was most likely related to the fragmentation caused by barriers, since the results from migration indicated that gene flow between groups without impermeable barriers was higher bidirectionally. Isolation by impermeable barriers played a more important role than hydrological distance in determining the genetic structure. Most of the genetic groups showed small effective population sizes. Genetic analysis at the river network scale provides evidence for the role of barriers in determining genetic diversity patterns, highlighting the importance of maintaining and restoring river longitudinal connectivity.
Similar content being viewed by others

Introduction
Rivers are dynamic and hierarchical ecosystems interacting along four dimensions (longitudinal, vertical, lateral and temporal; Ward 1989), in which connectivity is considered one of the fundamental properties determining biodiversity and ecosystem functioning patterns (Moore 2015; Tonkin et al. 2018). Connectivity can be divided into structural or functional connectivity (Baguette and Van Dyck 2007). On the one hand, structural connectivity refers to the physical relationship among landscape elements or patches (Eros et al. 2012). Functional connectivity, on the other hand, refers to the response of biological elements (genes, individuals, populations or communities) to landscape elements (Tischendorf et al. 2000). Changes in the structural connectivity of dendritic riverscapes are of great importance for the population demographic and genetic dynamics of riverine species (Fagan 2002; Campbell Grant et al. 2007) due to the branching structure, nested watersheds and stream segments connected by flow in river systems (Fagan 2002). Although river networks are naturally fragmented longitudinally (e.g., waterfalls), human actions have further divided these ecosystems (Fuller et al. 2015), and the fragmentation of rivers is increasing (Nilsson et al. 2005). One of the most important human impacts in this regard is the presence of longitudinal barriers, which may be physical (e.g., dams), chemical (e.g., pollution plumes), biological (e.g., diseases) or thermal (e.g., stream temperature) and, may change in permeability, location and abundance (Fuller et al. 2015). The fragmentation of river networks by longitudinal barriers can potentially alter the population demographic dynamics of aquatic organisms by reducing upstream and downstream migration and changing water quality or habitat conditions (Poff and Hart 2002). Moreover, fragmented ecosystems might alter population genetic dynamics by directly influencing gene flow and genetic drift (Dixo et al. 2009). These genetic consequences can make populations more vulnerable to stochastic events because the long-term persistence of populations depends on sufficient genetic diversity to adapt and survive in changing environments (Hughes et al. 2008).
Landscape genetics allow us to examine the relationship between structural and functional connectivity as an efficient tool to investigate the effects of landscape characteristics on population connectivity (Manel et al. 2003; Nevill et al. 2019). In river networks, freshwater species gene flow is constrained by a number of factors, such as catchment geomorphology, the dendritic structure of the river network (Chaput-Bardy et al. 2008) and species dispersal traits (Tonkin et al. 2018). Fragmentation leads to a decline in gene flow, causing a loss of genetic diversity, an increase in inbreeding and an increase in the risk of local population extinctions (Keyghobadi 2007; Pavlova et al. 2017). The permeability of longitudinal barriers to gene flow (which is often not available or difficult to evaluate), the time period of the barrier (Yamamoto et al. 2004) or the quantity and quality of patches can determine the severity of the effects on the genetic structure of riverine populations. Even in the absence of longitudinal barriers, distance itself is also likely to have an effect on functional connectivity, some populations can be isolated by distance (Wright 1943), and gene flow can decrease with distance among locations (e.g., Sotola et al. 2017).
Organisms that disperse through water are more likely to be more influenced by the structure of the river network than are those that disperse over land (Tonkin et al. 2018). Most fish species need to occupy multiple habitats over their life cycle (spawning, feeding, etc.), and migratory fishes cover long distances. Thus, river network structure and the loss of structural connectivity might be more important to fish species (Sheer and Steel 2006) than to other riverine organisms. Salmonid species are considered interesting models for investigating the influence of riverscape characteristics on their population genetic structure (e.g., Neville et al. 2006) due to its life history and their importance from an ecological and socioeconomic point of view, with many species characterized as the top-predator fish in many rivers (Sánchez-Hernández 2016) and/or important sport-fishing species (Almodovar and Nicola 1998).
The within-river population genetic structure of salmonid populations has been explained by contrasting evolutionary models: member-vagrant (Primmer et al. 2006), metapopulation (Dunham and Rieman 1999), a mix of both (Garant et al. 2000) or panmixia (Addison and Wilson 2010). The member-vagrant model proposes that spawning areas are key determinants of population genetic structure and that local adaptation and selection favor individuals who return to their natal spawning areas, where individuals who complete the process are considered members and those who do not return are considered vagrants. Evolution is indicated by a strong and temporally stable pattern of population genetic structure with clear isolation by distance (Garant et al. 2000). The metapopulation hypothesis proposes a set of local populations largely independent but interconnected by migration and subjected to extinction-recolonization processes (Hanski 1998). Evolution is indicated by weak genetic differentiation, absence of temporal stability and absence of isolation by distance (Garant et al. 2000). Panmixia is the absence of a genetic structure due to the existence of gene flow and unrestricted spawning migration (Griffiths et al. 2009), representing the null model with which metapopulation and member-vagrant are often compared to detect them. Information about the evolutionary model that explains genetic differentiation in salmonid populations is important to understand how river network fragmentation and loss of structural connectivity may influence population persistence.
Brown trout populations are composed of both stationary (low mobility and dispersal linked to the home range) and mobile (high mobility) individuals (Rodríguez 2002; Aparicio et al. 2018). Moreover, brown trout exhibit very plastic behavior, with different population types (lake-dwelling, stream-resident and sea-migrating) coexisting in the same river network (Klemetsen et al. 2003). Iberian populations located on the southern limits of the species' distribution, due to their drainage isolation, contain a large proportion of the genetic, life-history patterns, and habitat preference diversity of the species (Almodóvar et al. 2006; Lobón-Cerviá and Sanz 2017). The impacts of hydrologic alterations and climate change can have consequences, reducing population sizes and functional connectivity (e.g., Almodóvar et al. 2012). In addition, because of their higher vulnerability to stochastic demographic and environmental events, the risk of local extinction is expected to increase (Rieman et al. 1993). Understanding the impacts of barriers and the loss of structural connectivity on brown trout genetic patterns at a river network scale is essential to advance our understanding of how populations are genetically structured and to implement appropriate and efficient management and conservation strategies. However, to our knowledge, no detailed empirical research exists on the responses of native brown trout to habitat fragmentation over a whole river network (from headwaters to the river mouth) considering river geometry and longitudinal connectivity (natural and anthropogenic barriers).
The present study aimed to investigate and describe the population genetic structure of native brown trout in the Deva–Cares River network and quantify the strength of the genetic patterns with those of the structural connectivity patterns. We investigate the following: (1) genetic diversity of brown trout in the catchment, (2) genetic differentiation and genetic structure, (3) migration rates and effective population size, and (4) genetic differentiation and riverscape characteristics. We hypothesized that changes in the structural connectivity by longitudinal impermeable obstacles may alter the functional connectivity and would reduce gene flow and genetic diversity, resulting in decreasing effective population size and increasing genetic differentiation in the isolated study sites above barriers. We also considered different conservation and management strategies for the brown trout in the Deva–Cares catchment based on the obtained results.
Materials and methods
Study area
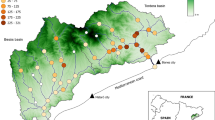
The study area comprises the Deva–Cares catchment (northern Spain; 1200 km2), which drains into the Cantabrian Sea (Fig. 1). Its geomorphology and hydrogeology are conditioned by the presence of the karst massif of the Picos de Europa, which acts partly as a natural division between the Deva River (main axis; length = 64 km) and its main tributary, the Cares River (length = 54 km). The total channel length of the river network is 1675 km (662.5 km of perennial rivers and 1012.5 km of temporary rivers; González-Ferreras and Barquín 2017). Previous studies have described the Deva–Cares catchment in detail (Estevez et al. 2017; González-Ferreras 2019; González-Ferreras et al. 2019), and only information relevant to this study is provided here. The study area includes several longitudinal barriers, both anthropogenic and natural (Fig. 1), but other major significant pressures, such as stream habitat alteration, hydrological regime alteration or exotic species, are mostly absent. This situation makes this catchment a suitable study area to achieve the objectives of the study.

Location of surveyed study sites between August and October 2014 and longitudinal barriers in the Deva–Cares catchment (Deva subcatchment S12, S11, S10, S9, S8 and S7; Cares subcatchment S6, S5, S4, S3, S2 and S1), northern Spain. Numbers with asterisks represent the barriers to which there is evidence of the date of their construction (1* Cordiñanes Dam-1995. 2* Cain Dam-1921, 3* Poncebos Dam-1958 and 4* Niserias Dam-1973)
Previous considerations: barrier information and stocking
The river network that connects all study sites includes 29 longitudinal barriers (6 are natural barriers; waterfalls and/or cascades and the rest are anthropogenic barriers; dams and/or weirs). Permeable and impermeable barriers are defined according to their characteristics, following a national standard for evaluating fish pass permeability (MAGRAMA 2015). Permeable barriers were considered obstacles with a maximum height of jump ≤ 1 m, a minimum depth of the pool ≥ 1.25 m height of jump, and a maximum width crest ≤ 0.5 m. Obstacles with functional fish passage structures were also considered permeable. Moreover, all obstacles present in the zone with the presence of the anadromous species Salmo salar, Linnaeus 1758 (river network from the outlet until the first impermeable barriers in Deva, Cares and Casaño Rivers; Fig. 1) were considered permeable for brown trout. The presence of S. salar was based on field data (González-Ferreras et al. 2019 and data from Government of Cantabria) and on the bibliography of the limits of its current distribution (Alvarez et al. 2010). We consider that obstacles were permeable for both species, as salmonid swimming performance curves are given as a function of their length rather than species (Larinier (2002) based on Beach (1984)).
Due to the absence of data collected before fragmentation, we must take certain considerations into account when interpreting the results of this study. Of the 23 anthropogenic barriers, we have proof of the date of construction of only four of them (see Fig. 1; information obtained from Gutiérrez et al. 2020), which represent the barriers dedicated to hydroelectric power. There is no record of the date of construction of the remaining obstacles. Before the construction of these impermeable barriers, we are not aware of any impermeable natural barriers in these study sites (beyond the natural barriers inventoried in this study: two permeable “downstream of study sites S5 and S11” and four impermeable “downstream of study sites S3, S5, S7 and S8”). In addition, the presence of historical records of S. salar upstream of the anthropogenic barriers that limit their current distribution may be an indication that some of the barriers contribute to the loss of functional and structural connectivity and a reduction of accessible habitat. It should also be added that the rivers of the Deva–Cares catchment are characterized by high channel gradients and short channels with the presence of rapid zones that could present a certain degree of temporal impermeability in some areas (i.e., in summer with low flows or high flow seasons).
Regarding the stocking activities, it is important to note that the Deva–Cares catchment is divided into three different administrative regions [Castilla y León (≈ 150 km2), Principado de Asturias (≈ 415 km2) and Cantabria (≈ 640 km2); see Fig. 1]. Cantabria first carried out stocking in the Deva–Cares catchment in 2021 (after the sampling date of this study) by releasing native stock (personal communication from the Cantabria Regional Government). Principado de Asturias (in collaboration with fishermen’s associations) has carried out stocking since the 1980s using fry releases of native stock from 1995 onward (personal communication from the Principado de Asturias Government). We are not aware of the data from Castilla y León, but releases of foreign stock have been prohibited for more than 15 years throughout the study area (Law 42/2007 of Natural Patrimony and Biodiversity). The introgression of foreign genomes into native gene pools are absent in most of the previous genetic studies performed in the Deva–Cares catchment (see Moran et al. 1993; Izquierdo et al. 2006; Almodóvar et al. 2008), except for the study by Horreo and Garcia–Vazquez (2011). These authors found a small percentage of alleles of nonnative origin presence, but their results suggest that it was introduced several generations ago, assigning the individuals to the population from which they were sampled, not to a hatchery. Thus, previous studies demonstrate that the targeted brown trout population in this study may be considered generally unaffected by past restocking.
Fish survey
Between August and October 2014, 197 brown trout were collected by electrofishing from 13 study sites in the Deva–Cares catchment (Fig. 1). Although trout are present in a larger area of the fluvial network (e.g., potential habitat in perennial fluvial network not connecting sample sites; see Fig. 1), we believe that the choice of these study sites is appropriate to achieve our objectives. The selection of these study sites was performed to cover the whole river network (from headwaters to the river mouth), and the selection includes reaches above and below permeable and impermeable barriers (both natural and anthropogenic). Study sites are located in the main rivers of the subcatchments (Cares and Deva Rivers) and in three tributaries of the main rivers (Duje, Casaño and Salvoron Rivers). Moreover, one site in the Deva River after its confluence with the Cares River was included (S13; Strahler order 7). The tributaries study sites (S3, S5 and S7) and the most upstream study sites of the main rivers (S1 and S8) have a Strahler order ranging from 3 to 4, while the remaining study sites have a Strahler order ranging from 5 to 6. Each sampling was carried out in a minimum area that included a sequence of mesohabitats that was representative of the river reach. Natural barriers or nets were used to block the river reach in the upstream and downstream directions at each study site. Individuals were sedated with eugenol, and the adipose fin was removed and preserved in individually labeled tubes with 96% ethanol and stored at – 16 °C for subsequent DNA analysis. Fish were placed into holding boxes until they recovered, and then they were returned alive to the stream near the point of capture.
River network and spatial data
The river network of the study area was obtained following a Virtual Watershed Approach (Barquín et al. 2015). The river network (divided into reaches; mean length of 500 m) is a digital representation of the surface water drainage network derived from a 25-m Digital Elevation Model “DEM” (see González-Ferreras and Barquín 2017 for more information) that incorporates all the spatial information used to test for relationships between genetic differentiation and riverscape characteristics. This spatial information refers to the geographical locations of the study sites and barriers in the river network as well as topographical variables such as slope and elevation data (derived from the DEM) in the reaches of the river network where the study sites are located. Data used for testing relationships between genetic differentiation and riverscape characteristics referring to the hydrological distances (length of the river network) and the number of barriers (permeable, impermeable and total) between the study sites and the differences in slope and elevation were calculated using ArcGIS Desktop 10.2 and ESRI’s ArcPy Python module (ESRI 2014).
Molecular analyses
Total DNA was extracted from adipose fins using a DNeasy Tissue Kit (QIAGEN, IZASA, Spain) and then stored at −20 °C. The quality and concentration of DNA were determined by spectrophotometry and verified by 0.8% agarose gel electrophoresis. Twelve microsatellite loci were amplified in three separate multiplex reactions (Table S1) with forward primers labeled with a fluorescent dye. For each multiplex reaction, PCR multiplex was carried out in a final volume of 20 μl containing 10 μl of DNA AmpliTools Multiplex Master Mix (Biotools, Spain), 0.15–0.30 μM of each primer and 50–100 ng genomic DNA template. Amplification reactions were performed using the following conditions: 95 °C for 15 min, 30–35 cycles of 95 °C for 30 s, 58 °C for 1 min and 30 s and 72 °C for 1 min, with a final extension at 60 °C for 30 min. Amplified PCR products were separated and visualized on an ABI Prism 3730 sequencer (Applied Biosystems, USA), and allele scoring was determined manually using PEAKSCANNER v1.0 (Applied Biosystems, USA).
Genetic analyses
The frequency of null alleles and scoring errors caused by stuttering or large allelic dropping-out were measured using MICRO-CHECKER v2.2.3 (Van Oosterhout et al. 2004). In addition, two different methods were performed to avoid false-positive and false-negative rates of null alleles (Dąbrowski et al. 2014) using CERVUS v3.0.3 (Kalinowski et al. 2007) and ML-NullFreq (Kalinowski and Taper 2006).
Genetic diversity within study sites was estimated by the number of alleles per locus (A) and observed (Ho) and expected (He) heterozygosity using GENETIX v4.05.2 (Belkhir et al. 2004), and allelic richness (AR) corrected for different sample sizes was calculated with FSTAT v2.9.3 (Goudet 1995). Tests for Hardy–Weinberg equilibrium and linkage disequilibrium were estimated for each locus and study site with GENEPOP v4.1 (Rousset 2008). Significance was evaluated using Bonferroni corrections. The estimation of Wright’s fixation indices for sample deviation from Hardy–Weinberg expectations for heterozygote disequilibrium (FIS) were estimated following Weir and Cockerham (1984) using GENETIX v4.05.2. Ho, He, A and AR average values for all loci within each study site were correlated with hydrologic distance to the most downstream study site (S13; Fig. 1) to evaluate the existence of a dendritic population in a fragmented area (Morrissey and De Kerckhove 2009; Pilger et al. 2017) using R software (R Core Team 2020). Genetic diversity among groups defined by STRUCTURE and DAPC (see below) was also estimated.
Genetic differentiation (FST) among pairs of study sites and groups defined by STRUCTURE and DAPC (see below) from the Deva–Cares catchment were estimated in FSTAT v2.9.3. We performed a discriminant analysis of principal components (DAPC) to explore intrapopulation genomic variation using the package adegenet v.3.3.3 (Jombart 2008) for R. The best-supported number of genetic clusters was estimated by Bayesian information criterion using the find cluster function. The optimal number of principal components for the DAPC was calculated by the cross-validation method (Jombart and Collins 2015). Genetic structure was further explored using the Bayesian clustering method implemented in STRUCTURE v.2.3.4 (Pritchard et al. 2000). Structure analysis was run for 1–10 clusters (K) with ten replicates for each simulated cluster. An admixture model with correlated allele frequencies was used. The number of Markov chains (MCMC) and the burn-in length were set to 1,000,000 replicates and 250,000 steps, respectively. Estimation of the most likely K on ΔK (an ad hoc quantity associated with the second order rate of change of the log probability of data; Evanno et al. 2005) was determined using STRUCTURE HARVESTER v0.6.94 (Earl and vonHoldt 2012) as well as the log probability of data (lnPrX│K) given by STRUCTURE. Mean individual membership coefficients (Q) to each study site over the ten runs were obtained using CLUMP v1.1.2 (Jakobsson and Rosenberg 2007) and graphically displayed using DISTRUCT v1.1 (Rosenberg 2004).
To investigate whether the genetic differentiation among study sites reflected contemporary patterns of stream connectivity, we used STREAMTREE (Kalinowski et al. 2008). The relative genetic distances for each stream section among study sites were estimated from the matrix of pairwise FST values among pairs of study sites. The fit of the STREAMTREE model to this matrix was quantified using a coefficient of determination (R2).
Bottlenecks were performed for each group defined by STRUCTURE and DAPC associated with fragmentation using two approaches. First, we tested whether the observed diversity exceeded that expected at mutation-drift equilibrium for heterozygosity excess using BOTTLENECK v1.2.02 (Cornuet and Luikart 1996; Piry et al. 1999). We assumed a two-phase mutation model (TPM) with 70, 80 and 90% stepwise mutations (SMMs) and 10,000 iterations. We tested statistical significance for heterozygote deficiency by one-tailed Wilcoxon’s signed-rank test. Second, we calculated the relationship between the number of alleles and the range in allele size M-ratio using M_P_VAL (Garza and Williamson 2001). The M-ratio was compared to a critical value of M (Mc) from the theoretical population in mutation-drift equilibrium by CRITICAL_M (Garza and Williamson 2001) assuming a pre-bottleneck effective population size of 50, 100, 500 and 1000 and a mutation rate (µ) of 5 × 10–4. The frequency of one-step mutations (pg) was fixed to 0.22, and the mean size of non-one-step mutations (Δg) was fixed to 3.1 following Peery et al. (2012).
Migration and effective population size
Migration rates between the groups defined by STRUCTURE and DAPC were explored using the coalescent method in MIGRATE-N 4.4.3 (Beerli and Palczewski 2010). Estimations of mutation-scaled migration rates M (M = m/µ) and Θ (Θ = 4Neµ) were calculated using a Brownian motion microsatellite model and Bayesian search strategy. Parameter space was searched using 10 short chains and 1 long chain with 3 replicates for 10,000,000 generations, an increment step of 1000 and a burn-in of 1,000,000. Parameter space was searched using four chains with an adaptive heating scheme (four heated chains, interval of one between swapping trees) to ensure that run results do not reflect local likelihood peaks. Estimations of M and Θ were used to calculate the effective number of migrants per generation Nm (Nm = ΘM/4). One migrant per generation was used as reference value to evaluate the migration. One migrant per generation has been suggested as sufficient gen flow to minimize losses in genetic diversity and maintaining genetic connectivity (Mills and Allendorf 1996).
Estimates of census size Nc were calculated directly from the adult individuals captured in the first electrofishing pass from the fish surveys. Nc was estimated from the sampling area (m2) of the fish surveys assuming a constant mean width (measured in the fish survey) in a river length of 1 km (i.e., extrapolating individuals/m2 from the field survey to this larger area − width × 1 km). The effective number of breeders over a reproductive year Nb was estimated for each group defined by STRUCTURE and DPAC using the linkage disequilibrium (LD) method (Waples and Do 2008) implemented in NeEstimator v.2.1 (Do et al. 2014). A minimum allele frequency cutoff value of 0.02 was employed, and 95% confidence intervals were obtained using the jackknife method. Migrants for each study site were identified in GENECLASS2 (Piry et al. 2004) and removed because the LD method is constrained on the assumption of a closed population model (Waples and Do 2008). The Bayesian statistical approach (Rannala and Mountain 1997) with the Monte Carlo resampling method (Paetkau et al. 2004) of 1000 simulated individuals and an alpha of 0.01 was used to identify individuals as migrants (Peel et al. 2013; Gilbert and Whitlock 2015). To avoid bias due to age structure, Nb was adjusted NbAdj using the ratio between adult lifespan (AL), age at maturity (α) and a coefficient of variation of age-specific fecundity (CVf), following the equation proposed by Waples et al. (2014): NbAdj = Nb/(0.991–0.206 × Log(AL) + 0.256 × Log(α) + 0.137CVf). The age at maturity for brown trout in the Deva–Cares catchment was 2 years. The AL value was calculated as described in Waples et al. (2014), with a maximum breeding age (ω) of 5 years (personal communication from the Cantabria Regional Government). CVf was computed using recruitment data for both sexes with an estimated value of 0.196. Likewise, NeAdj was estimated by means of the equation proposed by Waples et al. (2014): NeAdj = NbAdj/(0.833 + 0.637 × Log(AL) − 0.793 × Log(α) − 0.423 × CVf). The effective population size ratios NbAdj/Nc and NeAdj/Nc (Waples 2006) were also calculated following Perrier et al. (2016).
Genetic differentiation and riverscape characteristics
The relationships between genetic differentiation and riverscape characteristics were examined using two approaches. First, the Mantel test (Mantel 1967) was used to test for the significance of the correlation between linearized FST values (FST/(1 − FST)) and the pairwise study site of five variables (hydrological distance, difference in slope, elevation and number of total, permeable and impermeable barriers). Mantel tests with 9999 permutations were conducted in R v.3.3.3. package ade4 v.1.7-11 (Chessel et al. 2004). Decomposed pairwise regression analysis was used after each Mantel test to identify and remove potential outlier populations, which could mask the effects of the tested riverscape variable (Koizumi et al. 2006). Afterward, partial Mantel tests (Smouse et al. 1986) of only significant variables in the Mantel test were estimated. The Mantel test estimates the correlation between two matrices, whereas the partial Mantel test estimates the correlation between two matrices while controlling for the effects of a third matrix. These analyses have been widely used in population genetic studies to test for the effects of habitat fragmentation and isolation by distance (e.g., Stelkens et al. 2012).
Spatial analysis applications of the Mantel test in landscape genetics have been recently debated, raising concerns about the low statistical power and high type I error rates (Guillot and Rousset 2013; Legendre et al. 2015, among others). To compare the performance with an alternative method, correlations between genetic differentiation and riverscape variables were tested using distance-based canonical redundancy analysis (dbRDA) of pairwise differentiation, implemented in the R package vegan v.2.4-6 (Oksanen et al. 2013). Riverscape variables were tested after transforming from a Euclidean matrix to a continuous rectangular vector by principal coordinates analyses (PCoA). The significance of the predictors was assessed using multivariate F-statistics. We first analyzed the relationship between the linearized FST and each variable separately. Finally, we performed a partial dbRDA for each riverscape variable, controlling for the influence of hydrological distance (fitted as a covariate).
Results
Genetic diversity
Twelve microsatellite loci were analyzed, with an average success rate ranging from 86 to 100% depending on the locus. The locus Sssp1605 proved difficult to amplify, with allelic information in 86% of the individuals. Moreover, this locus showed reduced peaks and/or extra peaks of nonspecific binding or contamination origin. Therefore, the locus Sssp1605 was excluded from further analyses. Furthermore, evidence of null alleles for SSOSL311 was observed in most of the analyzed sample sites (MICRO-CHECKER, CERVUS, F(null) ≥ 0.340; ML-NullFreq, F(null) ≥ 0.200, p < 0.050), which justified the elimination of this locus from the data analysis.
The genetic diversity was high (Table S2). All 10 out of 12 loci successfully analyzed were polymorphic, with 120 alleles detected. The study sites exhibited on average number of alleles ranging from 1.900 in an tributary of the Cares River (S3) to 6.600 in a middle course of the Deva River (S9), an average allelic richness between 1.859 (S3) and 6.235 (S9) and an average expected and observed heterozygosity between 0.224 (S3)–0.708 (S2) in the upper course of the Cares River and 0.222 (S3)–0.710 (S9), respectively. Only 3 study sites presented fixed alleles (S3 = 4, S5 = 1 and S7 = 2). No evidence of linkage disequilibrium among pairs of loci was observed at any study site (p > 0.05). Finally, significant deviation from Hardy–Weinberg equilibrium was observed in 17.69% of the loci and in 31% of the study sites, with a decline in 3% of the loci and 0% of the study sites after Bonferroni corrections (Table S2).
The genetic diversity was significantly different among genetic groups defined by the STRUCTURE and DAPC results (see genetic differentiation) from the Deva–Cares catchment (Table 1). The average number of alleles was 2.3 in the tributaries and 6.8 in the main rivers. The average allelic richness varied between 2.241 (tributaries) and 5.650 (main rivers), and the average observed and expected heterozygosity for tributaries and main rivers was 0.290 and 0.602 and 0.283 and 0.644, respectively (Table 1).
Genetic differentiation
Correlations between A, AR, Ho and He with distance to the most downstream study site (S13) were observed only for A (p = 0.048, see Table 2 and Fig. 2). In the cases of Ho and AR, the values were marginally significant (p < 0.100), while for He, correlations were not observed.

Linear regression between A, AR, Ho and He with distance to the most downstream study site (S13) and 95% confidence intervals (results are represented in Table 2)
Overall genetic differentiation between study sites (FST = 0.182) was high and variable (Table S3). Tributaries (S3, S5, and S7) were markedly different from each other and had genetic distinctiveness in comparison to the rest of the study sites (FST = 0.573). The remaining study sites showed a moderate FST mean value (FST = 0.079). Study sites located in tributaries and in upper main rivers exhibit very great genetic differentiation (FST mean value = 0.380; ranging from 0.145 to 0.664), while study sites situated in middle and lower courses of the Deva and Cares rivers exhibited moderate genetic differentiation (FST mean value = 0.052: ranging from 0.002 to 0.142).
DAPC analysis showed that individuals of study sites in upper courses were partitioned into four groups (Fig. 3; Table 3): group 1 upper course of the Cares River (S1 and S2), group 2 upper course of the Duje river (tributary of the Cares River; S3), group 3 upper course of the Casaño River (tributary of the Cares River; S5) and group 4 upper course of the Deva subcatchment (Salvorón River and upper course of the Deva River; S7 and S8). The localities in the middle and lower courses could join in two additional groups: group 5 the middle course study sites of the Cares River (S4–S6) and group 6 the middle and lower course study sites of the Deva River (S9–S10–S11–S12–S13). However, groups 5 and 6 were not included clearly in different groups because they presented individuals with an admixture parental origin.

Scatterplots for the first two discriminant axes of the DAPC analysis in the Deva–Cares catchment. Each individual is represented as a dot, and the groups (K = 6) are represented as inertia ellipses
The results of the Evanno test suggested that K = 5 was the most likely cluster number for the STRUCTURE analysis (Fig. 4). The distribution of genetic differentiation was similar to that observed by DAPC. All tributaries and the upper study site of Cares River were genetically different (average Q > 0.900), while most of the remaining downstream study sites showed admixed ancestry between clusters. Thus, S1 and S2 were included in the first cluster (Q = 0.983). S3 had the highest Q value (Q = 0.996) and was assigned to the second cluster. S5 was assigned to the third cluster (Q = 0.961), and S7 and S8 were included in the fourth cluster (Q = 0.730). S4–S6–S9–S10–S11–S12 were assigned to the fifth cluster (Q = 0.784). However, given the high admixture in downstream study sites, we compared the results obtained applying different “cutoff” probabilities. Individuals were assigned to a cluster when their Q values > 0.500, 0.700 and 0.800; otherwise, they were considered admixed. When a membership probability of Q value > 0.500 was used, S8 exhibited high admixture between two clusters and was considered an independent group between the fourth (Q = 0.479) and fifth (Q = 0.329) clusters. S4 and S6 showed admixture genotypes between the third (Q = 0.263) and the fifth (Q = 0.626) clusters, with a cutoff probability of 0.700, and were considered an independent group for subsequent analysis. Last, the study site after the confluence of the Cares and Deva Rivers (S13) was assigned separately to the middle course (S9, S10, S11, S12) when the Q value > 0.800 because it showed an admixture between the first (average Q value = 0.186) and fifth clusters (average Q value = 0.776).

Geographical distribution of genetic clusters identified by STRUCTURE and clustering analysis results for K = 5 (PP1, PP2, PP3, PP4 and PP5 represent parental populations). Colored bars represent proportions of membership of each individual to each cluster (color figure online)
Therefore, eight groups were defined after considering the DAPC and STRUCTURE results for subsequent analysis: group 1 S1 and S2; group 2 S3; group 3 S5; group 4 S4 and S6; group 5 S7; group 6 S8; group 7 S9, S10, S11 and S12; and group 8 S13. Four groups were located in the Cares subcatchment (S1–S2, S3, S5 and S4–S6), three in the Deva subcatchment (S7, S8 and S9–S10–S11–S12) and one after the confluence of the Cares and Deva rivers (S13). Moreover, the S1–S2, S3, S5, S7 and S8 groups were located in upper courses, S4–S6, S9–S10–S11–S12 were in middle courses and the S13 group was in the lower course of the catchment.
The lowest genetic differentiation obtained from FST values between groups defined by STRUCTURE and DAPC from the Deva–Cares catchment (Table 4) was observed between the middle course of the Cares River (S4 and S6) and the Deva River (S9–S10–S11–S12) groups (FST = 0.018), whereas the highest differentiation was observed between tributaries from both rivers (S3 and S7; FST = 0.664).
Genetic distances for each stream section among study sites were estimated by STREAMTREE (Fig. 5). The coefficient of determination (R2 = 0.972) indicated a good fit to the observed data. The results were useful in visualizing and quantifying the resistance to migration in the Deva–Cares catchment. The STREAMTREE analysis was consistent with previous results, thus showing the highest values between the upper course tributaries S3, S5 and S7 and the rest of the downstream study sites. Genetic distances at or close to zero were assigned to stream sections without barriers or to those with a low density of permeable barriers (Fig. 5).

Graphical representation of migration estimates in the Deva–Cares catchment using MIGRATE (arrows represent the gene flow direction, and values represent the number of migrants per generation; “Nm” and 95% CI) and genetic distances associated with stream sections calculated using STREAMTREE analysis (italic values). Nm > 1 are indicated in bold. Dashed ellipses represent groups defined by STRUCTURE and DAPC from the Deva–Cares catchment
The BOTTLENECK and M-ratio analyses did not show evidence for recent bottlenecks in the studied groups (Table S4).
Migration and effective population size
Only migration results between contiguous upstream/downstream groups and the confluence of the Deva and Cares Rivers were analyzed to determine if there was migration between fragmented groups. The effective number of migrants per generation between groups ranged from 0.06 between the middle course of the Cares River (S4–S6) and the upper course of the Duje River (S3) to 1.82 from the upper (S8) to the middle course of the Deva River (S9–S10–S11–S12) (Fig. 5). Two pairwise comparisons suggested bidirectional gene flow with values Nm > 1 between the middle and lower courses of the Deva catchment and between the main rivers Deva and Cares (S9–S10–S11–S12 to S4–S6; S13 to S9–S10–S11–S12 and vice versa). Moreover, migrations between the lower part of the catchment and the middle course of the Cares River had values of Nm near 1 for upstream (0.96) and downstream (0.93) migration (S13 to S4–S6). Pairwise comparisons of the upper to middle courses of the Cares and Deva main rivers (S1–S2 to S4–S6 and S8 to S9–S10–S11–S12) had only downstream gene flow with Nm > 1. The rest of the analyzed groups presented values Nm < 1 both upstream and downstream.
The Deva–Cares catchment exhibited high differences in the effective number of breeders Nb estimated with NeLD among genetic groups (Table 5). The highest values were observed in the upper course of the Cares River (S1 and S2) and the middle course of the Deva River (S9–S10–S11–S12) (Nb > 50). The middle course of the Deva River also showed high genetic diversity and was located below an impermeable barrier, although the upper course of the Cares River, with a lower genetic diversity value, was located above an impermeable barrier. However, the upper course of the Duje River (S3) and the upper course of Salvoron and Deva Rivers (S7 and S8) showed low Nb and genetic diversity values and were located above impermeable barriers. The exception was the upper course tributary of the Cares River (Casaño River; S5), which showed an infinite value due to its very high density and was not included in subsequent analyses. The study site in the lower course of the Deva–Cares catchment had a low value. The NbAdj estimates were approximately 5% higher than the non-adjusted values and showed the same tendency (Table 5). NbAdj showed high values in the upper (S1–S2) and middle courses of the Cares and Deva Rivers (S4–S6 and S9–S10–S11–S12) (> 50), in contrast with the low values estimated in the other groups, which were mostly placed above the barriers. The NeAdj estimates yield values and patterns similar to those of Nb. The estimated ratio between the adjusted effective number of breeders NbAdj and census size Nc varied from 0.053–0.619. Likewise, the estimated ratio between the adjusted effective population size and census size NeAdj/Nc changed similarly.
Genetic differentiation and riverscape characteristics
There was a significant pattern of isolation by distance-IBD (Mantel test; r2 = 0.232, p < 0.050; Table 6). Moreover, the presence of total barriers and impermeable barriers showed a significant pattern of isolation by barriers (Mantel test; r2 = 0.322, p < 0.050 and r2 = 0.700, p < 0.050, respectively). In contrast, elevation, slope and the presence of permeable barriers were not significant. The dbRDA analysis confirmed the above findings by the Mantel test. The dbRDA results showed a significant pattern of isolation by distance and isolation by impermeable barriers (r2 = 0.951, p < 0.050 and r2 = 0.640, p < 0.050, respectively). The partial Mantel test showed a significant pattern of isolation by impermeable barriers after controlling for the effects of hydrological distance and total barriers (partial Mantel test; r2 = 0.611, p < 0.050 and r2 = 0.558, p < 0.050, respectively), showing that the influence of impermeable barriers was higher than that of hydrological distance and total barriers.
Discussion
The study revealed a high degree of genetic differentiation among study sites and groups defined within the catchment and a significant isolation effect by barriers. Our results are consistent with our initial hypothesis, and they also agree with other studies showing the importance of longitudinal barriers in river networks and loss of structural connectivity on fish population genetic structure (Yamamoto et al. 2004; Griffiths et al. 2009) and the possible effect associated with genetic drift, loss of genetic diversity or isolation (Yamamoto et al. 2004; Horreo et al. 2011; Stelkens et al. 2012).
Genetic diversity and genetic structure
Microsatellite loci have been used in multiple studies as an efficient resource in the description of the genetic diversity and population genetic structure of fish populations within watersheds (Wofford et al. 2005; Griffiths et al. 2009; McGlashan et al. 2011) and among watersheds (Huey et al. 2010). Allelic richness presented a broader range (1.859–6.235) than that obtained by Horreo et al. (2011) in a previous study in the Deva–Cares catchment (1.394–4.778); these authors considered 12 study sites and only 4 barriers and underestimated the real number of barriers present in the catchment. Additionally, the observed and expected heterozygosity had a slightly narrower range (He = 0.224–0.708, Ho = 0.222–0.710) than that in the previous study (He = 0.130–0.805, Ho = 0.154–0.742). The global genetic differentiation across all 13 study sites (FST = 0.181) was considered great (Hartl et al. 1997), as in the results by Horreo et al. (2011) (FST = 0.204).
Genetic diversity is distributed heterogeneously, revealing the existence of several genetically differentiated study sites. This result indicates that drift and gene flow have interplayed in shaping the genetic constitution of the brown trout at the study sites, as has been shown in other studies (e.g., Massa-Gallucci et al. 2010). In our study, below-barrier study sites had high genetic diversity and lower FST values. In contrast, headwater and above-barrier study sites had low genetic diversity and high FST values, evidence that could support fast rates of genetic drift due to isolation. These study sites were the only ones presenting fixed alleles (S3 = 4, S5 = 1 and S7 = 2). This result has also been found for above-barrier locations in other studies of salmonids (Neville et al. 2009). Moreover, we found concordance between impermeable barriers and the identified genetic groups in DAPC and STRUCTURE analysis, which together with the STREAMTREE results, suggest the role of impermeable barriers in shaping an altered genetic structure, specifically acting as an isolation mechanism. The absence of data before fragmentation does not allow establishing casuality that the observed results were due to the presence of anthropogenic fragmentation. However, based on the information presented in the methodology regarding barriers, the data suggest that barriers may be related to population genetic structure. All upper study sites separated by one or more impermeable barriers belonged to different genetic groups, except between the two upper course study sites from the Cares River (S1 and S2). This result may be because the barrier between both study sites is the most recently built dam in the catchment (constructed in 1995). Quite likely, not enough time has passed to cause genetic divergence among these study sites because genetic differentiation is correlated with the time since physical isolation by a barrier started (Yamamoto et al. 2004). Study sites separated by barriers with an older construction date (Cain Dam-1921 and Poncebos Dam-1958) belonged to different genetic groups. Moreover, these dams are both located between the upper course of the Cares River (S1 and S2) and the middle course of the Cares River (S4–S6), denoting that fragmentation by anthropogenic barriers may have occurred nearly 100 years ago between these groups. Poncebos Dam represent the limit of the current S. salar distribution (Alvarez et al. 2010; González-Ferreras et al. 2019), and the river network until the Cain Dam is considered a potential habitat for the anadromous species. Regarding this barrier, the DIVAQUA project (LIFE18 NAT/ES/000121), which is currently in its execution phase, contemplates fish pass improvements for some dams (included Poncebos) and some barriers removal in the Deva–Cares catchment. Future studies after the execution of the project could help to corroborate the results obtained in this study. Similarly, future studies focusing on S1 and S2 could demonstrate if the populations may diverge.
The low FST values among groups where dispersal is possible (by the presence of permeable barriers or absence of obstacles) indicate that minor genetic structure exists, and allele frequencies differed throughout the permeable area. These differentiations could be explained by a certain degree of reduction in gene flow caused by permeable barriers. In the case of groups where there is no barrier between study sites (e.g., S6–S4), genetic differentiation could be caused by environmental factors such as geological substrate (Perrier et al. 2011) or temperature (Dionne et al. 2008) that could influence gene flow. It should also be noted that due to the hydro-geomorphological characteristics of the catchment, the presence of rapids in some areas may denote a certain degree of temporal impermeability during low flow or high flow seasons.
Gene flow and riverscape genetics: influence on genetic differentiation
Study sites showed a significant IBD pattern, which is common in stream salmonids with mobile and stationary individuals (Kanno et al. 2011) and other fish species (Sotola et al. 2017). In addition, we reported a decrease in genetic diversity (number of alleles) in upstream study sites and a downstream-biased gene flow. Similar tendencies have been found in other fish species (e.g., Poecilia reticulata; Barson et al. 2009; Cottus gobio; Junker et al. 2012; Oncorhynchus mykiss; Winans et al. 2015). Evidence of a correlation between the distance to most downstream study sites and the number of alleles but not heterozygosity could be explained because the number of alleles and allelic richness approach equilibrium more rapidly than heterozygosity (Epps and Keyghobadi 2015; Salisbury et al. 2016). Barson et al. (2009) reported that populations with downstream-biased gene flow may act as sinks in lowland populations and as sources in upland populations, contributing to gene flow into downstream populations. Moreover, previous studies found that genetic diversity and allelic richness in dendritic networks increase with increasing downstream locations related to the alleles fixed in upstream locations collected in downstream confluences (e.g., Morrissey and De Kerckhove 2009; Paz-Vinas et al. 2015). In our study, we found characteristics of both dendritic metapopulation (i.e., downstream-biased gene flow) and member-vagrant models (i.e., significant effect of IBD). According to the study of S. salar realized by Garant et al. (2000), both models (metapopulation and member-vagrant) were not mutually exclusive, and their combined use may help us better understand the dynamics of the genetic structure in unstable environments. Thus, the member-vagrant hypothesis is useful to explain the number of subpopulations given the life history of a species and habitat structure, while the metapopulation model is useful to explain the temporal persistence and extent of genetic divergence among subpopulations (Garant et al. 2000).
The results from migration in our study indicate that gene flow was higher (bidirectional Nm > 1) between downstream groups located in the middle and lower course of the Deva–Cares Rivers, matching groups without impermeable barriers. We emphasize that this is the current distribution area of S. salar linking our assumptions prior to the study. Previous studies have suggested symmetric migration in the absence of barriers (Gomez-Uchida et al. 2009; Horreo et al. 2011). Likewise, previous studies have shown that asymmetric migration occurs often in river networks and that barriers amplify the dispersal asymmetry because of negligible upstream dispersal (Junker et al. 2012). In the Deva–Cares catchment, downstream migration (unidirectional Nm > 1) was reported in some upper course groups above impermeable barriers (from S1–S2 and S8 to downstream groups), while there was an almost absence of migration (values Nm < 1) from other groups above impermeable barriers (S3, S5 and S7), although values of Nm downstream are higher than upstream. In relation to these groups, S1–S2 and S8 are situated in the main rivers (Cares and Deva Rivers, respectively), while S3, S5 and S7 are located in their tributaries (Duje, Casaño and Salvorón Rivers). These results are in agreement with previous salmonid studies related to riverscape genetic frameworks, indicating that resistance to gene flow is lower in the main rivers and that relative migration rates in the main rivers are greater than those in the tributaries (White 2019). However, future studies will be necessary to determine the relative influence of barriers or tributary-main river conditions on low gene flow. Moreover, reaches below the barriers contained a high proportion of mixed genetic pools that could be the result of immigrants from above (see Fig. 4). Isolation by barriers appeared to provide more support for generating the observed population genetic structure of the study sites and groups in the studied catchment than isolation by distance alone. This pattern has also been found in previous studies analyzing fragmentation in river systems (e.g., Meldgaard et al. 2003; Leclerc et al. 2008). Furthermore, neither slope nor elevation was found to influence genetic structure, suggesting that the barriers may be acting as a bias against upstream dispersal.
Bottleneck and effective population sizes
Bottlenecks have been associated with fragmentation in previous studies (e.g., Coleman et al. 2018) and can contribute to the loss of genetic diversity and increases in genetic differentiation. Despite the reduction in allelic diversity and the presence of monomorphic loci in upstream study sites, no evidence of bottlenecks was found. The low statistical power of bottleneck detection tests has been previously observed as a result of an insufficient sample size due to a limited number of loci analyzed and an underestimation of microsatellite mutation rates (Cornuet and Luikart 1996; Piry et al. 1999; Peery et al. 2012). Single sampling period bottleneck detection methods assume that a population is close to migration. In downstream study sites, the effectiveness of bottleneck tests can also be limited by the presence of immigration, which can remove signs of bottlenecks in two to three generations (Keller et al. 2001).
All groups showed adjusted effective population size NeAdj values below 100, the limit required to avoid inbreeding depression (Frankham et al. 2014). In addition, the study sites from upper course tributaries isolated by impermeable barriers coincided with those with extremely low NeAdj values, the lowest genetic diversity and a high percentage of monomorphic loci. Groups with small effective population sizes are expected to reach intense genetic drift and suffer a higher probability of extinction (Newman and Pilson 1997), in addition to an increased likelihood of fixation of deleterious alleles and reduced selection effectiveness (Hare et al. 2011). However, there were differences among genetic groups; thus, the effective number of breeders NbAdj in the upper and middle course of the Cares and Deva Rivers located below barriers showed values above the recommended minimum of 50 to avoid inbreeding in the short term (Franklin 1980). In contrast, the upper course tributaries of the Cares and Deva Rivers and the upper course of the Deva River located above-barrier showed low values (< 50). The upper course tributary of the Cares River (S5) showed infinite Ne estimate values, possibly due to a larger sample size, which might result in more difficult reliable estimates (Waples and Do 2010). S5 study site is characterized by natural high densities of brown trout (no stocking activities were performed in S5; personal communication of Principado de Asturias Government), and it was located in an unaltered habitat where fishing is prohibited. Therefore, these conditions may lead to higher population densities (Sánchez-Hernández et al. 2011).
The estimated ratio between the effective and census sizes in salmonids is approximately 0.1–0.2 (Campos et al. 2006), but higher values have also been reported (Ardren and Kapuscinski 2003). This ratio is important for monitoring changes in genetic diversity and predicting the rate of genetic loss (Ardren and Kapuscinski 2003). In our study, it had relatively high values in the middle course of the Deva and Cares Rivers (0.300–0.488) below the impermeable barriers. The lack of relationship between NeAdj/Nc and Nc could be explained by the fact that mechanisms of genetic compensation may counteract NeAdj reductions and thus increase NeAdj/Nc (Palstra and Ruzzante 2008). This genetic compensation may produce a buffering effect against the loss of genetic diversity at low Nc. Another possible explanation is the difficulty of sampling these study sites, leading to the underestimation of their census sizes. However, the difficulty of delimiting a population in this scenario means that the area used to estimate census size could have been insufficient. In some studies, Nc was obtained by extrapolating to the length of the stream without barriers (Ruzzante et al. 2016) or by extrapolating to the occupied stream length (Peacock and Dochtermann 2012), but there is no universal method. This lack of consensus highlights the need for further research on the definition and delimitation of study sites or populations for census size estimation for mobile organisms that inhabit river networks.
Conservation strategies and management implications
The obtained results have strong implications for the management and conservation strategies of native brown trout. The genetically distinct study sites and groups that were identified could be recognized as genetically independent management units in the Deva–Cares catchment. Moreover, it is possible that a considerable number of distinctive genetic groups are yet to be recognized due to the presence of several barriers in other tributaries: a more detailed genetic study (i.e., other headstreams or tributaries) could identify additional distinct genetic groups using an approach similar to the present study.
This work provides strong support for the critical role of structural connectivity loss and gene flow on the persistence of brown trout, securing their genetic diversity for the future. To achieve that goal, the ideal option would be to rewild the ecosystem (by completely removing all anthropogenic barriers), but due to the high number of anthropogenic barriers present in the catchment, it is currently a very unfeasible option. For this reason, it would be appropriate to identify key locations or groups to conserve and key areas to increase the genetic flow by removing barriers or increasing the efficiency of fish passes. For those locations in which connectivity cannot be improved, the habitat should be enhanced considering its positive relation with genetic diversity and genetic differentiation (Whiteley et al. 2013). In our study, the S8 group contained a mixed genetic pool and constituted an important genetic reservoir that should be considered a priority area for conservation. In addition, isolated study sites situated in upper courses presenting low genetic diversity that provide downstream gene flow to other locations should be given special consideration in conservation efforts (Kelson et al. 2015). In our catchment, these locations were study sites S1, S2 and S8, and all of them are located above anthropogenic impermeable barriers (a natural impermeable barrier is also located near S8). Moreover, upstream study sites located in the tributaries (S3, S5 and S7) are important to conserve, despite the low gene flow, because of the presence of fixed alleles in these study sites.
The consideration of populations upstream of natural barriers is also important for conservation and management implications. There are two main possible causes for the presence of fish populations above natural impermeable barriers: human transfer of fish to fishless streams (Rahel 2007) or geological processes (Currens et al. 1990). Regarding human transfer, it is unlikely that past stocking activities with foreign trout were carried out in these study sites due to the difficult accessibility and previous information of stocking in the catchment (see the materials and methods section). However, historically, the human transfer of downstream trout by anglers could have been carried out. However, there is currently a lack of knowledge about the origin of brown trout above natural barriers in the Deva–Cares catchment. Future in-depth research on these locations could reveal whether these rivers have been previously fishless to determine the best conservation strategy. In the case of finding evidence of human transfer, should these locations be protected, or should the stream return to being fishless? We cannot answer these questions based on the results of the current study, indicating that for appropriate management and conservation, future studies will be necessary.
In conclusion, this study suggests that impermeable barriers have a large effect on the genetic variation of the native brown trout inhabiting the Deva–Cares catchment, demonstrating high differentiation between study sites and groups above and below barriers and an isolation by distance pattern. The presented analysis at a river network scale provides evidence regarding the role of barriers in influencing patterns of genetic diversity, highlighting the importance of maintaining and restoring the connectivity of freshwater ecosystems for conserving native brown trout. Such results suggest that measures of management and conservation should be taken for longer-term viability of brown trout inhabiting the Deva–Cares catchment.
Availability of data and material
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
References
Addison PA, Wilson CC (2010) Matching management to biological scale: connectivity among lacustrine brook trout populations. North Am J Fish Manag 30:1132–1142. https://doi.org/10.1577/M09-146.1
Almodovar A, Nicola GG (1998) Assessment of a brown trout Salmo trutta population in the River Gallo (central Spain): angling effects and management implications (Salmonidae). Ital J Zool 65:539–543. https://doi.org/10.1080/11250009809386881
Almodóvar A, Nicola GG, Elvira B (2006) Spatial variation in brown trout production: the role of environmental factors. Trans Am Fish Soc 135:1348–1360. https://doi.org/10.1577/T05-189.1
Almodóvar A, Nicola GG, Elviera B, Leal S (2008) Analysis of the genetic variability of trout in Cantabria and applications to the management of its populations. Report of collaboration between the regional ministry of livestock, agriculture and fisheries of the government of Cantabria and the UCM, Spain
Almodóvar A, Nicola GG, Ayllón D, Elvira B (2012) Global warming threatens the persistence of Mediterranean brown trout. Glob Change Biol 18:1549–1560. https://doi.org/10.1111/j.1365-2486.2011.02608.x
Alvarez J, Antón A, Azpiroz I et al (2010) Atlas de los Ríos Salmoneros de la Península Ibérica. Iberian Peninsula Salmon Rivers Atlas, Spain
Aparicio E, Rocaspana R, de Sostoa A et al (2018) Movements and dispersal of brown trout (Salmo trutta Linnaeus, 1758) in Mediterranean streams: influence of habitat and biotic factors. PeerJ 6:e5730. https://doi.org/10.7717/peerj.5730
Ardren WR, Kapuscinski AR (2003) Demographic and genetic estimates of effective population size (Ne) reveals genetic compensation in steelhead trout. Mol Ecol 12:35–49. https://doi.org/10.1046/j.1365-294X.2003.01705.x
Baguette M, Van Dyck H (2007) Landscape connectivity and animal behavior: functional grain as a key determinant for dispersal. Landsc Ecol 22:1117–1129. https://doi.org/10.1007/S10980-007-9108-4/FIGURES/2
Barquín J, Benda LE, Villa F et al (2015) Coupling virtual watersheds with ecosystem services assessment: a 21st century platform to support river research and management. Wiley Interdiscip Rev Water 2:609–621. https://doi.org/10.1002/wat2.1106
Barson NJ, Cable J, Van Oosterhout C (2009) Population genetic analysis of microsatellite variation of guppies (Poecilia reticulata) in Trinidad and Tobago: evidence for a dynamic source-sink metapopulation structure, founder events and population bottlenecks. J Evol Biol 22:485–497. https://doi.org/10.1111/J.1420-9101.2008.01675.X
Beach MH (1984) Fish pass design-criteria for the design and approval of fish passes and other structures to facilitate the passage of migratory fish in rivers. Ministry of Agriculture, Fisheries and Food, Lowestoft
Beerli P, Palczewski M (2010) Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185:313–326. https://doi.org/10.1534/genetics.109.112532
Belkhir K, Borsa P, Chikhi L et al (2004) GENETIX 4.05, logiciel sous Windows TM pour la génétiquedes populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier
Campbell Grant EH, Lowe WH, Fagan WF (2007) Living in the branches: population dynamics and ecological processes in dendritic networks. Ecol Lett 10:165–175. https://doi.org/10.1111/j.1461-0248.2006.01007.x
Campos JL, Posada D, Morán P (2006) Genetic variation at MHC, mitochondrial and microsatellite loci in isolated populations of Brown trout (Salmo trutta). Conserv Genet 7:515–530. https://doi.org/10.1007/S10592-005-9063-Z
Chaput-Bardy A, Lemaire C, Picard D, Secondi J (2008) In-stream and overland dispersal across a river network influences gene flow in a freshwater insect, Calopteryx splendens. Mol Ecol 17:3496–3505. https://doi.org/10.1111/j.1365-294X.2008.03856.x
Chessel D, Dufour A-B, Thioulouse J (2004) The ade4 package—I : one-table methods. R News 4(1):5–10
Coleman RA, Gauffre B, Pavlova A et al (2018) Artificial barriers prevent genetic recovery of small isolated populations of a low-mobility freshwater fish. Heredity (edinb) 120:515–532. https://doi.org/10.1038/S41437-017-0008-3
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Currens KP, Schreck CB, Li HW (1990) Allozyme and morphological divergence of rainbow trout (Oncorhynchus mykiss) above and below Waterfalls in the Deschutes River, Oregon. Copeia 1990:730–746. https://doi.org/10.2307/1446439
Dąbrowski MJ, Pilot M, Kruczyk M et al (2014) Reliability assessment of null allele detection: inconsistencies between and within different methods. Mol Ecol Resour 14:361–373. https://doi.org/10.1111/1755-0998.12177
Dionne M, Caron F, Dodson JJ, Bernatchez L (2008) Landscape genetics and hierarchical genetic structure in Atlantic salmon: the interaction of gene flow and local adaptation. Mol Ecol 17:2382–2396. https://doi.org/10.1111/j.1365-294X.2008.03771.x
Dixo M, Metzger JP, Morgante JS, Zamudio KR (2009) Habitat fragmentation reduces genetic diversity and connectivity among toad populations in the Brazilian Atlantic Coastal Forest. Biol Conserv 142:1560–1569. https://doi.org/10.1016/j.biocon.2008.11.016
Do C, Waples RS, Peel D et al (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214. https://doi.org/10.1111/1755-0998.12157
Dunham JB, Rieman BE (1999) Metapopulation structure of bull trout: influences of physical, biotic, and geometrical landscape characteristics. Ecol Appl 9:642–655. https://doi.org/10.1890/1051-0761(1999)009[0642:MSOBTI]2.0.CO;2
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Epps CW, Keyghobadi N (2015) Landscape genetics in a changing world: disentangling historical and contemporary influences and inferring change. Mol Ecol 24:6021–6040. https://doi.org/10.1111/mec.13454
Eros T, Olden JD, Schick RS et al (2012) Characterizing connectivity relationships in freshwaters using patch-based graphs. Landsc Ecol 27:303–317. https://doi.org/10.1007/S10980-011-9659-2/FIGURES/6
ESRI (2014) ArcGIS Desktop 10.2.1 (GIS software)
Estevez E, Rodríguez-Castillo T, Álvarez-Cabria M et al (2017) Analysis of structural and functional indicators for assessing the health state of mountain streams. Ecol Indic 72:553–564. https://doi.org/10.1016/j.ecolind.2016.08.052
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fagan WF (2002) Connectivity, fragmentation, and extinction risk in dendritic metapopulations. Ecology 83:3243–3249. https://doi.org/10.1890/0012-9658(2002)083[3243:CFAERI]2.0.CO;2
Frankham R, Bradshaw CJA, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol Conserv 170:56–63. https://doi.org/10.1016/j.biocon.2013.12.036
Franklin IR (1980) Evolutionary change in small populations. In: Soulé ME, Wilcox BA (eds) Conservation biology: an evolutionary-ecological perspective. Sinauer Associates, Sunderland, pp 135–149
Fuller MR, Doyle MW, Strayer DL (2015) Causes and consequences of habitat fragmentation in river networks. Ann N Y Acad Sci 1355:31–51. https://doi.org/10.1111/nyas.12853
Garant D, Dodson JJ, Bernatchez L (2000) Ecological determinants and temporal stability of the within-river population structure in Atlantic salmon (Salmo salar L.). Mol Ecol 9:615–628. https://doi.org/10.1046/j.1365-294X.2000.00909.x
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318. https://doi.org/10.1046/j.1365-294X.2001.01190.x
Gilbert KJ, Whitlock MC (2015) Evaluating methods for estimating local effective population size with and without migration. Evolution (n y) 69:2154–2166. https://doi.org/10.1111/evo.12713
Gomez-Uchida D, Knight TW, Ruzzante DE (2009) Interaction of landscape and life history attributes on genetic diversity, neutral divergence and gene flow in a pristine community of salmonids. Mol Ecol 18:4854–4869. https://doi.org/10.1111/J.1365-294X.2009.04409.X
González-Ferreras AM (2019) Determination and modeling of spatial patterns of brown trout (Salmo trutta Linnaeus, 1758) in the Deva–Cares catchment: the role of connectivity and the niche. University of Cantabria, Santander
González-Ferreras AM, Barquín J (2017) Mapping the temporary and perennial character of whole river networks. Water Resour Res 53:6709–6724. https://doi.org/10.1002/2017WR020390
González-Ferreras AM, Bertuzzo E, Barquín J et al (2019) Effects of altered river network connectivity on the distribution of Salmo trutta: insights from a metapopulation model. Freshw Biol 64:1877–1895. https://doi.org/10.1111/fwb.13379
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Griffiths AM, Koizumi I, Bright D, Stevens JR (2009) A case of isolation by distance and short-term temporal stability of population structure in brown trout (Salmo trutta) within the River Dart, southwest England. Evol Appl 2:537–554. https://doi.org/10.1111/j.1752-4571.2009.00092.x
Guillot G, Rousset F (2013) Dismantling the Mantel tests. Methods Ecol Evol 4:336–344. https://doi.org/10.1111/2041-210x.12018
Gutiérrez FR, Fernández RM, Diáz CP (2020) La identidad geográfica del Parque Nacional de los Picos de Europa: la dificultad de su gestión. Boletín La Asoc Geógrafos Españoles. https://doi.org/10.21138/bage.2957
Hanski I (1998) Metapopulation dynamics. Nature 396:41–49
Hare MP, Nunney L, Schwartz MK et al (2011) Understanding and estimating effective population size for practical application in marine species management. Conserv Biol 25:438–449. https://doi.org/10.1111/j.1523-1739.2010.01637.x
Hartl DL, Clark AG, Clark AG (1997) Principles of population genetics. Sinauer Associates, Sunderland
Horreo JL, Garcia-Vazquez E (2011) Foreign brown trout in protected landscapes as a consequence of connectivity. Fish Manag Ecol 18:431–436. https://doi.org/10.1111/j.1365-2400.2011.00803.x
Horreo JL, Martinez JL, Ayllon F et al (2011) Impact of habitat fragmentation on the genetics of populations in dendritic landscapes. Freshw Biol 56:2567–2579. https://doi.org/10.1111/J.1365-2427.2011.02682.X
Huey JA, Baker AM, Hughes JM (2010) High levels of genetic structure in the Australian freshwater fish, Ambassis macleayi. J N Am Benthol Soc 29:1148–1160. https://doi.org/10.1899/09-093.1
Hughes AR, Inouye BD, Johnson MTJ et al (2008) Ecological consequences of genetic diversity. Ecol Lett 11:609–623. https://doi.org/10.1111/j.1461-0248.2008.01179.x
Izquierdo JI, Castillo AGF, Ayllon F et al (2006) Stock transfers in Spanish brown trout populations: a long-term assessment. Environ Biol Fishes 75:153–157. https://doi.org/10.1007/s10641-005-5068-1
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Collins C (2015) A tutorial for discriminant analysis of principal components (DAPC) using adegenet 2.0. 0. Imp Coll Lond MRC Cent Outbreak Anal Model 43:1–43
Junker J, Peter A, Wagner CE et al (2012) River fragmentation increases localized population genetic structure and enhances asymmetry of dispersal in bullhead (Cottus gobio). Conserv Genet 13:545–556. https://doi.org/10.1007/s10592-011-0306-x
Kalinowski ST, Taper ML (2006) Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv Genet 7:991–995. https://doi.org/10.1007/s10592-006-9134-9
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106. https://doi.org/10.1111/j.1365-294X.2007.03089.x
Kalinowski ST, Meeuwig MH, Narum SR, Taper ML (2008) Stream trees: a statistical method for mapping genetic differences between populations of freshwater organisms to the sections of streams that connect them. Can J Fish Aquat Sci 65:2752–2760. https://doi.org/10.1139/F08-171
Kanno Y, Vokoun JC, Letcher BH (2011) Fine-scale population structure and riverscape genetics of brook trout (Salvelinus fontinalis) distributed continuously along headwater channel networks. Mol Ecol 20:3711–3729. https://doi.org/10.1111/J.1365-294X.2011.05210.X
Keller LF, Jeffery KJ, Arcese P et al (2001) Immigration and the ephemerality of a natural population bottleneck: evidence from molecular markers. Proc R Soc Lond Ser B Biol Sci 268:1387–1394. https://doi.org/10.1098/rspb.2001.1607
Kelson SJ, Kapuscinski AR, Timmins D, Ardren WR (2015) Fine-scale genetic structure of brook trout in a dendritic stream network. Conserv Genet 16:31–42. https://doi.org/10.1007/S10592-014-0637-5/TABLES/3
Keyghobadi N (2007) The genetic implications of habitat fragmentation for animals. Can J Zool 85:1049–1064. https://doi.org/10.1139/Z07-095
Klemetsen A, Amundsen PA, Dempson JB et al (2003) Atlantic salmon Salmo salar L., brown trout Salmo trutta L. and Arctic charr Salvelinus alpinus (L.): a review of aspects of their life histories. Ecol Freshw Fish 12:1–59. https://doi.org/10.1034/j.1600-0633.2003.00010.x
Koizumi I, Yamamoto S, Maekawa K (2006) Decomposed pairwise regression analysis of genetic and geographic distances reveals a metapopulation structure of stream-dwelling Dolly Varden charr. Mol Ecol 15:3175–3189. https://doi.org/10.1111/J.1365-294X.2006.03019.X
Larinier M (2002) Biological factors to be taken into account in the design of fishways, the concept of obstructions to upstream migration. Bull Fr Pêche Piscic 364:28–38
Leclerc É, Mailhot Y, Mingelbier M, Bernatchez L (2008) The landscape genetics of yellow perch (Perca flavescens) in a large fluvial ecosystem. Mol Ecol 17:1702–1717. https://doi.org/10.1111/J.1365-294X.2008.03710.X
Legendre P, Fortin M-J, Borcard D (2015) Should the Mantel test be used in spatial analysis? Methods Ecol Evol 6:1239–1247. https://doi.org/10.1111/2041-210x.12425
Lobón-Cerviá J, Sanz N (2017) Brown trout: biology, ecology and management. Wiley, Hoboken
MAGRAMA (2015) Protocolo de caracterización hidromorfológica de masas de agua de la categoría ríos. M-R-HMF-2015 (versión 0 consulta pública), Spain
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197. https://doi.org/10.1016/S0169-5347(03)00008-9
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Massa-Gallucci A, Coscia I, O’Grady M et al (2010) Patterns of genetic structuring in a brown trout (Salmo trutta L.) metapopulation. Conserv Genet 11:1689–1699. https://doi.org/10.1007/s10592-010-0061-4
McGlashan DJ, Hughes JM, Bunn SE (2011) Within-drainage population genetic structure of the freshwater fish Pseudomugil signifer (Pseudomugilidae) in northern Australia. Can J Fish Aquat Sci 58:1842–1852. https://doi.org/10.1139/F01-113
Meldgaard T, Nielsen EE, Loeschcke V (2003) Fragmentation by weirs in a riverine system: a study of genetic variation in time and space among populations of European grayling (Thymallus thymallus) in a Danish river system. Conserv Genet 4:735–747. https://doi.org/10.1023/B:COGE.0000006115.14106.DE
Mills LS, Allendorf FW (1996) The one-migrant-per-generation rule in conservation and management. Conserv Biol 10:1509–1518. https://doi.org/10.1046/J.1523-1739.1996.10061509
Moore JW (2015) Bidirectional connectivity in rivers and implications for watershed stability and management. Can J Fish Aquat Sci. https://doi.org/10.1139/cjfas-2014-0478
Moran P, Garcia-Vazquez E, Pendas AM et al (1993) Status of genetic conservation in salmonid populations from Asturian Rivers (North of Spain). In: Cloud JG, Thorgaard GH (eds) Genetic conservation of salmonid fishes. Springer, Boston, pp 213–218
Morrissey MB, De Kerckhove DT (2009) The maintenance of genetic variation due to asymmetric gene flow in dendritic metapopulations. Am Nat 174:875–889. https://doi.org/10.1086/648311/0
Nevill PG, Robinson TP, Di Virgilio G, Wardell-Johnson G (2019) Beyond isolation by distance: what best explains functional connectivity among populations of three sympatric plant species in an ancient terrestrial island system? Divers Distrib 25:1551–1563. https://doi.org/10.1111/DDI.12959
Neville HM, Dunham JB, Peacock MM (2006) Landscape attributes and life history variability shape genetic structure of trout populations in a stream network. Landsc Ecol 21:901–916. https://doi.org/10.1007/s10980-005-5221-4
Neville HM, Dunham J, Rosenberger A et al (2009) Influences of wildfire, habitat size, and connectivity on trout in headwater streams revealed by patterns of genetic diversity. Trans Am Fish Soc 138:1314–1327. https://doi.org/10.1577/t08-162.1
Newman D, Pilson D (1997) Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchella. Evolution (n y) 51:354–362. https://doi.org/10.1111/j.1558-5646.1997.tb02422.x
Nilsson C, Reidy CA, Dynesius M, Revenga C (2005) Fragmentation and flow regulation of the world’s large river systems. Science 308:405–408. https://doi.org/10.1126/science.1107887
Oksanen J, Blanchet FG, Kindt R et al (2013) vegan: Community ecology package. R package version 2.0-9
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65. https://doi.org/10.1046/j.1365-294X.2004.02008.x
Palstra FP, Ruzzante DE (2008) Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17:3428–3447. https://doi.org/10.1111/j.1365-294X.2008.03842.x
Pavlova A, Beheregaray LB, Coleman R et al (2017) Severe consequences of habitat fragmentation on genetic diversity of an endangered Australian freshwater fish: a call for assisted gene flow. Evol Appl 10:531–550. https://doi.org/10.1111/eva.12484
Paz-Vinas I, Loot G, Stevens VM, Blanchet S (2015) Evolutionary processes driving spatial patterns of intraspecific genetic diversity in river ecosystems. Mol Ecol 24:4586–4604. https://doi.org/10.1111/mec.13345
Peacock MM, Dochtermann NA (2012) Evolutionary potential but not extinction risk of Lahontan cutthroat trout (Oncorhynchus clarkii henshawi) is associated with stream characteristics. Can J Fish Aquat Sci 69:615–626. https://doi.org/10.1139/f2012-006
Peel D, Waples RS, Macbeth GM et al (2013) Accounting for missing data in the estimation of contemporary genetic effective population size (Ne). Mol Ecol Resour 13:243–253. https://doi.org/10.1111/1755-0998.12049
Peery MZ, Kirby R, Reid BN et al (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418. https://doi.org/10.1111/j.1365-294X.2012.05635.x
Perrier C, Guyomard R, Bagliniere JL, Evanno G (2011) Determinants of hierarchical genetic structure in Atlantic salmon populations: environmental factors vs. anthropogenic influences. Mol Ecol 20:4231–4245. https://doi.org/10.1111/J.1365-294X.2011.05266.X
Perrier C, April J, Cote G et al (2016) Effective number of breeders in relation to census size as management tools for Atlantic salmon conservation in a context of stocked populations. Conserv Genet 17:31–44. https://doi.org/10.1007/s10592-015-0758-5
Pilger TJ, Gido KB, Propst DL et al (2017) River network architecture, genetic effective size and distributional patterns predict differences in genetic structure across species in a dryland stream fish community. Mol Ecol 26:2687–2697. https://doi.org/10.1111/mec.14079
Piry S, Luikart G, Cornuet JM (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90:502–503. https://doi.org/10.1093/jhered/90.4.502
Piry S, Alapetite A, Cornuet J-M et al (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Poff NL, Hart DD (2002) How dams vary and why it matters for the emerging science of dam removal. Bioscience 52:659–668. https://doi.org/10.1641/0006-3568(2002)052[0659:HDVAWI]2.0.CO;2
Primmer CR, Veselov AJ, Zubchenko A et al (2006) Isolation by distance within a river system: genetic population structuring of Atlantic salmon, Salmo salar, in tributaries of the Varzuga River in northwest Russia. Mol Ecol 15:653–666. https://doi.org/10.1111/J.1365-294X.2005.02844.X
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Core Team (2020) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rahel FJ (2007) Biogeographic barriers, connectivity and homogenization of freshwater faunas: it’s a small world after all. Freshw Biol 52:696–710. https://doi.org/10.1111/j.1365-2427.2006.01708.x
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201
Rieman B, Lee D, McIntyre J et al (1993) Consideration of extinction risks for salmonids. Fish Habitat Relationships Tech Bull 14 Boise, Idaho: USDA Forest Service Intermountain Research Station, pp 1–12
Rodríguez MA (2002) Restricted movement in stream fish: the paradigm is incomplete, not lost. Ecology 83:1–13. https://doi.org/10.1890/0012-9658(2002)083[0001:RMISFT]2.0.CO;2
Rosenberg NA (2004) distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Ruzzante DE, McCracken GR, Parmelee S et al (2016) Effective number of breeders, effective population size and their relationship with census size in an iteroparous species, Salvelinus fontinalis. Proc R Soc B Biol Sci 283:20152601. https://doi.org/10.1098/rspb.2015.2601
Salisbury SJ, McCracken GR, Keefe D et al (2016) A portrait of a sucker using landscape genetics: how colonization and life history undermine the idealized dendritic metapopulation. Mol Ecol 25:4126–4145. https://doi.org/10.1111/mec.13757
Sánchez-Hernández J (2016) Do age-related changes in feeding habits of brown trout alter structural properties of food webs? Aquat Ecol 50:685–695. https://doi.org/10.1007/s10452-016-9586-z
Sánchez-Hernández J, Servia MJ, Vieira R et al (2011) References data on the growth and population parameters of brown trout in siliceous rivers of Galicia (NW Spain). Limnetica 29:273–288. https://doi.org/10.23818/limn.31.24
Sheer MB, Steel EA (2006) Lost watersheds: barriers, aquatic habitat connectivity, and salmon persistence in the Willamette and Lower Columbia River Basins. Trans Am Fish Soc 135:1654–1669. https://doi.org/10.1577/T05-221.1
Smouse PE, Long JC, Sokal RR (1986) Multiple regression and correlation extensions of the mantel test of matrix correspondence. Syst Zool 35:627–632. https://doi.org/10.2307/2413122
Sotola VA, Schrey AW, Ragsdale AK et al (2017) Genetic evidence of isolation by distance and impact of impoundments on genetic diversity of riverine channel catfish. Trans Am Fish Soc 146:1204–1211. https://doi.org/10.1080/00028487.2017.1362471
Stelkens RB, Jaffuel G, Escher M, Wedekind C (2012) Genetic and phenotypic population divergence on a microgeographic scale in brown trout. Mol Ecol 21:2896–2915. https://doi.org/10.1111/j.1365-294X.2012.05581.x
Tischendorf L, Fahrig L, Tischendorf L, Fahrig L (2000) On the usage and measurement of landscape connectivity. Oikos 90:7–19. https://doi.org/10.1034/J.1600-0706.2000.900102.X
Tonkin JD, Altermatt F, Finn DS et al (2018) The role of dispersal in river network metacommunities: patterns, processes, and pathways. Freshw Biol 63:141–163. https://doi.org/10.1111/fwb.13037
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci*. Conserv Genet 7:167. https://doi.org/10.1007/s10592-005-9100-y
Waples RS, Do CHI (2008) ldne: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756. https://doi.org/10.1111/j.1755-0998.2007.02061.x
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262. https://doi.org/10.1111/J.1752-4571.2009.00104.X
Waples RS, Antao T, Luikart G (2014) Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 197:769–780. https://doi.org/10.1534/genetics.114.164822
Ward JV (1989) The four-dimensional nature of lotic ecosystems. J N Am Benthol Soc 8:2–8. https://doi.org/10.2307/1467397
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution (n y) 38:1358–1370. https://doi.org/10.2307/2408641
White SL (2019) The significance of intraspecific diversity for the conservation of an imperiled fish species. The Pennsylvania State University, State College
Whiteley AR, Coombs JA, Hudy M et al (2013) Fragmentation and patch size shape genetic structure of brook trout populations. Can J Fish Aquat Sci 70:678–688. https://doi.org/10.1139/CJFAS-2012-0493
Winans GA, Gayeski N, Timmins-Schiffman E (2015) All dam-affected trout populations are not alike: fine scale geographic variability in resident rainbow trout in Icicle Creek, WA, USA. Conserv Genet 16:301–315. https://doi.org/10.1007/S10592-014-0659-Z
Wofford JEB, Gresswell RE, Banks MA (2005) Influence of barriers to movement on within-watershed genetic variation of coastal cutthroat trout. Ecol Appl 15:628–637. https://doi.org/10.1890/04-0095
Wright S (1943) Isolation by distance. Genetics 28:114–138
Yamamoto S, Morita K, Koizumi I, Maekawa K (2004) Genetic differentiation of white-spotted charr (Salvelinus leucomaenis) populations after habitat fragmentation: spatial-temporal changes in gene frequencies. Conserv Genet 5:529–538. https://doi.org/10.1023/B:COGE.0000041029.38961.A0
Acknowledgements
This study was partly funded by the Spanish Ministry of Economy and Competitiveness as part of the RIVERLANDS project (Ref: BIA-2012-33572). A.M.G.F. was supported by a predoctoral research grant (Ref: BES-2013-065770) from the Spanish Ministry of Economy and Competitiveness. To carry out the study, biological field sampling permits were requested from the regional governments of the study area and the Picos de Europa National Park, and the corresponding authorization was received. We would like to thank the Government of Cantabria and Government of Principado de Asturias for providing useful information, and we thank all the people involved in the field data collection. Finally, we would like to acknowledge the Interautonomic Consortium of the Picos de Europa National Park and the Biodiversity Foundation from the Ministry of Agriculture, Food and Environment for their advice and project support.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This study was partly funded by the Spanish Ministry of Economy and Competitiveness as part of the RIVERLANDS project (Ref: BIA-2012–33572). AMGF was supported by a predoctoral research grant (Ref: BES-2013–065770) from the Spanish Ministry of Economy and Competitiveness.
Author information
Authors and Affiliations
Contributions
All authors conceived the ideas and designed the methodology. AMGF and JB collected the data. SL, AA and AMGF analyzed the data. AMGF led the writing of the manuscript with input from all coauthors. All authors contributed critically to the drafts and gave final approval for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
González-Ferreras, A.M., Leal, S., Barquín, J. et al. Patterns of genetic diversity of brown trout in a northern Spanish catchment linked to structural connectivity. Aquat Sci 84, 48 (2022). https://doi.org/10.1007/s00027-022-00877-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00027-022-00877-0